
ATM-Dependent Phosphorylation of All Three Members of the MRN Complex: From Sensor to Adaptor


Abstract
:1. Introduction
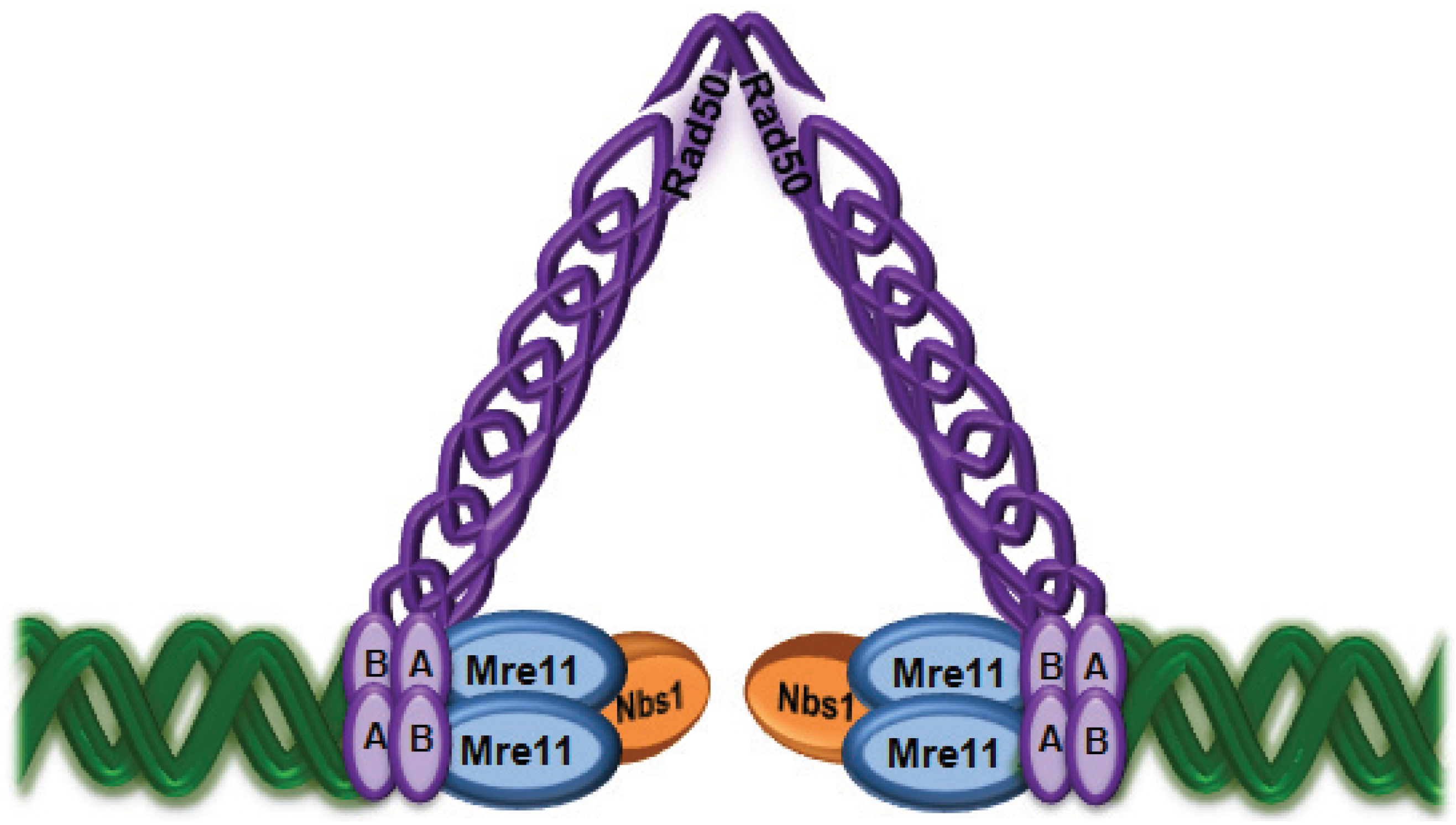
2. Sensing DNA Double Strand Breaks

{kind=link}
{kind=link}
{kind=link}
{kind=link}
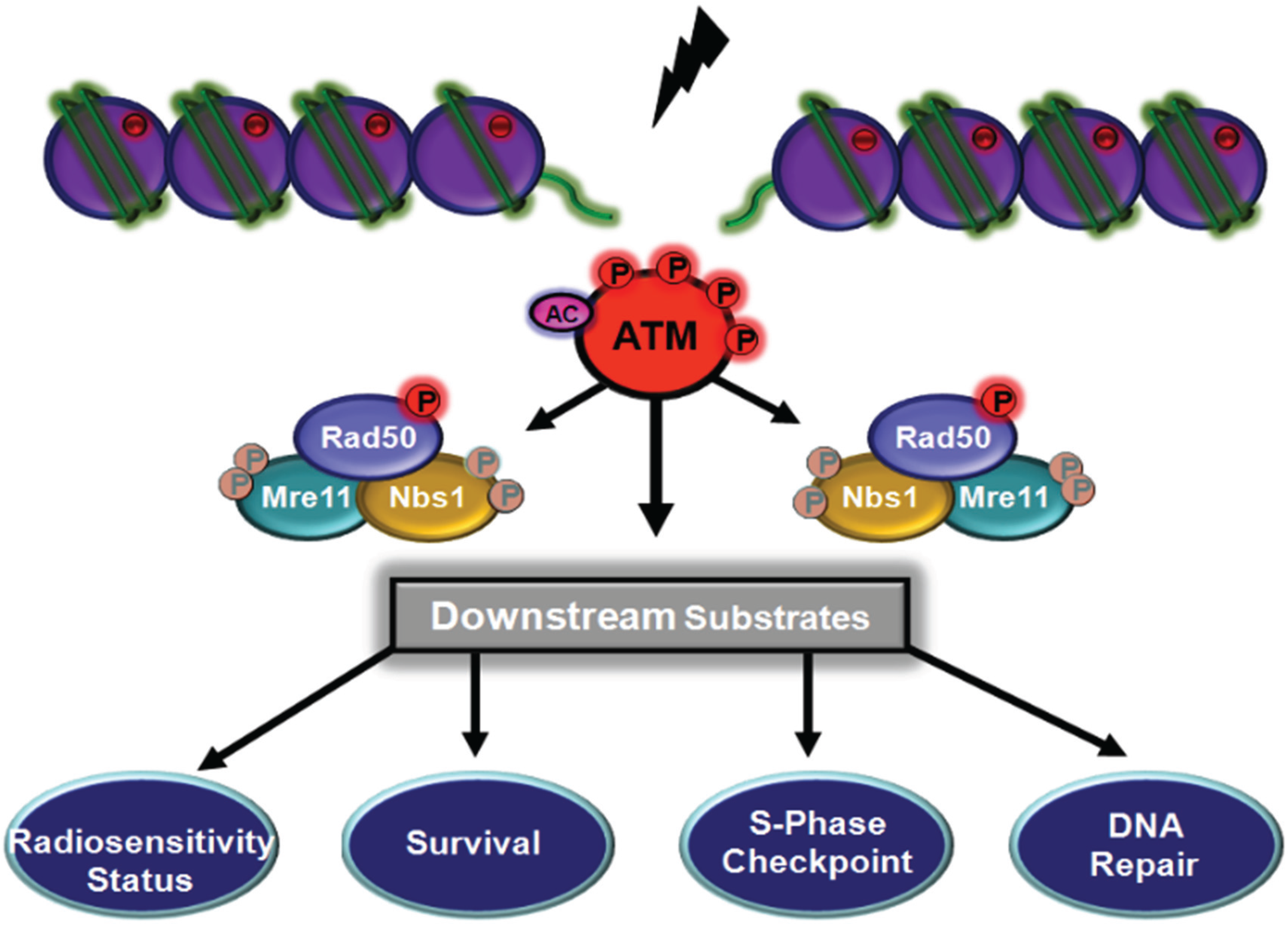{kind=link}
{kind=link}
{kind=link}
| Genetic disorder | Abbreviation | Gene/Protein | Neurological features | Cancer pre-disposition | Chromosomal instability | Radio-sensitivity | Cell cycle checkpoint defect | DNA DSB repair defect |
|---|---|---|---|---|---|---|---|---|
| Nijmegen breakage syndrome | NBS | NBN/Nbs1 | Microcephaly; Mental retardation | Yes | Yes | ++++ | Yes | Yes |
| Nijmegen breakage-like disorder | NBLD | Rad50 | Microcephaly; Mental retardation | ? | Yes | ++ | Yes | Yes |
| Ataxia-telangiectasia-like disorder | ATLD | Mre11 | Cerebellar ataxia; Ocular apraxia; microcephaly (2 patients) | Yes | Yes | ++ | Yes | Yes |
| Ataxia telangiectasia | A-T | ATM | Cerebellar ataxia; Ocular apraxia | Yes | Yes | ++++ | Yes | Yes |

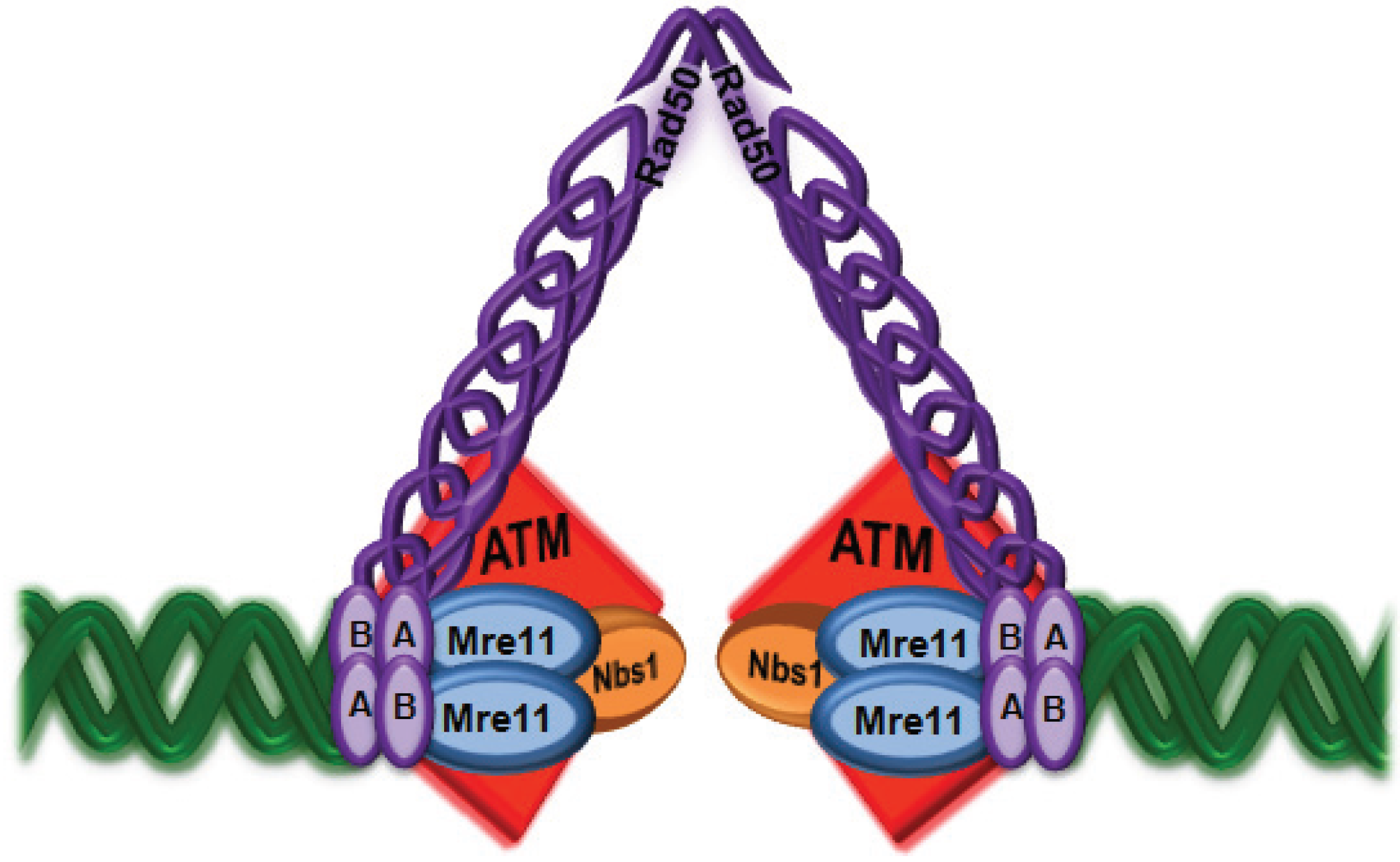
3. Importance of the MRN Complex in ATM Activation

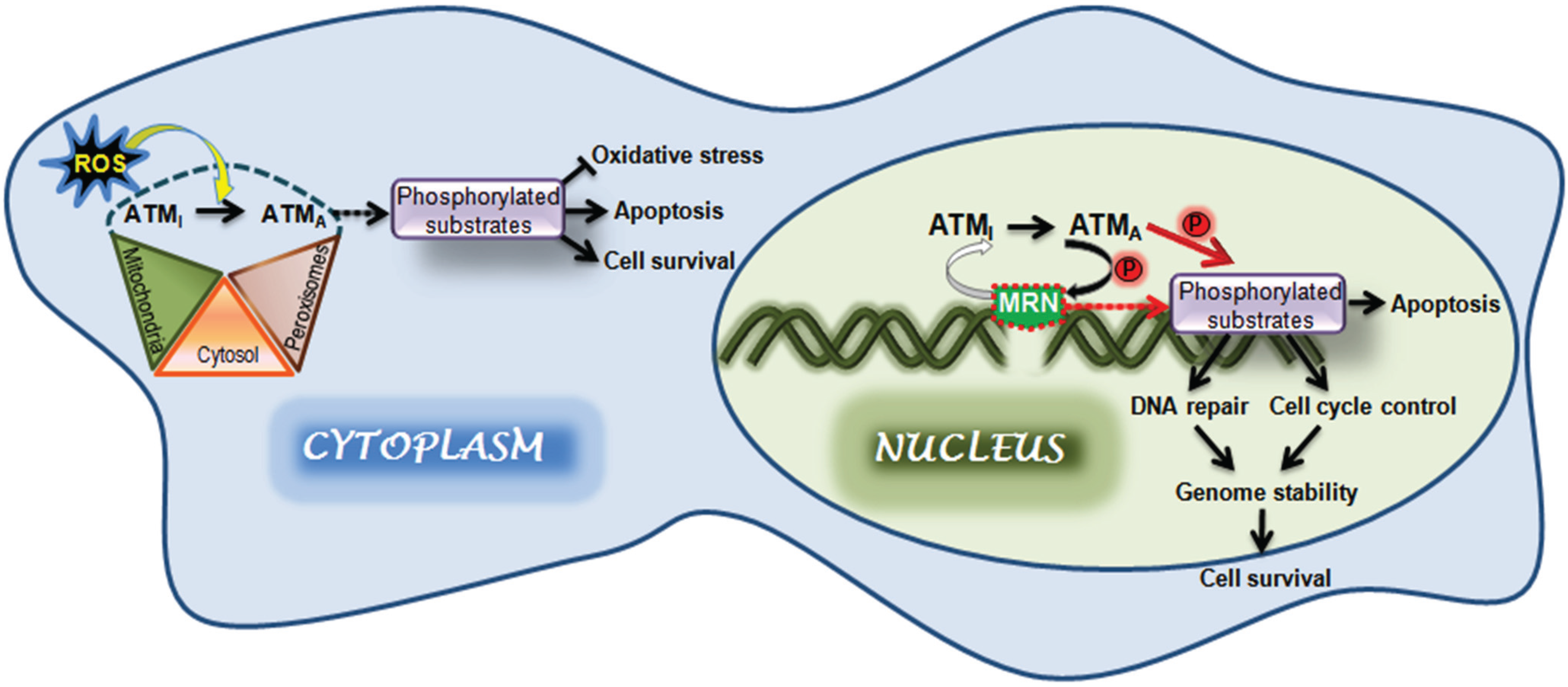
4. ATM Activation by Oxidative Stress
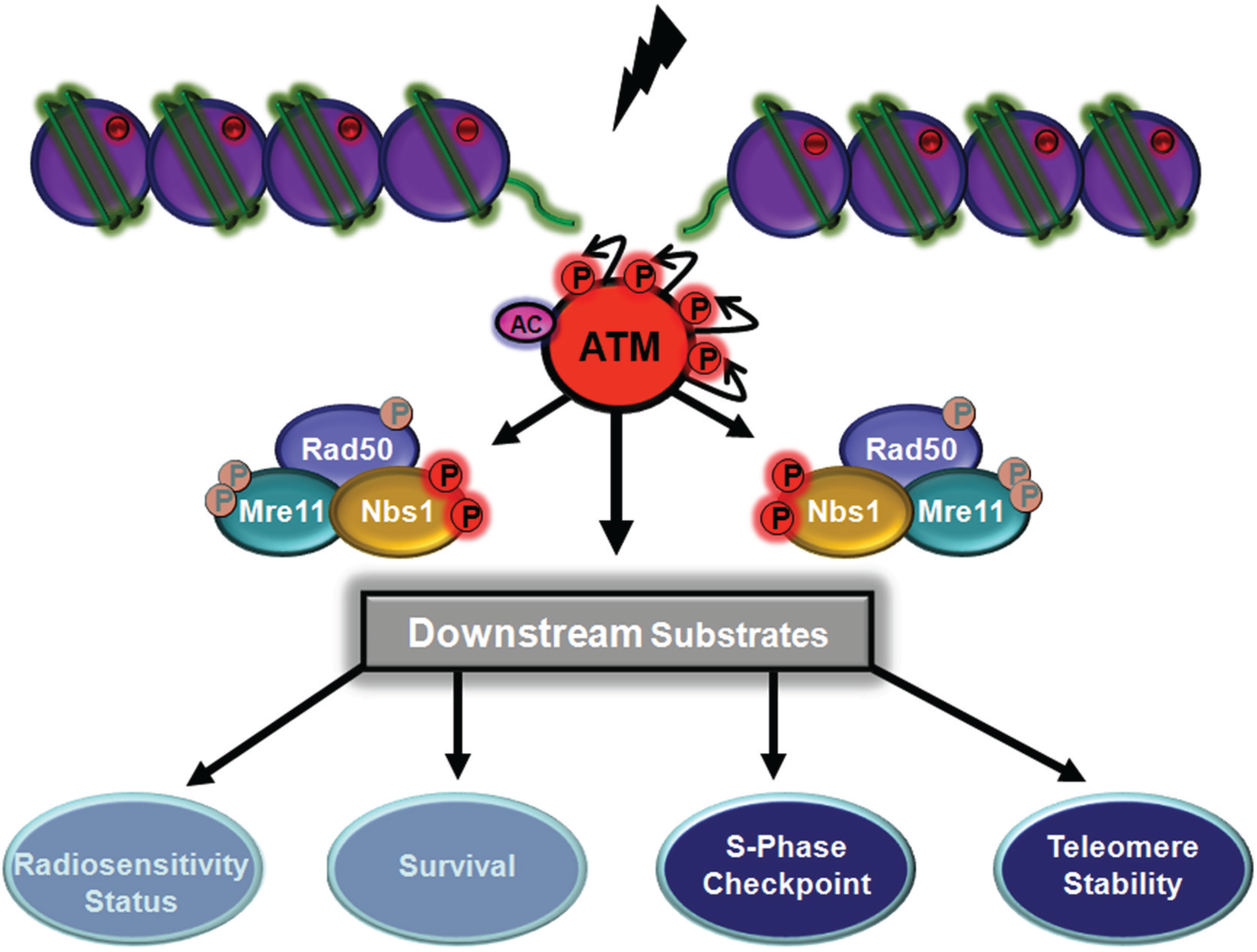
5. Functional role of MRN Complex Members as Substrates for ATM

5.1. ATM-Dependent Signalling by Nbs1 Phosphorylation
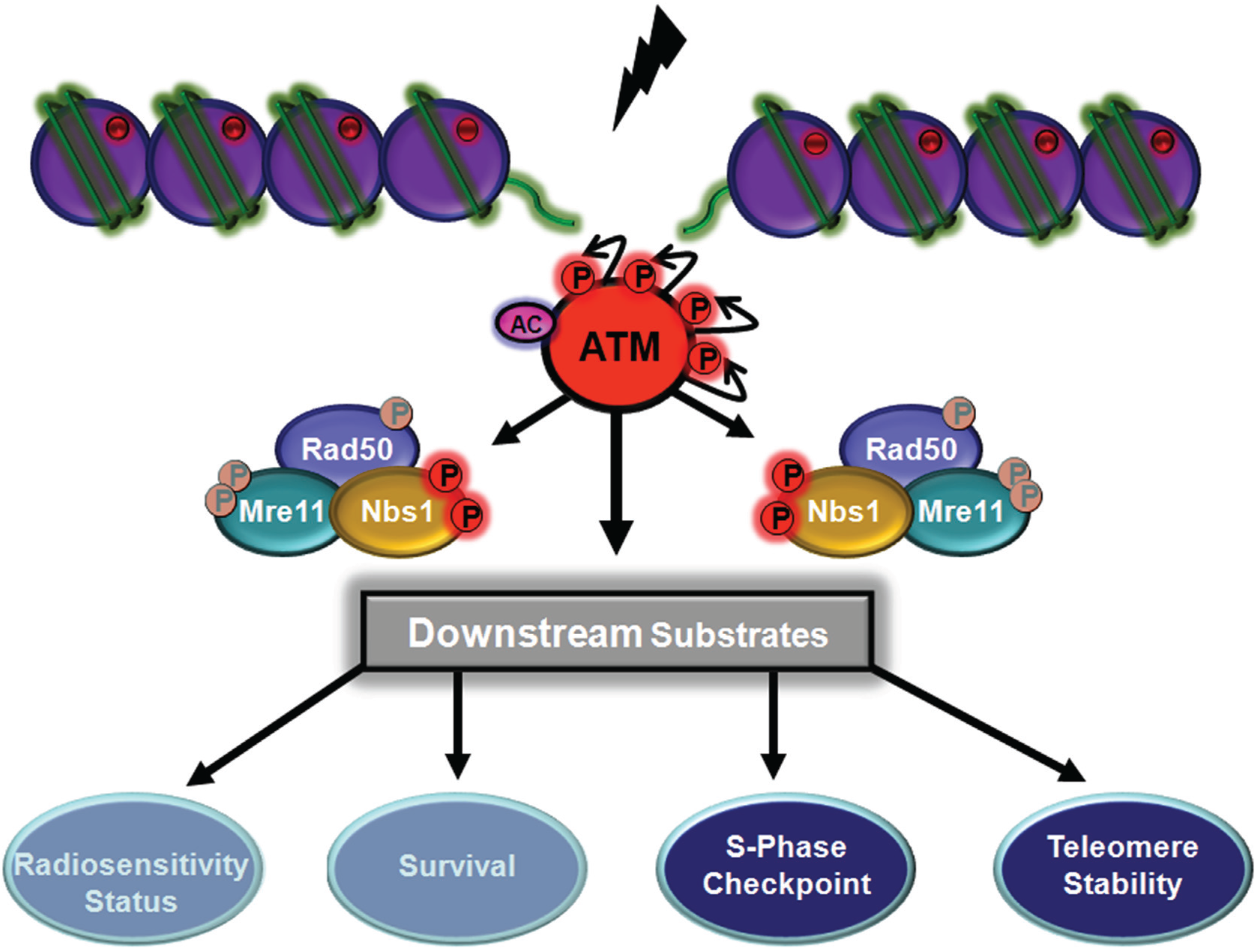
5.2. ATM-Dependent Signalling Through Rad50

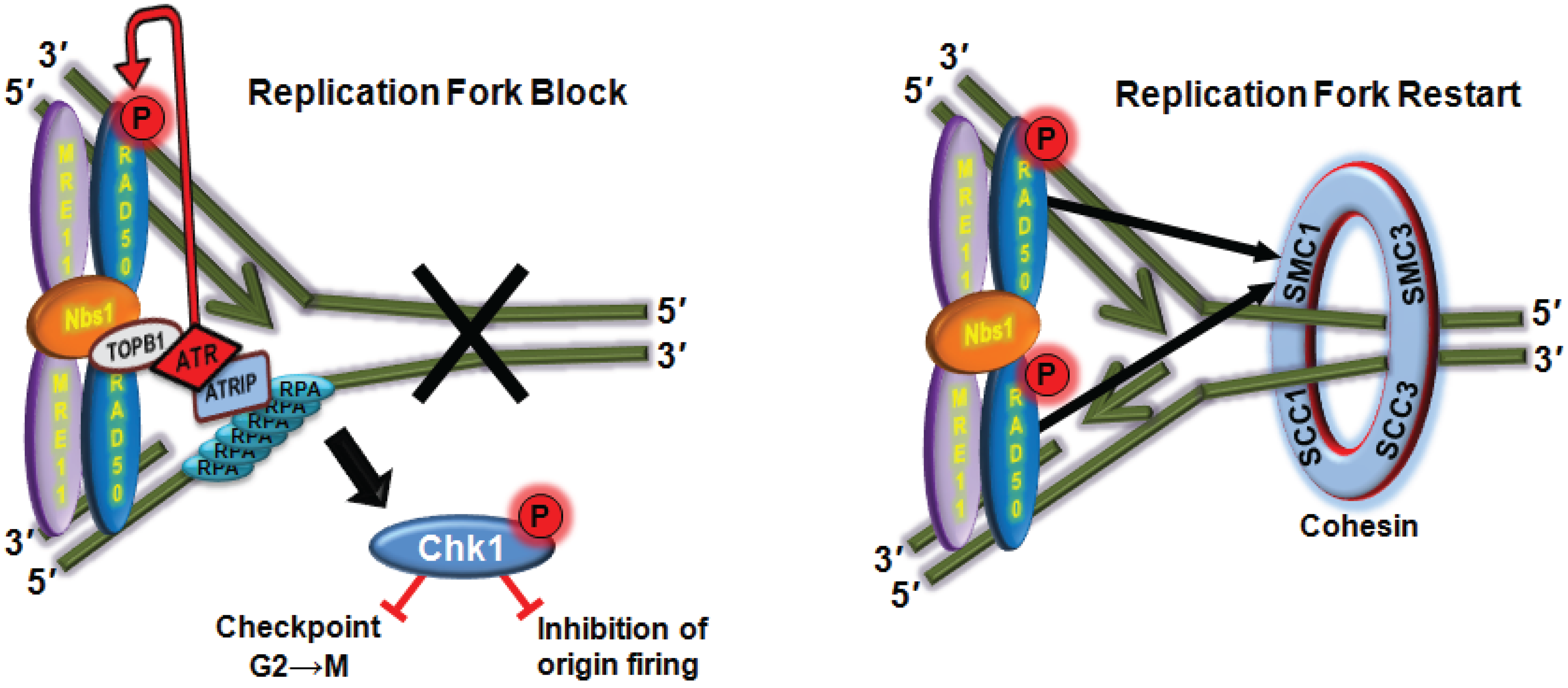
5.3. ATR-Dependent Signalling Through Rad50

5.4. ATM-Dependent Phosphorylation of Mre11 Controls Resection

6. Conclusions
| Member of complex | Number of sites | Consensus sequence | Phosphorylation Site | DNA damaging agent | Functional role |
|---|---|---|---|---|---|
| Nbs1 | 2 | SQ | S278, S343 | Irradiation | S-phase checkpoint |
| Mre11 | 2 | SQ | S676, S678 | Irradiation; Camptothecin; Etoposide | HDR repair (extent of resection); Survival |
| Rad50 | 1 | SQ | S635 | Irradiation; Ultraviolet; Hydroxyurea | DNA repair (HR and NHEJ); Survival; S-phase checkpoint |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rodriguez, Y.; Hinz, J.M.; Smerdon, M.J. Accessing DNA damage in chromatin: Preparing the chromatin landscape for base excision repair. DNA Repair 2015, 32, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Brickner, J.R.; Majid, M.C.; Mosammaparast, N. Crosstalk between ubiquitin and other post-translational modifications on chromatin during double-strand break repair. Trends Cell Biol. 2014, 24, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, A.T.; Milner, A.M.; Gordon, D.G.; Schwartz, J.L. Interaction between ionizing radiation and supercoiled DNA within human tumor cells. Cancer Res. 1991, 51, 3857–3861. [Google Scholar] [PubMed]
- Ahel, D.; Horejsi, Z.; Wiechens, N.; Polo, S.E.; Garcia-Wilson, E.; Ahel, I.; Flynn, H.; Skehel, M.; West, S.C.; Jackson, S.P.; et al. Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science 2009, 325, 1240–1243. [Google Scholar] [CrossRef] [PubMed]
- Gottschalk, A.J.; Timinszky, G.; Kong, S.E.; Jin, J.; Cai, Y.; Swanson, S.K.; Washburn, M.P.; Florens, L.; Ladurner, A.G.; Conaway, J.W.; et al. Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler. Proc. Natl. Acad. Sci. USA 2009, 106, 13770–13774. [Google Scholar] [CrossRef] [PubMed]
- Larsen, D.H.; Poinsignon, C.; Gudjonsson, T.; Dinant, C.; Payne, M.R.; Hari, F.J.; Danielsen, J.M.R.; Menard, P.; Sand, J.C.; Stucki, M.; et al. The chromatin-remodeling factor CHD4 coordinates signaling and repair after DNA damage. J. Cell Biol. 2010, 190, 731–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukas, J.; Lukas, C.; Bartek, J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat. Cell Biol. 2011, 13, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Kakarougkas, A.; Ismail, A.; Chambers, A.L.; Riballo, E.; Herbert, A.D.; Kunzel, J.; Lobrich, M.; Jeggo, P.A.; Downs, J.A. Requirement for PBAF in transcriptional repression and repair at DNA breaks in actively transcribed regions of chromatin. Mol. Cell 2014, 55, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Shanbhag, N.M.; Rafalska-Metcalf, I.U.; Balane-Bolivar, C.; Janicki, S.M.; Greenberg, R.A. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 2010, 141, 970–981. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Celeste, A.; Fernandez-Capetillo, O.; Kruhlak, M.J.; Pilch, D.R.; Staudt, D.W.; Lee, A.; Bonner, R.F.; Bonner, W.M.; Nussenzweig, A. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat. Cell Biol. 2003, 5, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Cook, P.J.; Ju, B.G.; Telese, F.; Wang, X.; Glass, C.K.; Rosenfeld, M.G. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature 2009, 458, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Xiao, A.; Li, H.; Shechter, D.; Ahn, S.H.; Fabrizio, L.A.; Erdjument-Bromage, H.; Ishibe-Murakami, S.; Wang, B.; Tempst, P.; Hofmann, K.; et al. Wstf regulates the H2AX DNA damage response via a novel tyrosine kinase activity. Nature 2009, 457, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Polo, S.E.; Jackson, S.P. Dynamics of DNA damage response proteins at DNA breaks: A focus on protein modifications. Genes Dev. 2011, 25, 409–433. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y. ATM: Expanding roles as a chief guardian of genome stability. Exp. Cell. Res. 2014, 329, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Savic, V.; Yin, B.; Maas, N.L.; Bredemeyer, A.L.; Carpenter, A.C.; Helmink, B.A.; Yang-Iott, K.S.; Sleckman, B.P.; Bassing, C.H. Formation of dynamic gamma-H2AX domains along broken DNA strands is distinctly regulated by ATM and MDC1 and dependent upon H2AX densities in chromatin. Mol. Cell 2009, 34, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jiang, X.; Price, B.D. Tip60: Connecting chromatin to DNA damage signaling. Cell Cycle 2010, 9, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Ayrapetov, M.K.; Gursoy-Yuzugullu, O.; Xu, C.; Xu, Y.; Price, B.D. DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proc. Natl. Acad. Sci. USA 2014, 111, 9169–9174. [Google Scholar] [CrossRef] [PubMed]
- Huyen, Y.; Zgheib, O.; Ditullio, R.A., Jr.; Gorgoulis, V.G.; Zacharatos, P.; Petty, T.J.; Sheston, E.A.; Mellert, H.S.; Stavridi, E.S.; Halazonetis, T.D. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature 2004, 432, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Pei, H.; Zhang, L.; Luo, K.; Qin, Y.; Chesi, M.; Fei, F.; Bergsagel, P.L.; Wang, L.; You, Z.; Lou, Z. Mmset regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature 2011, 470, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Bekker-Jensen, S.; Mailand, N. The ubiquitin- and sumo-dependent signaling response to DNA double-strand breaks. FEBS Lett. 2011, 585, 2914–2919. [Google Scholar] [CrossRef] [PubMed]
- Mailand, N.; Bekker-Jensen, S.; Faustrup, H.; Melander, F.; Bartek, J.; Lukas, C.; Lukas, J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 2007, 131, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.S.; Jackson, S.P. Ubiquitylation, neddylation and the DNA damage response. Open Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, A.A.; Jeggo, P.; Lobrich, M. The influence of heterochromatin on DNA double strand break repair: Getting the strong, silent type to relax. DNA Repair 2010, 9, 1273–1282. [Google Scholar] [CrossRef] [PubMed]
- Cejka, P. DNA end resection: Nucleases team up with the right partners to initiate homologous recombination. J. Biol. Chem. 2015. [Google Scholar] [CrossRef] [PubMed]
- Truong, L.N.; Li, Y.; Shi, L.Z.; Hwang, P.Y.; He, J.; Wang, H.; Razavian, N.; Berns, M.W.; Wu, X. Microhomology-mediated end joining and homologous recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, 7720–7725. [Google Scholar] [CrossRef] [PubMed]
- Merkle, D.; Douglas, P.; Moorhead, G.B.; Leonenko, Z.; Yu, Y.; Cramb, D.; Bazett-Jones, D.P.; Lees-Miller, S.P. The DNA-dependent protein kinase interacts with DNA to form a protein-DNA complex that is disrupted by phosphorylation. Biochemistry 2002, 41, 12706–12714. [Google Scholar] [CrossRef] [PubMed]
- Ochi, T.; Blackford, A.N.; Coates, J.; Jhujh, S.; Mehmood, S.; Tamura, N.; Travers, J.; Wu, Q.; Draviam, V.M.; Robinson, C.V.; et al. PAXX, a paralog of XRCC4 and Xlf, interacts with Ku to promote DNA double-strand break repair. Science 2015, 347, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Paull, T.T. DNA-dependent protein kinase regulates DNA end resection in concert with mre11-rad50-nbs1 (MRN) and ataxia telangiectasia-mutated (ATM). J. Biol. Chem. 2013, 288, 37112–37125. [Google Scholar] [CrossRef] [PubMed]
- Richard, D.J.; Cubeddu, L.; Urquhart, A.J.; Bain, A.; Bolderson, E.; Menon, D.; White, M.F.; Khanna, K.K. hSSB1 interacts directly with the mrn complex stimulating its recruitment to DNA double-strand breaks and its endo-nuclease activity. Nucleic Acids Res. 2011, 39, 3643–3651. [Google Scholar] [CrossRef] [PubMed]
