
Peptidylprolyl Isomerases as In Vivo Carriers for Drugs That Target Various Intracellular Entities

Abstract
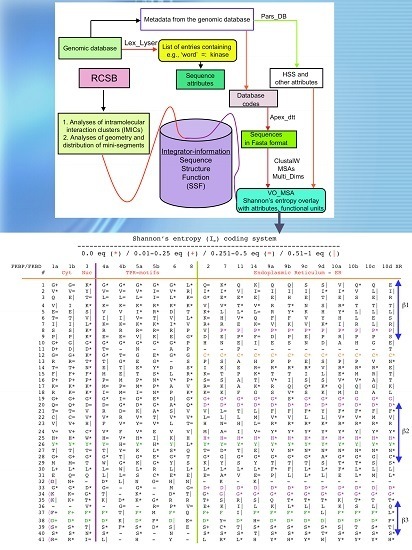
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
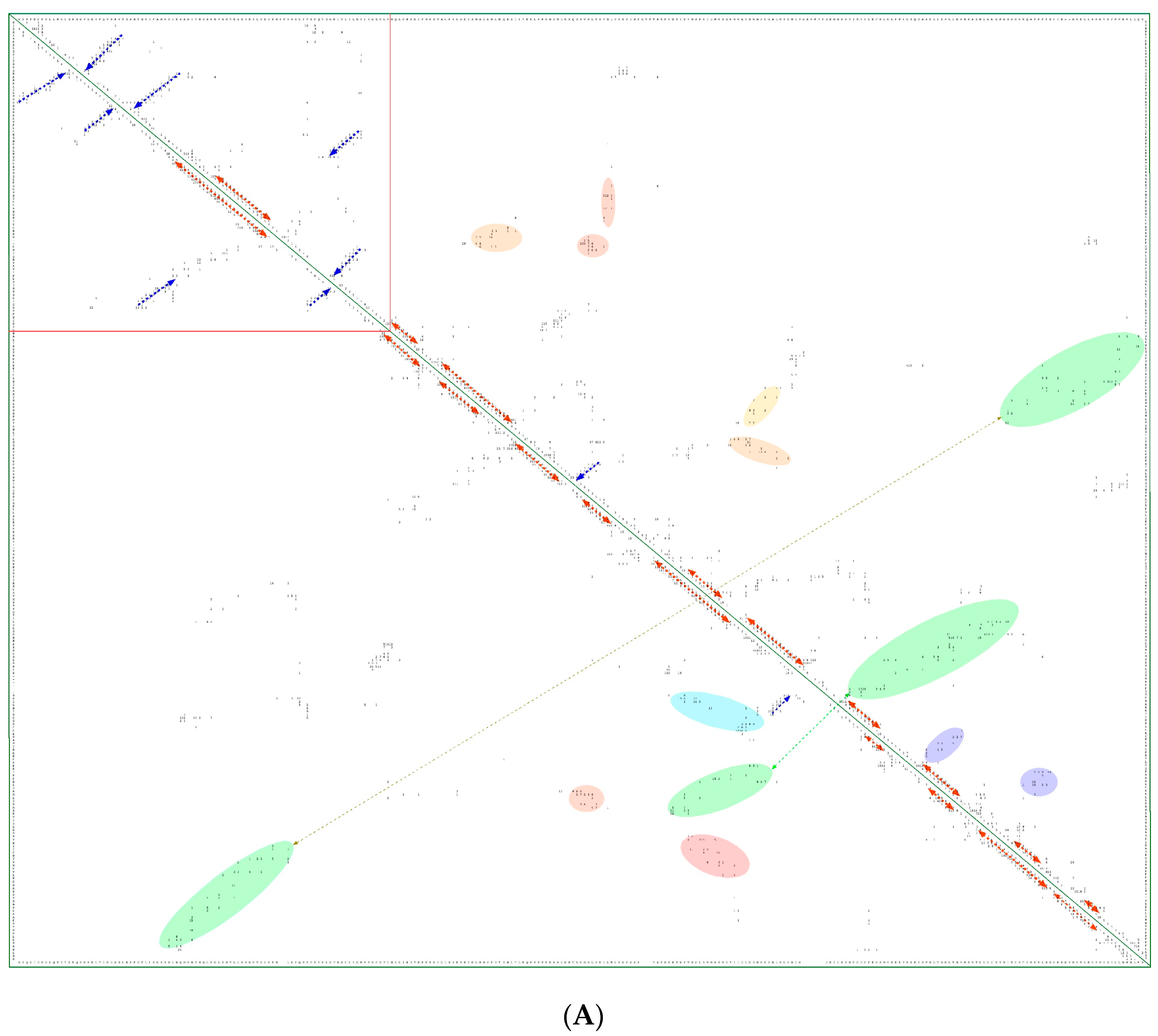{kind=link}
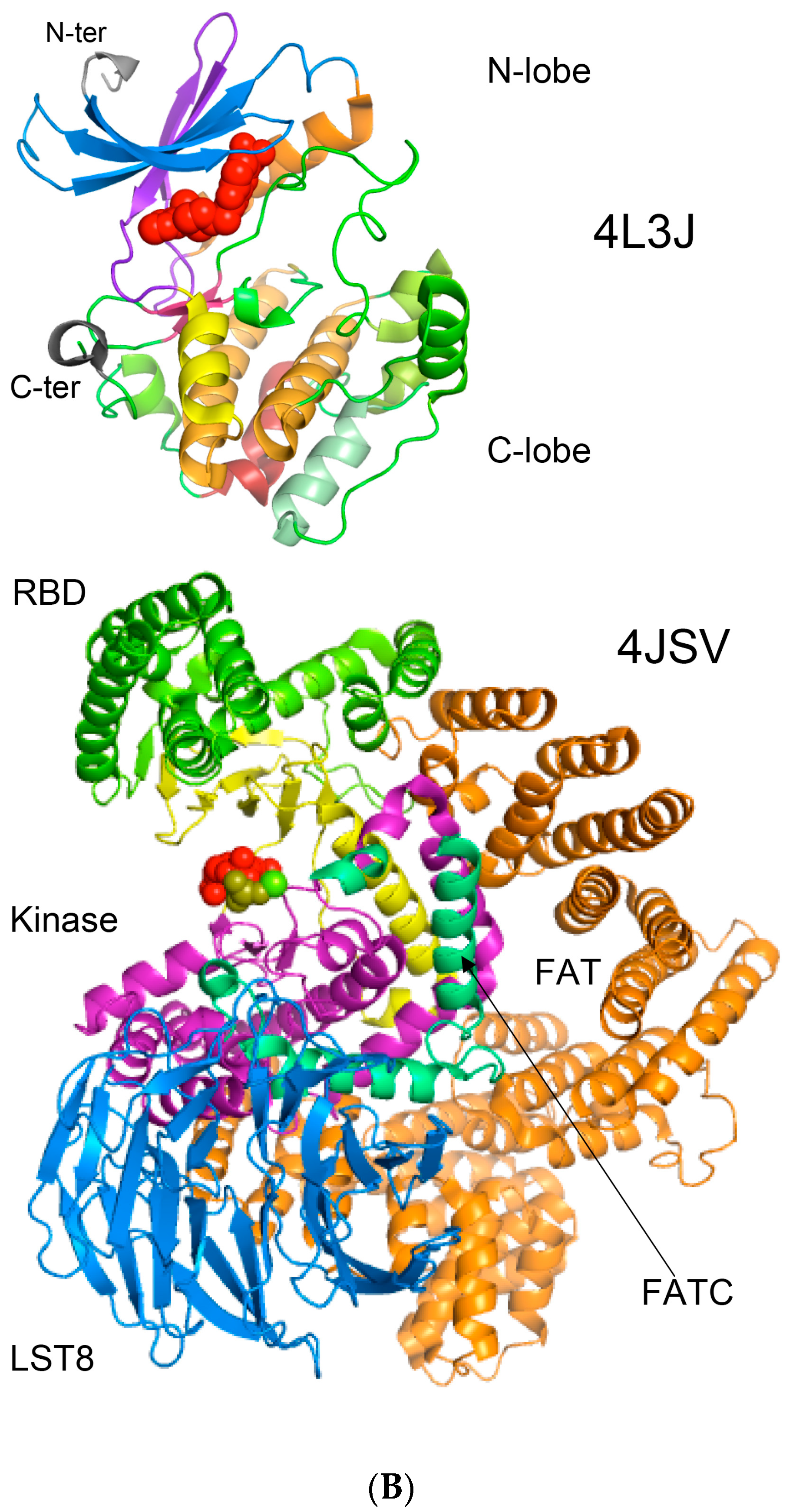
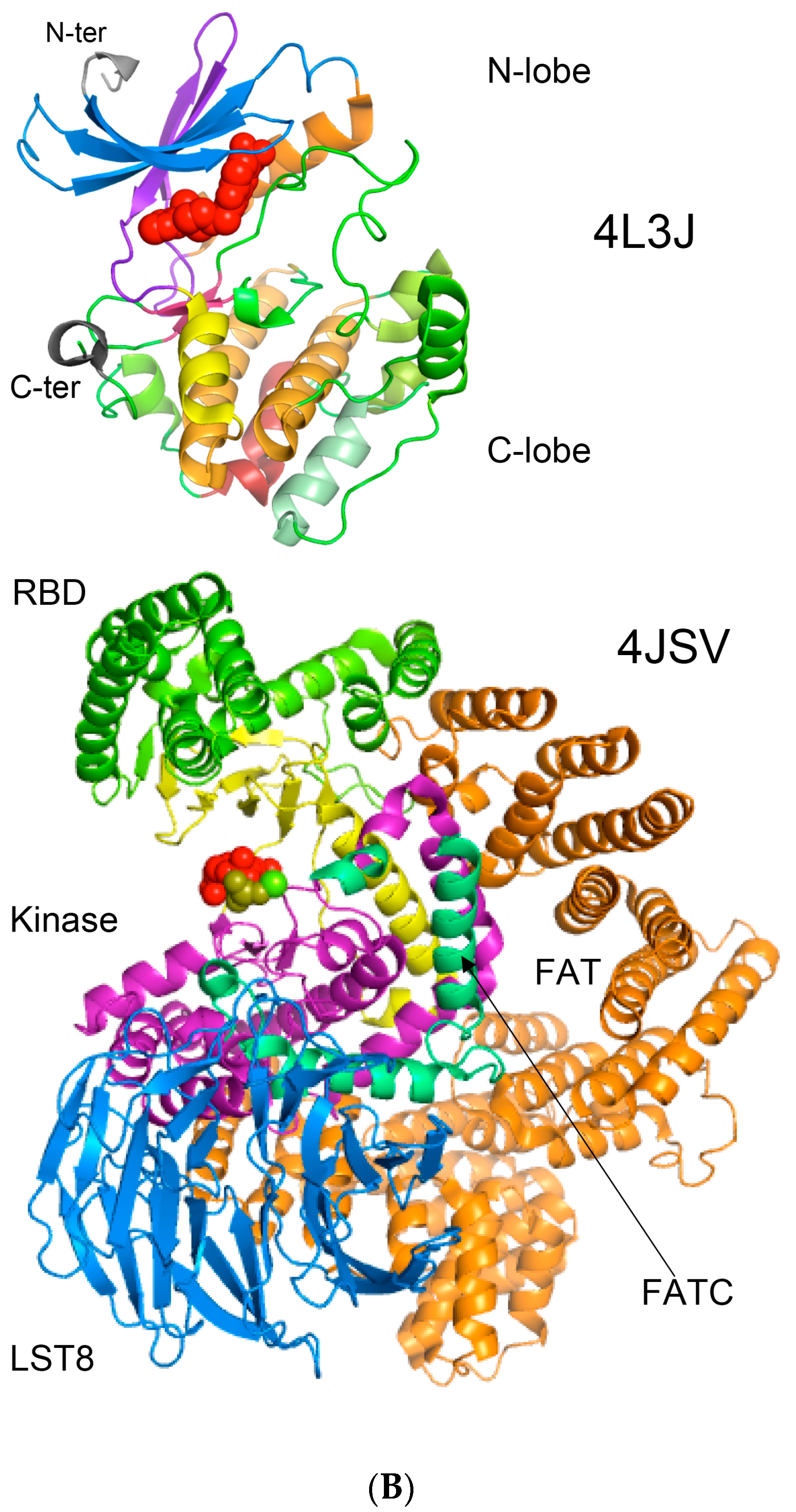{kind=link}
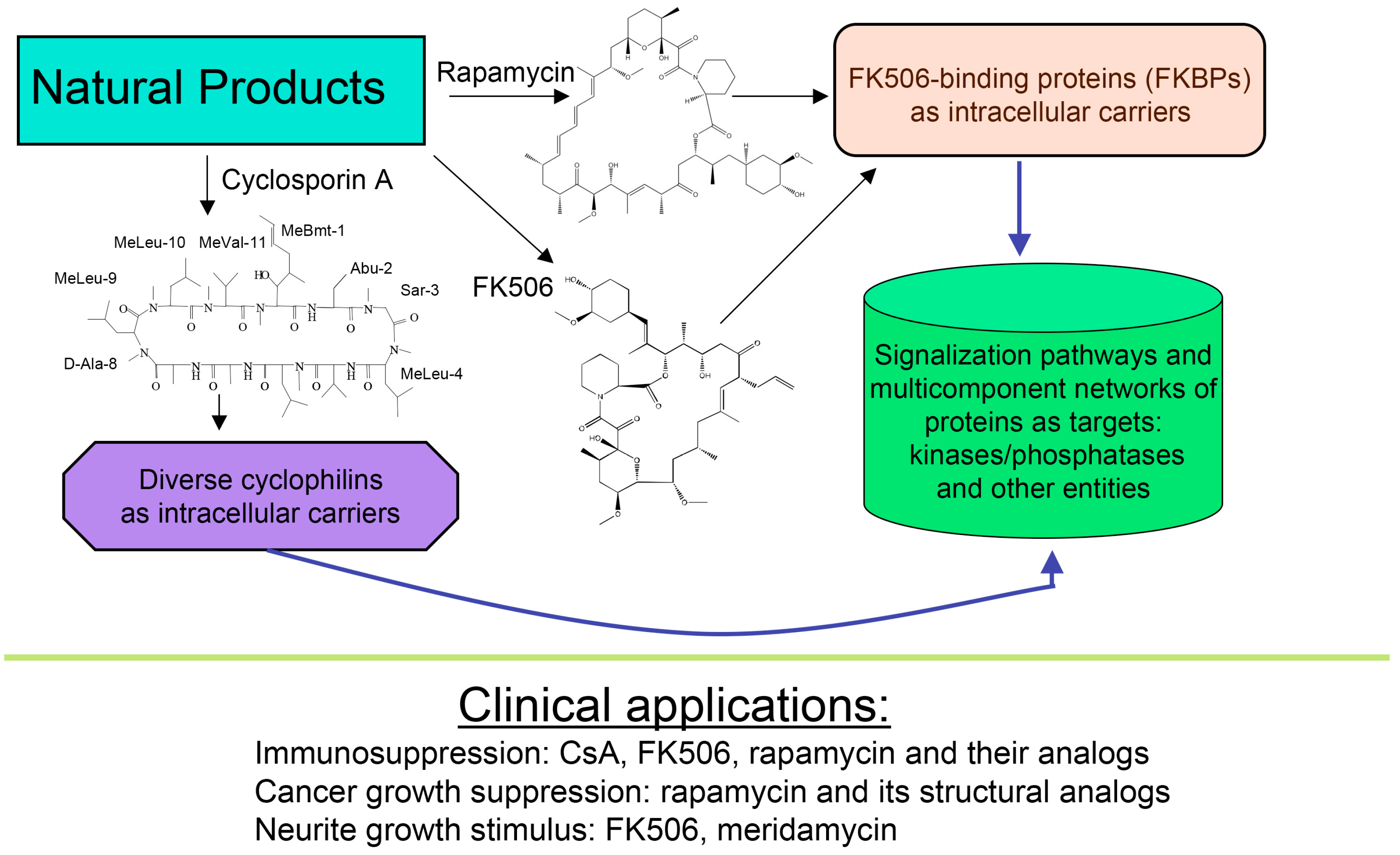
1. Introduction
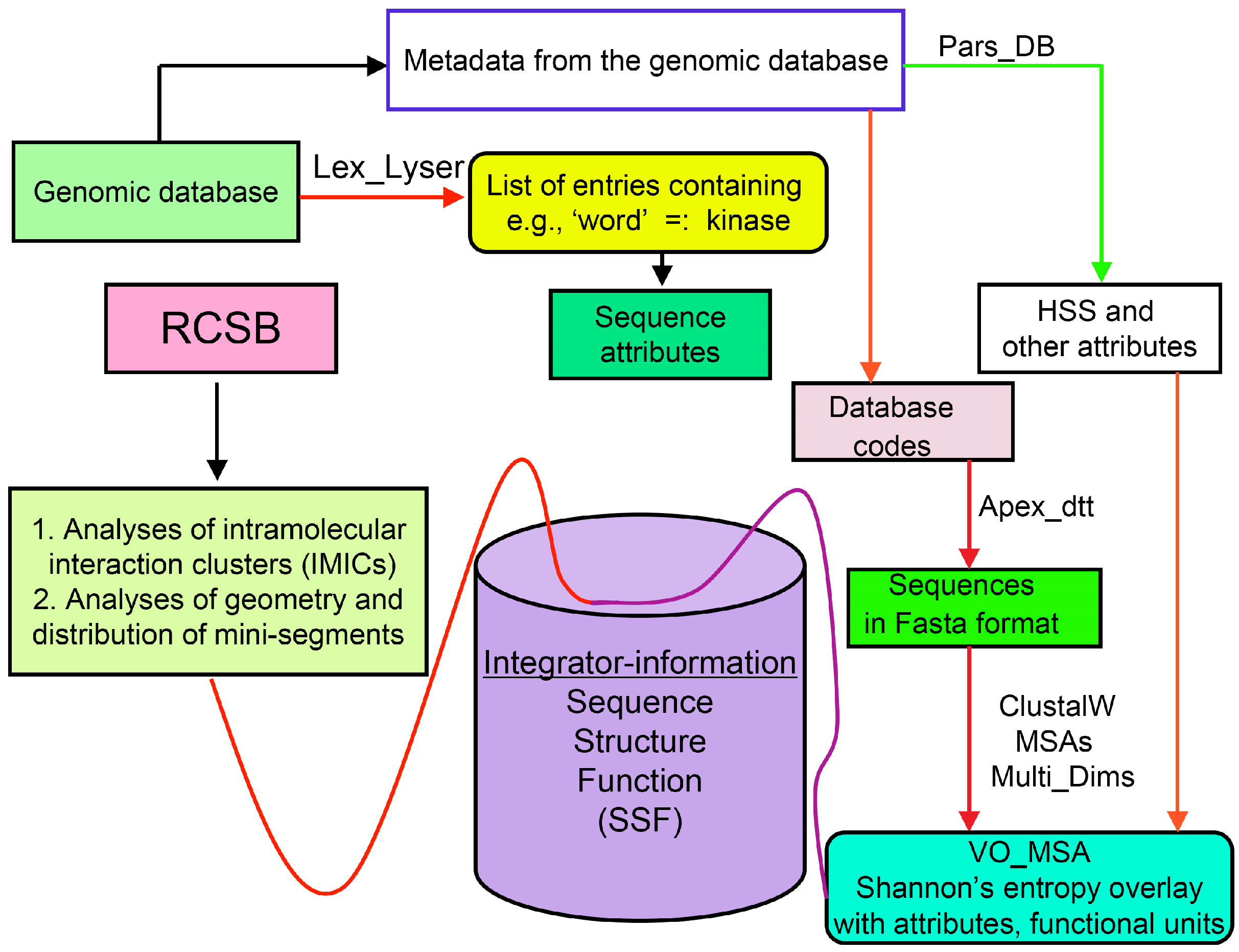
2. Strategy for Analyses of Sequences and Structures Used in This Review
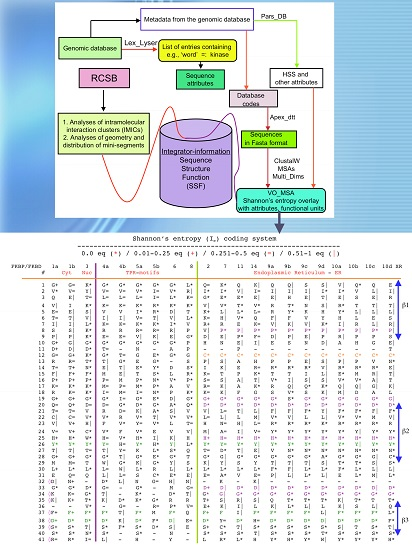
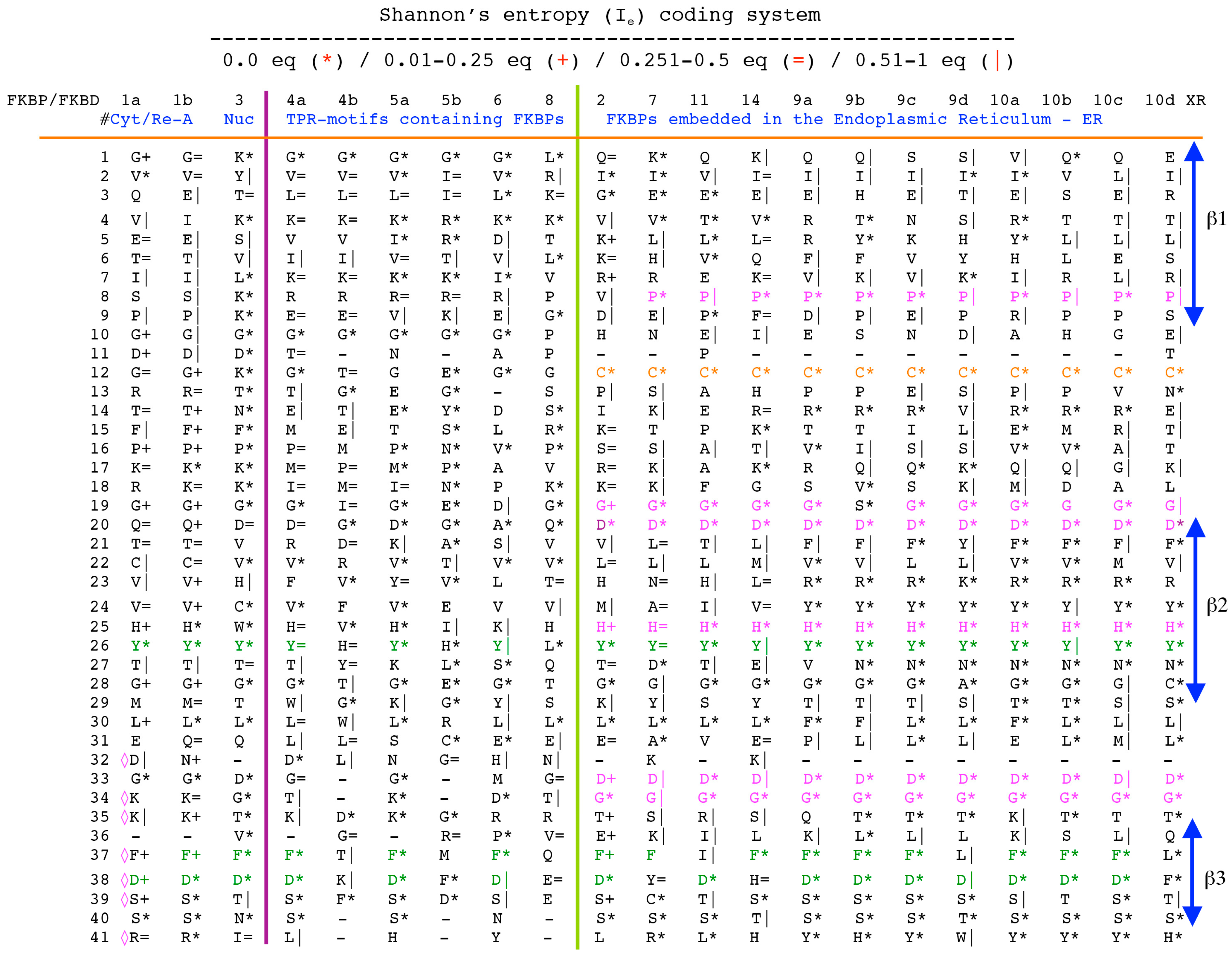
2.1. Analyses of Sequences
2.2. Analyses of Structural Data
3. Physical-Chemical Attributes of Human Cyclophilins and FKBPs
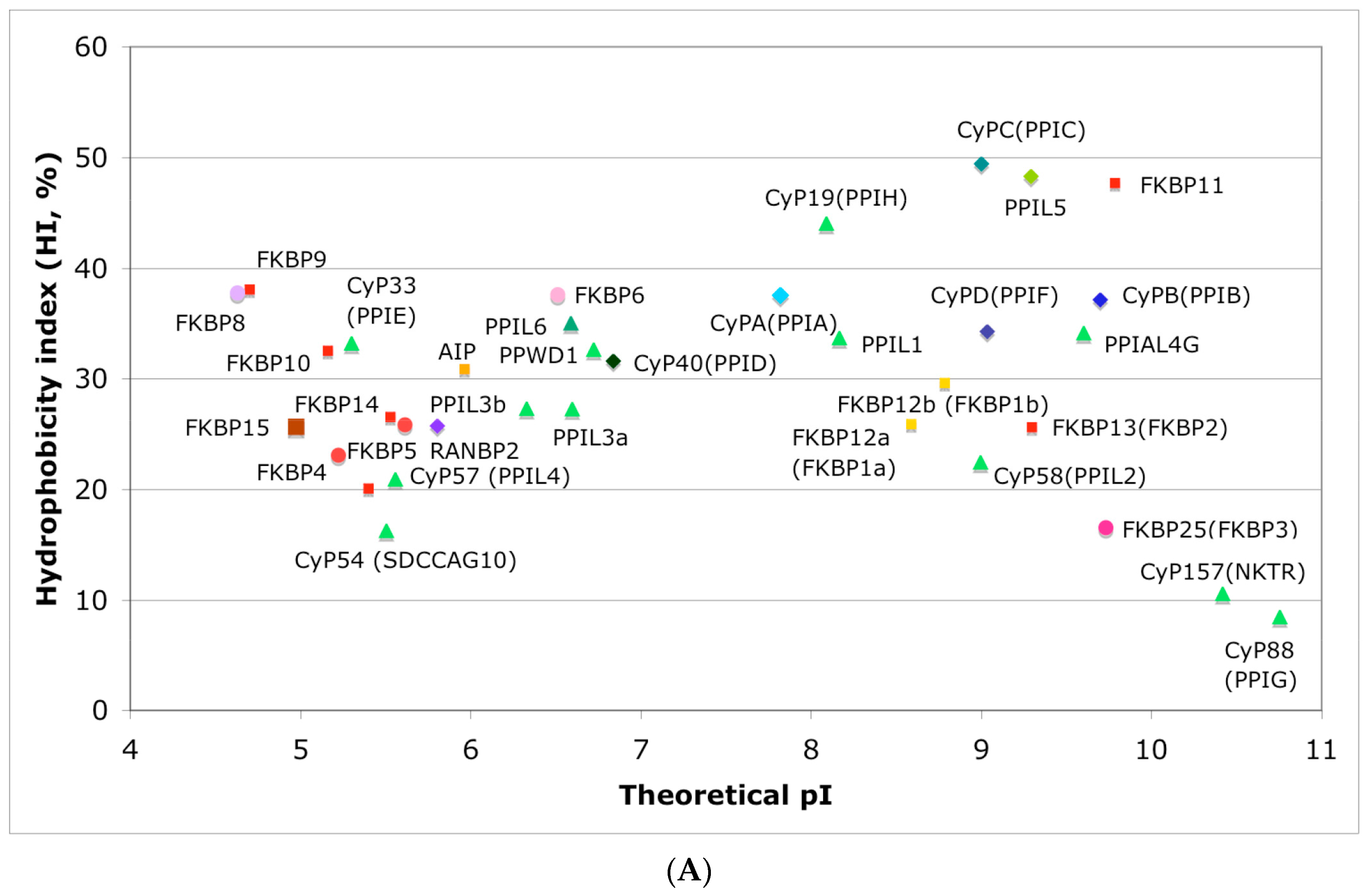
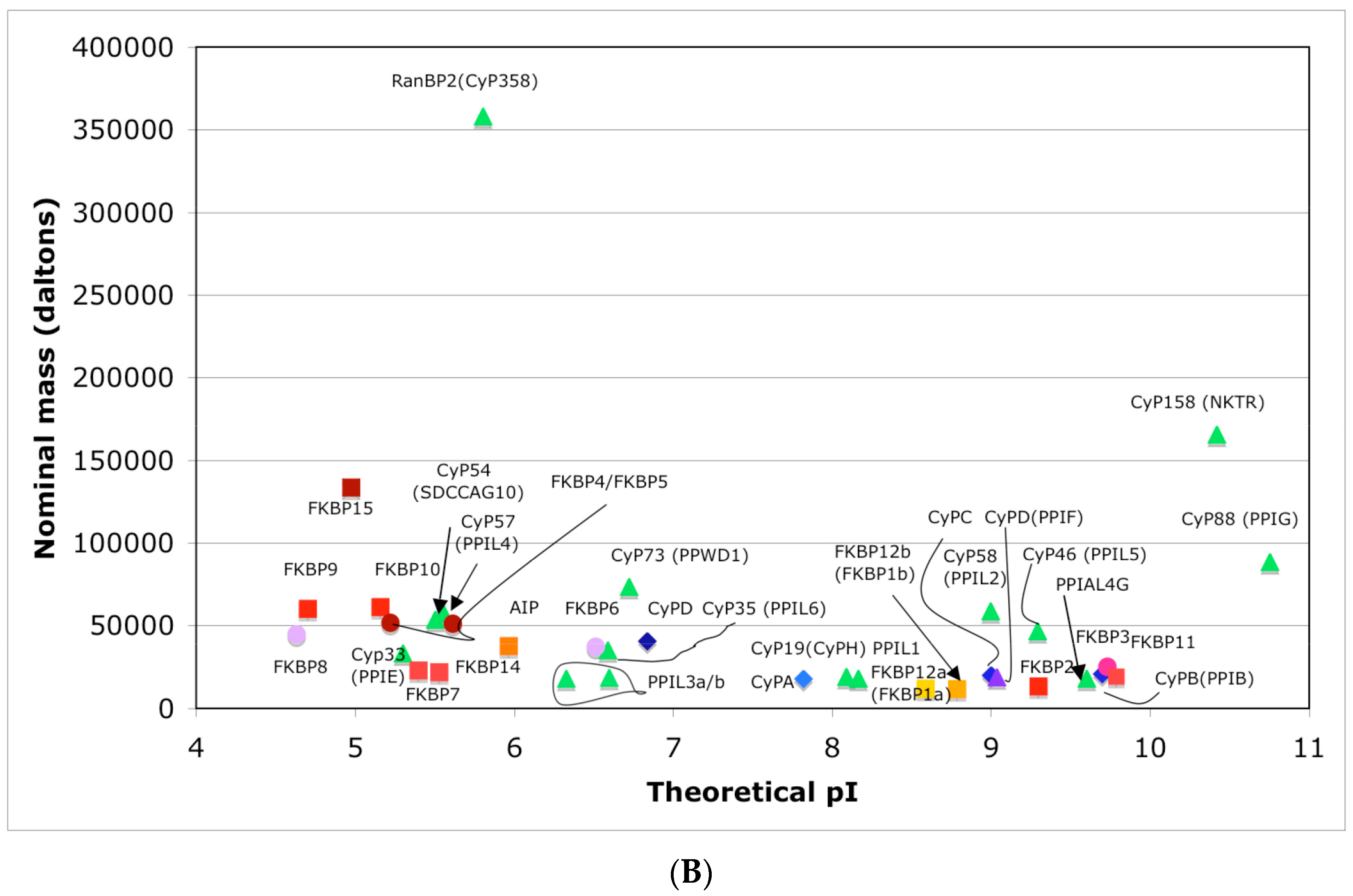