- Falck, J.; Forment, J.V.; Coates, J.; Mistrik, M.; Lukas, J.; Bartek, J.; Jackson, S.P. CDK targeting of nbs1 promotes DNA-end resection, replication restart and homologous recombination. EMBO Rep. 2012, 13, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Clouaire, T.; Legube, G. DNA double strand break repair pathway choice: A chromatin based decision? Nucleus 2015, 6, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Sarangi, P.; Steinacher, R.; Altmannova, V.; Fu, Q.; Paull, T.T.; Krejci, L.; Whitby, M.C.; Zhao, X. Sumoylation influences DNA break repair partly by increasing the solubility of a conserved end resection protein. PLoS Genet. 2015. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Hunt, C.R.; Hegde, M.L.; Chakraborty, S.; Chakraborty, S.; Udayakumar, D.; Horikoshi, N.; Singh, M.; Ramnarain, D.B.; Hittelman, W.N.; et al. Mof phosphorylation by ATM regulates 53BP1-mediated double-strand break repair pathway choice. Cell Rep. 2014, 8, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Escribano-Diaz, C.; Orthwein, A.; Fradet-Turcotte, A.; Xing, M.; Young, J.T.; Tkac, J.; Cook, M.A.; Rosebrock, A.P.; Munro, M.; Canny, M.D.; et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. cell 2013, 49, 872–883. [Google Scholar] [CrossRef] [PubMed]
- D’Amours, D.; Jackson, S.P. The Mre11 complex: At the crossroads of DNA repair and checkpoint signalling. Nat. Rev. Mol. Cell Biol. 2002, 3, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Van den Bosch, M.; Bree, R.T.; Lowndes, N.F. The mrn complex: Coordinating and mediating the response to broken chromosomes. EMBO Rep. 2003, 4, 844–849. [Google Scholar] [CrossRef] [PubMed]
- Waltes, R.; Kalb, R.; Gatei, M.; Kijas, A.W.; Stumm, M.; Sobeck, A.; Wieland, B.; Varon, R.; Lerenthal, Y.; Lavin, M.F.; et al. Human RAD50 deficiency in a nijmegen breakage syndrome-like disorder. Am. J. Hum. Genet. 2009, 84, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.S.; Maser, R.S.; Stankovic, T.; Bressan, D.A.; Kaplan, M.I.; Jaspers, N.G.; Raams, A.; Byrd, P.J.; Petrini, J.H.; Taylor, A.M. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell 1999, 99, 577–587. [Google Scholar] [CrossRef]
- Carney, J.P.; Maser, R.S.; Olivares, H.; Davis, E.M.; le Beau, M.; Yates, J.R., 3rd; Hays, L.; Morgan, W.F.; Petrini, J.H. The hMRE11/hRAD50 protein complex and Nijmegen breakage syndrome: Linkage of double-strand break repair to the cellular DNA damage response. Cell 1998, 93, 477–486. [Google Scholar] [CrossRef]
- Hohl, M.; Kochanczyk, T.; Tous, C.; Aguilera, A.; Krezel, A.; Petrini, J.H. Interdependence of the RAD50 hook and globular domain functions. Mol. Cell 2015, 57, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Lafrance-Vanasse, J.; Williams, G.J.; Tainer, J.A. Envisioning the dynamics and flexibility of MRE11-RAD50-NBS1 complex to decipher its roles in DNA replication and repair. Prog. Biophys. Mol. Biol. 2015, 117, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, R.A.; Williams, G.J.; Limbo, O.; Williams, R.S.; Kuhnlein, J.; Lee, J.H.; Classen, S.; Guenther, G.; Russell, P.; Tainer, J.A.; et al. ATP-driven RAD50 conformations regulate DNA tethering, end resection, and ATM checkpoint signaling. EMBO J. 2014, 33, 482–500. [Google Scholar] [CrossRef] [PubMed]
- Buis, J.; Wu, Y.; Deng, Y.; Leddon, J.; Westfield, G.; Eckersdorff, M.; Sekiguchi, J.M.; Chang, S.; Ferguson, D.O. MRE11 nuclease activity has essential roles in DNA repair and genomic stability distinct from ATM activation. Cell 2008, 135, 85–96. [Google Scholar] [PubMed]
- Lee, J.H.; Mand, M.R.; Deshpande, R.A.; Kinoshita, E.; Yang, S.H.; Wyman, C.; Paull, T.T. Ataxia telangiectasia-mutated (ATM) kinase activity is regulated by ATP-driven conformational changes in the MRE11/RAD50/NBS1 (MRN) complex. J. Biol. Chem. 2013, 288, 12840–12851. [Google Scholar] [CrossRef] [PubMed]
- Paull, T.T. Mechanisms of ATM activation. Annu. Rev. Biochem. 2015, 84, 711–738. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.F. Ataxia-telangiectasia: From a rare disorder to a paradigm for cell signalling and cancer. Nat. Rev. Mol. Cell Biol. 2008, 9, 759–769. [Google Scholar] [PubMed]
- Mirzoeva, O.K.; Petrini, J.H. DNA damage-dependent nuclear dynamics of the MRE11 complex. Mol. Cell. Biol. 2001, 21, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Uziel, T.; Lerenthal, Y.; Moyal, L.; Andegeko, Y.; Mittelman, L.; Shiloh, Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003, 22, 5612–5621. [Google Scholar] [CrossRef] [PubMed]
- Carson, C.T.; Schwartz, R.A.; Stracker, T.H.; Lilley, C.E.; Lee, D.V.; Weitzman, M.D. The MRE11 complex is required for ATM activation and the G2/M checkpoint. EMBO J. 2003, 22, 6610–6620. [Google Scholar] [CrossRef] [PubMed]
- Watters, D.; Kedar, P.; Spring, K.; Bjorkman, J.; Chen, P.; Gatei, M.; Birrell, G.; Garrone, B.; Srinivasa, P.; Crane, D.I.; et al. Localization of a portion of extranuclear ATM to peroxisomes. J. Biol. Chem. 1999, 274, 34277–34282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valentin-Vega, Y.A.; Kastan, M.B. A new role for ATM: Regulating mitochondrial function and mitophagy. Autophagy 2012, 8, 840–841. [Google Scholar] [CrossRef] [PubMed]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, S.V.; Graham, M.E.; Peng, C.; Chen, P.; Robinson, P.J.; Lavin, M.F. Involvement of novel autophosphorylation sites in ATM activation. EMBO J. 2006, 25, 3504–3514. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, S.V.; Graham, M.E.; Jakob, B.; Tobias, F.; Kijas, A.W.; Tanuji, M.; Chen, P.; Robinson, P.J.; Taucher-Scholz, G.; Suzuki, K.; et al. Autophosphorylation and ATM activation: Additional sites add to the complexity. J. Biol. Chem. 2011, 286, 9107–9119. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, M.; Celeste, A.; Difilippantonio, S.; Guo, R.; Wang, W.; Feigenbaum, L.; Nussenzweig, A. Autophosphorylation at serine 1987 is dispensable for murine ATM activation in vivo. Nature 2006, 443, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Daniel, J.A.; Pellegrini, M.; Lee, J.H.; Paull, T.T.; Feigenbaum, L.; Nussenzweig, A. Multiple autophosphorylation sites are dispensable for murine ATM activation in vivo. J. Biol. Chem. 2008, 183, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Dupre, A.; Boyer-Chatenet, L.; Gautier, J. Two-step activation of ATM by DNA and the MRE11-RAD50-NBS1 complex. Nat. Struct. Mol. Biol. 2006, 13, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Paull, T.T. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 2007, 26, 7741–7748. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Xu, Y.; Roy, K.; Price, B.D. DNA damage-induced acetylation of lysine 3016 of ATM activates ATM kinase activity. Mol. Cell. Biol. 2007, 27, 8502–8509. [Google Scholar] [CrossRef] [PubMed]
- Kaidi, A.; Jackson, S.P. KAT5 tyrosine phosphorylation couples chromatin sensing to ATM signalling. Nature 2013, 498, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Chen, Y.; Lu, L.Y.; Wu, Y.; Paulsen, M.T.; Ljungman, M.; Ferguson, D.O.; Yu, X. Chfr and RNF8 synergistically regulate ATM activation. Nat. Struct. Mol. Biol. 2011, 18, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Tresini, M.; Warmerdam, D.O.; Kolovos, P.; Snijder, L.; Vrouwe, M.G.; Demmers, J.A.; van IJcken, W.F.J.; Grosveld, F.G.; Medema, R.H.; Hoeijmakers, J.H.; et al. The core spliceosome as target and effector of non-canonical ATM signalling. Nature 2015, 523, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, M.; Gatti, R.A. Pathogenesis of ataxia-telangiectasia: The next generation of ATM functions. Blood 2013, 121, 4036–4045. [Google Scholar] [CrossRef] [PubMed]
- Biton, S.; Barzilai, A.; Shiloh, Y. The neurological phenotype of ataxia-telangiectasia: Solving a persistent puzzle. DNA Repair 2008, 7, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.F.; Gueven, N.; Bottle, S.; Gatti, R.A. Current and potential therapeutic strategies for the treatment of ataxia-telangiectasia. Br. Med. Bull. 2007. [Google Scholar] [CrossRef] [PubMed]
- Savitsky, K.; Sfez, S.; Tagle, D.A.; Ziv, Y.; Sartiel, A.; Collins, F.S.; Shiloh, Y.; Rotman, G. The complete sequence of the coding region of the ATM gene reveals similarity to cell cycle regulators in different species. Hum. Mol. Genet. 1995, 4, 2025–2032. [Google Scholar] [CrossRef] [PubMed]
- Rotman, G.; Shiloh, Y. Hypothesis: Ataxia-telangiectasia: Is ATM a sensor of oxidative damage and stress? Cell Mol. Biol. 1997, 19, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Broccoletti, T.; del Giudice, E.; Cirillo, E.; Vigliano, I.; Giardino, G.; Ginocchio, V.M.; Bruscoli, S.; Riccardi, C.; Pignata, C. Efficacy of very-low-dose betamethasone on neurological symptoms in ataxia-telangiectasia. Eur. J. Neurol. 2011, 18, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Broccoletti, T.; del Giudice, E.; Amorosi, S.; Russo, I.; di Bonito, M.; Imperati, F.; Romano, A.; Pignata, C. Steroid-induced improvement of neurological signs in ataxia-telangiectasia patients. Eur. J. Neurol. 2008, 15, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Chessa, L.; Leuzzi, V.; Plebani, A.; Soresina, A.; Micheli, R.; D’Agnano, D.; Venturi, T.; Molinaro, A.; Fazzi, E.; Marini, M.; et al. Intra-erythrocyte infusion of dexamethasone reduces neurological symptoms in ataxia teleangiectasia patients: Results of a phase 2 trial. Orphanet J. Rare Dis. 2014. [Google Scholar] [CrossRef] [PubMed]
- Quarantelli, M.; Giardino, G.; Prinster, A.; Aloj, G.; Carotenuto, B.; Cirillo, E.; Marsili, A.; Salvatore, E.; del Giudice, E.; Pignata, C. Steroid treatment in ataxia-telangiectasia induces alterations of functional magnetic resonance imaging during prono-supination task. Eur. J. Paediatr. Neurol. 2013, 17, 135–140. [Google Scholar] [CrossRef] [PubMed]