3.1. Hydrophobicity versus Charge Distribution
3.2. Spatial Hydrophobicity versus Polarity in the X-ray Structures of CyPA and FKBP12a
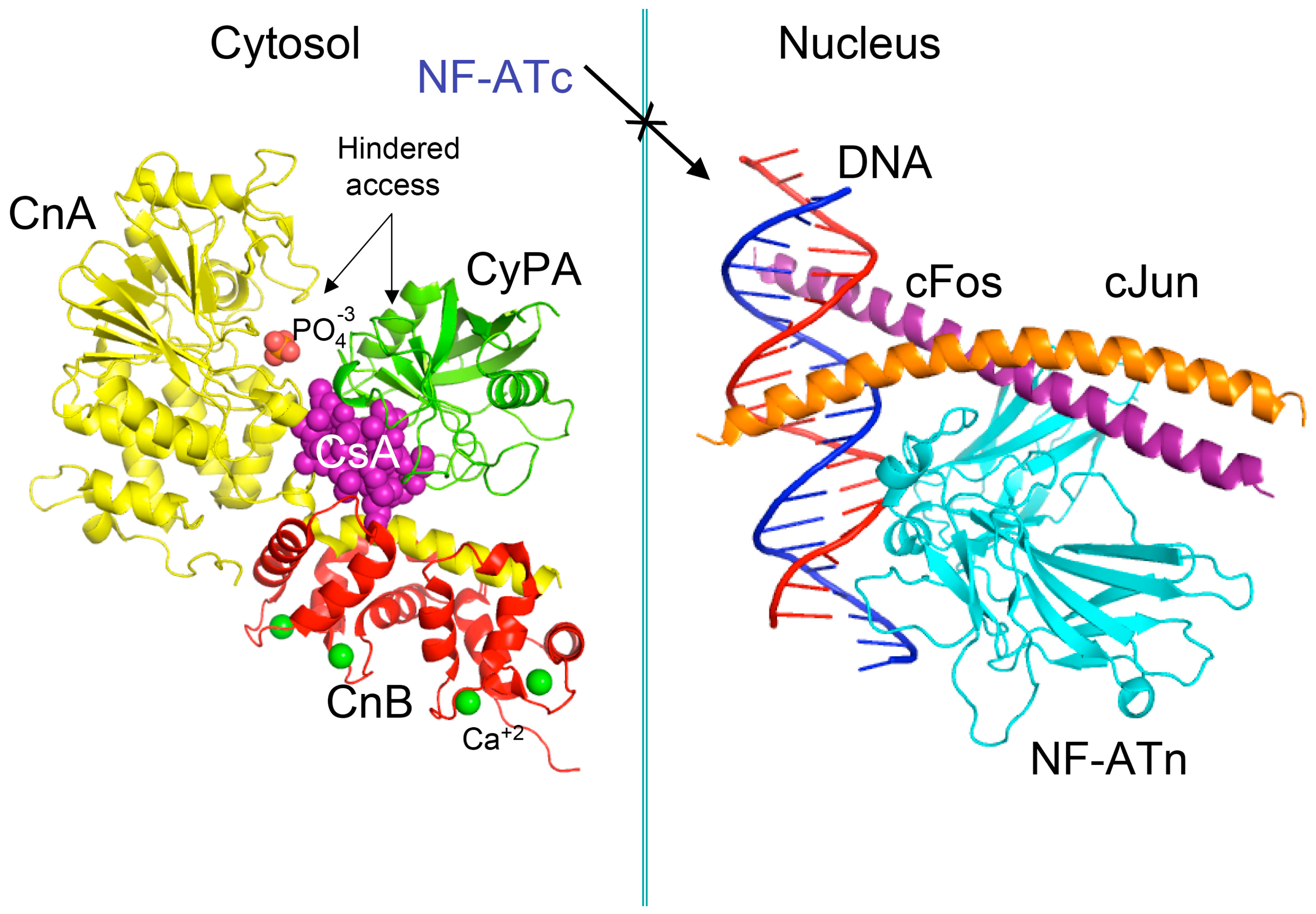
4. Some Reflections on the Mechanism of Action Induced by CsA, Tacrolimus and Sirolimus
5. Analyses of Sequence Attributes of Human Kinases and Phosphatases
6. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Heusler, K.; Pletscher, A. The