- McGrath-Morrow, S.A.; Gower, W.A.; Rothblum-Oviatt, C.; Brody, A.S.; Langston, C.; Fan, L.L.; Lefton-Greif, M.A.; Crawford, T.O.; Troche, M.; Sandlund, J.T.; et al. Evaluation and management of pulmonary disease in ataxia-telangiectasia. Pediatr. Pulm. 2010, 45, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Barzilai, A.; Rotman, G.; Shiloh, Y. ATM deficiency and oxidative stress: A new dimension of defective response to DNA damage. DNA Repair 2002, 1, 3–25. [Google Scholar] [CrossRef]
- Hartlova, A.; Erttmann, S.F.; Raffi, F.A.; Schmalz, A.M.; Resch, U.; Anugula, S.; Lienenklaus, S.; Nilsson, L.M.; Kroger, A.; Nilsson, J.A.; et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor sting to promote anti-microbial innate immunity. Immunity 2015, 42, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Katlinskaya, Y.V.; Carbone, C.J.; Zhao, B.; Katlinski, K.V.; Zheng, H.; Guha, M.; Li, N.; Chen, Q.; Yang, T.; et al. DNA-damage-induced type I interferon promotes senescence and inhibits stem cell function. Cell Rep. 2015, 11, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Barlow, C.; Dennery, P.A.; Shigenaga, M.K.; Smith, M.A.; Morrow, J.D.; Roberts, L.J., 2nd; Wynshaw-Boris, A.; Levine, R.L. Loss of the ataxia-telangiectasia gene product causes oxidative damage in target organs. Proc. Natl. Acad. Sci. USA 1999, 96, 9915–9919. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Peng, C.; Luff, J.; Spring, K.; Watters, D.; Bottle, S.; Furuya, S.; Lavin, M.F. Oxidative stress is responsible for deficient survival and dendritogenesis in purkinje neurons from ataxia-telangiectasia mutated mutant mice. J. Neurosci. 2003, 23, 11453–11460. [Google Scholar] [PubMed]
- Gueven, N.; Luff, J.; Peng, C.; Hosokawa, K.; Bottle, S.E.; Lavin, M.F. Dramatic extension of tumor latency and correction of neurobehavioral phenotype in ATM-mutant mice with a nitroxide antioxidant. Free Radical Biol. Med. 2006, 41, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Schubert, R.; Erker, L.; Barlow, C.; Yakushiji, H.; Larson, D.; Russo, A.; Mitchell, J.B.; Wynshaw-Boris, A. Cancer chemoprevention by the antioxidant tempol in ATM-deficient mice. Hum. Mol. Genet. 2004, 13, 1793–1802. [Google Scholar] [CrossRef] [PubMed]
- Reliene, R.; Schiestl, R.H. Antioxidants suppress lymphoma and increase longevity in ATM-deficient mice. J. Nutr. 2007, 137, S229–S232. [Google Scholar]
- Kuang, X.; Yan, M.; Ajmo, J.M.; Scofield, V.L.; Stoica, G.; Wong, P.K. Activation of amp-activated protein kinase in cerebella of ATM−/− mice is attributable to accumulation of reactive oxygen species. Biochem. Biophys. Res. Commun. 2012, 418, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Quick, K.L.; Dugan, L.L. Superoxide stress identifies neurons at risk in a model of ataxia-telangiectasia. Ann. Neurol. 2001, 49, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Khoronenkova, S.V.; Dianov, G.L. ATM prevents dsb formation by coordinating ssb repair and cell cycle progression. Proc. Natl. Acad. Sci. USA 2015, 112, 3997–4002. [Google Scholar] [CrossRef] [PubMed]
- Shackelford, R.E.; Innes, C.L.; Sieber, S.O.; Heinloth, A.N.; Leadon, S.A.; Paules, R.S. The ataxia telangiectasia gene product is required for oxidative stress-induced G1 and G2 checkpoint function in human fibroblasts. J. Biol. Chem. 2001, 276, 21951–21959. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Deshpande, R.; Paull, T.T. ATM activation in the presence of oxidative stress. Cell cycle 2010, 9, 4805–4811. [Google Scholar] [CrossRef] [PubMed]
- Stamnaes, J.; Pinkas, D.M.; Fleckenstein, B.; Khosla, C.; Sollid, L.M. Redox regulation of transglutaminase 2 activity. J. Biol. Chem. 2010, 285, 25402–25409. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.C.; Hsieh, Y.H.; Su, M.I.; Jiang, X.; Hsu, P.H.; Lo, W.T.; Weng, J.Y.; Jeng, Y.M.; Wang, J.M.; Chen, P.L.; et al. Loss of the oxidative stress sensor NPGPx compromises GRP78 chaperone activity and induces systemic disease. Mol. Cell 2012, 48, 747–759. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Kozlov, S.; Lavin, M.F.; Person, M.D.; Paull, T.T. ATM activation by oxidative stress. Science 2010, 330, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Guo, Z.; Myler, L.R.; Zheng, S.; Paull, T.T. Direct activation of ATM by resveratrol under oxidizing conditions. PLoS ONE 2014. [Google Scholar] [CrossRef] [PubMed]
- Young, D.B.; Jonnalagadda, J.; Gatei, M.; Jans, D.A.; Meyn, S.; Khanna, K.K. Identification of domains of ataxia-telangiectasia mutated required for nuclear localization and chromatin association. J. Biol. Chem. 2005, 280, 27587–27594. [Google Scholar] [CrossRef] [PubMed]
- Bencokova, Z.; Kaufmann, M.R.; Pires, I.M.; Lecane, P.S.; Giaccia, A.J.; Hammond, E.M. ATM activation and signaling under hypoxic conditions. Mol. Cell. Biol. 2009, 29, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tripathi, D.N.; Jing, J.; Alexander, A.; Kim, J.; Powell, R.T.; Dere, R.; Tait-Mulder, J.; Lee, J.H.; Paull, T.T.; et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat. Cell Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Bensimon, A.; Schmidt, A.; Ziv, Y.; Elkon, R.; Wang, S.Y.; Chen, D.J.; Aebersold, R.; Shiloh, Y. ATM-dependent and -independent dynamics of the nuclear phosphoproteome after DNA damage. Sci. Signal 2010. [Google Scholar] [CrossRef] [PubMed]
- Beli, P.; Lukashchuk, N.; Wagner, S.A.; Weinert, B.T.; Olsen, J.V.; Baskcomb, L.; Mann, M.; Jackson, S.P.; Choudhary, C. Proteomic investigations reveal a role for RNA processing factor THRAP3 in the DNA damage response. Mol. Cell 2012, 46, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Han, Y.R.; Plummer, M.R.; Herrup, K. Cytoplasmic ATM in neurons modulates synaptic function. Curr. Biol. 2009, 19, 2091–2096. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Li, L.; Xing, G.; Tian, C.; Yin, Y.; He, F.; Zhang, L. ATM-mediated nusap phosphorylation induces mitotic arrest. Biochem. Biophys. Res. Commun. 2011, 404, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Bensimon, A.; Aebersold, R.; Shiloh, Y. Beyond ATM: The protein kinase landscape of the DNA damage response. FEBS Lett. 2011, 585, 1625–1639. [Google Scholar] [CrossRef] [PubMed]
- Jette, N.; Lees-Miller, S.P. The DNA-dependent protein kinase: A multifunctional protein kinase with roles in DNA double strand break repair and mitosis. Prog. Biophys. Mol. Biol. 2015, 117, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.J.; Chen, B.P.; Chen, D.J. DNA-PK: A dynamic enzyme in a versatile DSB repair pathway. DNA Repair 2014, 17, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A. Role of SMG-1-mediated Upf1 phosphorylation in mammalian nonsense-mediated mRNA decay. Genes cells 2013, 18, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Roberts, T.L.; Richards, R.; Woods, R.; Birrell, G.; Lim, Y.C.; Ohno, S.; Yamashita, A.; Abraham, R.T.; Gueven, N.; et al. A novel role for hSMG-1 in stress granule formation. Mol. Cell. Biol. 2011, 31, 4417–4429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, T.L.; Ho, U.; Luff, J.; Lee, C.S.; Apte, S.H.; MacDonald, K.P.; Raggat, L.J.; Pettit, A.R.; Morrow, C.A.; Waters, M.J.; et al. SMG1 haploinsufficiency predisposes to tumor formation and inflammation. Proc. Natl. Acad. Sci. USA 2013, 110, E285–E294. [Google Scholar] [CrossRef] [PubMed]
- Showkat, M.; Beigh, M.A.; Andrabi, K.I. mTOR signaling in protein translation regulation: Implications in cancer genesis and therapeutic interventions. Biochem. Mol. Biol. Int. 2014. [Google Scholar] [CrossRef] [PubMed]
- Falck, J.; Coates, J.; Jackson, S.P. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005, 434, 605–611. [Google Scholar] [CrossRef] [PubMed]
- You, Z.; Chahwan, C.; Bailis, J.; Hunter, T.; Russell, P. ATM activation and its recruitment to damaged DNA require binding to the C terminus of nbs1. Mol. Cell. Biol. 2005, 25, 5363–5379. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.F.; Kozlov, S. ATM activation and DNA damage response. Cell Cycle 2007, 6, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Weng, Y.C.; Yuan, S.S.; Lin, Y.T.; Hsu, H.C.; Lin, S.C.; Gerbino, E.; Song, M.H.; Zdzienicka, M.Z.; Gatti, R.A.; et al. Functional link between ataxia-telangiectasia and Nijmegen breakage syndrome gene products. Nature 2000, 405, 473–477. [Google Scholar] [PubMed]
- Lim, D.S.; Kim, S.T.; Xu, B.; Maser, R.S.; Lin, J.; Petrini, J.H.; Kastan, M.B. ATM phosphorylates p95/nbs1 in an S-phase checkpoint pathway. Nature 2000, 404, 613–617. [Google Scholar] [PubMed]
- Wu, X.; Ranganathan, V.; Weisman, D.S.; Heine, W.F.; Ciccone, D.N.; O’Neill, T.B.; Crick, K.E.; Pierce, K.A.; Lane, W.S.; Rathbun, G.; et al. ATM phosphorylation of Nijmegen breakage syndrome protein is required in a DNA damage response. Nature 2000, 405, 477–482. [Google Scholar] [PubMed]
- Gatei, M.; Young, D.; Cerosaletti, K.M.; Desai-Mehta, A.; Spring, K.; Kozlov, S.; Lavin, M.F.; Gatti, R.A.; Concannon, P.; Khanna, K. ATM-dependent phosphorylation of nibrin in response to radiation exposure. Nat. Genet. 2000, 25, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Xu, B.; Lee, C.H.; Ahn, J.Y.; Song, M.S.; Lee, H.; Canman, C.E.; Lee, J.S.; Kastan, M.B.; Lim, D.S. Distinct functions of nijmegen breakage syndrome in ataxia telangiectasia mutated-dependent responses to DNA damage. Mol. Cancer Res. 2003, 1, 674–681. [Google Scholar] [PubMed]
- Li, T.; Wang, Z.Q. Point mutation at the Nbs1 threonine 278 site does not affect mouse development, but compromises the Chk2 and SMC1 phosphorylation after DNA damage. Mech. Ageing Dev. 2011, 132, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Huang, M.; Elledge, S.J. Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 1998, 282, 1893–1897. [Google Scholar] [CrossRef] [PubMed]
- Buscemi, G.; Savio, C.; Zannini, L.; Micciche, F.; Masnada, D.; Nakanishi, M.; Tauchi, H.; Komatsu, K.; Mizutani, S.; Khanna, K.; et al. Chk2 activation dependence on Nbs1 after DNA damage. Mol. Cell. Biol. 2001, 21, 5214–5222. [Google Scholar] [CrossRef] [PubMed]
- Yazdi, P.T.; Wang, Y.; Zhao, S.; Patel, N.; Lee, E.Y.; Qin, J. SMC1 is a downstream effector in the ATM/Nbs1 branch of the human S-phase checkpoint. Genes Dev. 2002, 16, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Li, Y.; Mu, J.J.; Zhang, J.; Tonaka, T.; Hamamori, Y.; Jung, S.Y.; Wang, Y.; Qin, J. Regulation of intra-S phase checkpoint by Ionizing Radiation (IR)-dependent and IR-independent phosphorylation of SMC3. J. Biol. Chem. 2008, 283, 19176–19183. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Murnane, J.P. Telomere instability in a human tumor cell line expressing Nbs1 with mutations at sites phosphorylated by ATM. Mol. Cancer Res. 2003, 1, 1058–1069. [Google Scholar] [PubMed]
- Zhang, Y.; Zhou, J.; Lim, C.U. The role of Nbs1 in DNA double strand break repair, telomere stability, and cell cycle checkpoint control. Cell Res. 2006, 16, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, V.; Heine, W.F.; Ciccone, D.N.; Rudolph, K.L.; Wu, X.; Chang, S.; Hai, H.; Ahearn, I.M.; Livingston, D.M.; Resnick, I.; et al. Rescue of a telomere length defect of Nijmegen breakage syndrome cells requires Nbs and telomerase catalytic subunit. Curr. Biol. 2001, 11, 962–966. [Google Scholar] [CrossRef]
- Difilippantonio, S.; Celeste, A.; Kruhlak, M.J.; Lee, Y.; Difilippantonio, M.J.; Feigenbaum, L.; Jackson, S.P.; McKinnon, P.J.; Nussenzweig, A. Distinct domains in Nbs1 regulate irradiation-induced checkpoints and apoptosis. J. Exp. Med. 2007, 204, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Barbi, G.; Scheres, J.M.; Schindler, D.; Taalman, R.D.; Rodens, K.; Mehnert, K.; Muller, M.; Seyschab, H. Chromosome instability and X-ray hypersensitivity in a microcephalic and growth-retarded child. Am. J. Med. Genet 1991, 40, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, E.; van Rossum-Fikkert, S.; Sanchez, H.; Kertokalio, A.; Wyman, C. Human RAD50 makes a functional DNA-binding complex. Biochim. 2015, 113, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Linding, R.; Jensen, L.J.; Ostheimer, G.J.; van Vugt, M.A.; Jorgensen, C.; Miron, I.M.; Diella, F.; Colwill, K.; Taylor, L.; Elder, K.; et al. Systematic discovery of in vivo phosphorylation networks. Cell 2007, 129, 1415–1426. [Google Scholar] [CrossRef] [PubMed]
- Gatei, M.; Jakob, B.; Chen, P.; Kijas, A.W.; Becherel, O.J.; Gueven, N.; Birrell, G.; Lee, J.H.; Paull, T.T.; Lerenthal, Y.; et al. ATM protein-dependent phosphorylation of RAD50 protein regulates DNA repair and cell cycle control. J. Biol. Chem. 2011, 286, 31542–31556. [Google Scholar] [CrossRef] [PubMed]
- Dinkelmann, M.; Spehalski, E.; Stoneham, T.; Buis, J.; Wu, Y.; Sekiguchi, J.M.; Ferguson, D.O. Multiple functions of MRN in end-joining pathways during isotype class switching. Nat. Struct. Mol. Biol. 2009, 16, 808–813. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Nakamura, K.; Taniguchi, Y.; Paull, T.T. Ctp1/CtIP and the mrn complex collaborate in the initial steps of homologous recombination. Mol. Cell 2007, 28, 351–352. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.; Nievera, C.J.; Lee, A.Y.; Chen, L.; Wu, X. The mre11-rad50-nbs1 complex acts both upstream and downstream of ataxia telangiectasia mutated and RAD3-related protein (ATR) to regulate the S-phase checkpoint following UV treatment. Nat. Struct. Mol. Biol. 2007, 282, 22939–22952. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.S.; Cortez, D. Rapid activation of ATR by ionizing radiation requires ATM and MRE11. J. Biol. Chem. 2006, 281, 9346–9350. [Google Scholar] [CrossRef] [PubMed]
- Stiff, T.; Reis, C.; Alderton, G.K.; Woodbine, L.; O’Driscoll, M.; Jeggo, P.A. Nbs1 is required for ATR-dependent phosphorylation events. EMBO J. 2005, 24, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Duursma, A.M.; Driscoll, R.; Elias, J.E.; Cimprich, K.A. A role for the mrn complex in ATR activation via TOPBP1 recruitment. Mol. Cell 2013, 50, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Gatei, M.; Kijas, A.W.; Biard, D.; Dork, T.; Lavin, M.F. RAD50 phosphorylation promotes ATR downstream signaling and DNA restart following replication stress. Hum. Mol. Genet. 2014, 23, 4232–4248. [Google Scholar] [CrossRef] [PubMed]
- Manthey, K.C.; Opiyo, S.; Glanzer, J.G.; Dimitrova, D.; Elliott, J.; Oakley, G.G. Nbs1 mediates ATR-dependent RPA hyperphosphorylation following replication-fork stall and collapse. J. Cell Sci. 2007, 120, 4221–4229. [Google Scholar] [CrossRef] [PubMed]
- Stracker, T.H.; Morales, M.; Couto, S.S.; Hussein, H.; Petrini, J.H. The carboxy terminus of nbs1 is required for induction of apoptosis by the Mre11 complex. Nature 2007, 447, 218–221. [Google Scholar] [CrossRef] [PubMed]
- Symington, L.S. DNA repair: Making the cut. Nature 2014, 514, 39–40. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.J.; Williams, R.S.; Williams, J.S.; Moncalian, G.; Arvai, A.S.; Limbo, O.; Guenther, G.; SilDas, S.; Hammel, M.; Russell, P.; et al. ABC ATPase signature helices in RAD50 link nucleotide state to MRE11 interface for DNA repair. Nat. Struct. Mol. Biol. 2011, 18, 423–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, H.S.; Kim, J.S.; Park, Y.B.; Gwon, G.H.; Cho, Y. Crystal structure of the MRE11-RAD50-ATPgammas complex: Understanding the interplay between MRE11 and RAD50. Genes Dev. 2011, 25, 1091–1104. [Google Scholar] [CrossRef] [PubMed]
- Bennetzen, M.V.; Cox, J.; Mann, M.; Andersen, J.S. Phosphositeanalyzer: A bioinformatic platform for deciphering phospho proteomes using kinase predictions retrieved from networkin. J. Proteome Res. 2012, 11, 3480–3486. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Zhong, Q.; Chen, P.L. The nijmegen breakage syndrome protein is essential for MRE11 phosphorylation upon DNA damage. J. Biol. Chem. 1999, 274, 19513–19516. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.S.; Chang, H.L.; Hou, M.F.; Chan, T.F.; Kao, Y.H.; Wu, Y.C.; Su, J.H. Neocarzinostatin induces mre11 phosphorylation and focus formation through an ATM- and Nbs1-dependent mechanism. Toxicol. 2002, 177, 123–130. [Google Scholar] [CrossRef]
- Di Virgilio, M.; Ying, C.Y.; Gautier, J. PIKK-dependent phosphorylation of MRE11 induces mrn complex inactivation by disassembly from chromatin. DNA Repair 2009, 8, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Kijas, A.W.; Lim, Y.C.; Bolderson, E.; Cerosaletti, K.; Gatei, M.; Jakob, B.; Tobias, F.; Taucher-Scholz, G.; Gueven, N.; Oakley, G.; et al. ATM-dependent phosphorylation of MRE11 controls extent of resection during homology directed repair by signalling through exonuclease 1. Nucleic Acids Res. 2015, 43, 8352–8367. [Google Scholar] [CrossRef] [PubMed]
- Nimonkar, A.V.; Genschel, J.; Kinoshita, E.; Polaczek, P.; Campbell, J.L.; Wyman, C.; Modrich, P.; Kowalczykowski, S.C. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011, 25, 350–362. [Google Scholar] [CrossRef] [PubMed]
- Garcia, V.; Phelps, S.E.; Gray, S.; Neale, M.J. Bidirectional resection of DNA double-strand breaks by MRE11 and EXO1. Nature 2011, 479, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Morin, I.; Ngo, H.P.; Greenall, A.; Zubko, M.K.; Morrice, N.; Lydall, D. Checkpoint-dependent phosphorylation of EXO1 modulates the DNA damage response. EMBO J. 2008, 27, 2400–2410. [Google Scholar] [CrossRef] [PubMed]
- Bolderson, E.; Tomimatsu, N.; Richard, D.J.; Boucher, D.; Kumar, R.; Pandita, T.K.; Burma, S.; Khanna, K.K. Phosphorylation of EXO1 modulates homologous recombination repair of DNA double-strand breaks. Nucleic Acids Res. 2010, 38, 1821–1831. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lavin, M.F.; Kozlov, S.; Gatei, M.; Kijas, A.W. ATM-Dependent Phosphorylation of All Three Members of the MRN Complex: From Sensor to Adaptor. Biomolecules 2015, 5, 2877-2902. https://doi.org/10.3390/biom5042877
Lavin MF, Kozlov S, Gatei M, Kijas AW. ATM-Dependent Phosphorylation of All Three Members of the MRN Complex: From Sensor to Adaptor. Biomolecules. 2015; 5(4):2877-2902. https://doi.org/10.3390/biom5042877
Chicago/Turabian StyleLavin, Martin F., Sergei Kozlov, Magtouf Gatei, and Amanda W. Kijas. 2015. "ATM-Dependent Phosphorylation of All Three Members of the MRN Complex: From Sensor to Adaptor" Biomolecules 5, no. 4: 2877-2902. https://doi.org/10.3390/biom5042877
APA StyleLavin, M. F., Kozlov, S., Gatei, M., & Kijas, A. W. (2015). ATM-Dependent Phosphorylation of All Three Members of the MRN Complex: From Sensor to Adaptor. Biomolecules, 5(4), 2877-2902. https://doi.org/10.3390/biom5042877