controversial early history of cyclosporine. Swiss Med. Wkly. 2001, 131, 299–302. [Google Scholar] [PubMed]
- Kino, T.; Hatanaka, H.; Hashimoto, M.; Nishiyama, M.; Goto, T.; Okuhara, M.; Kohsaka, M.; Aoki, H.; Imanaka, H. FK505, a novel immunosuppressant isolated from a Streptomyces. I. Fermentation, isolation, and physico-chemical and biological characteristics. J. Antibiot. (Tokyo) 1987, 40, 1249–1255. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, S.N.; Baker, H.; Vezina, C. Rapamycin (AY-22,989), a new antifungal antibiotic. II. Fermentation, isolation and characterization. J. Antibiot. (Tokyo) 1975, 28, 727–732. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.P.; Sehgal, S.N.; Vezina, C. Activity of rapamycin (AY-22,989) against transplanted tumors. J. Antibiot. (Tokyo) 1984, 37, 1231–1237. [Google Scholar] [CrossRef] [PubMed]
- Dar, A.C.; Das, T.K.; Shokat, K.M.; Cagan, R.L. Chemical genetic discovery of targets and anti-targets for cancer polypharmacology. Nature 2012, 486, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Wagle, N.; Grabiner, B.C.; Van Allen, E.M.; Amin-Mansour, A.; Taylor-Weiner, A.; Rosenberg, M.; Gray, N.; Barletta, J.A.; Guo, Y.; Swanson, S.J.; et al. Response and acquired resistance to everolimus in anaplastic thyroid cancer. N. Engl. J. Med. 2014, 371, 1426–1433. [Google Scholar] [CrossRef] [PubMed]
- Galat, A. Functional diversity and pharmacological profiles of the FKBPs and their complexes with small natural ligands. Cell. Mol. Life Sci. 2013, 70, 3243–3275. [Google Scholar] [CrossRef] [PubMed]
- Galat, A.; Bua, J. Molecular aspects of cyclophilins mediating therapeutic actions of their ligands. Cell. Mol. Life Sci. 2010, 67, 3467–3488. [Google Scholar] [CrossRef] [PubMed]
- Salituro, G.M.; Zink, D.L.; Dahl, A.; Nielsen, J.; Wu, L.; Huang, C.; Kastner, C.; Dumond, F.J. Meridamycin: A novel nonimmunosuppressive FKBP12 ligand from Streptomyces hydroscopicus. Tetrahedron Lett. 1995, 36, 997–1000. [Google Scholar] [CrossRef]
- Raju, R.; Piggott, A.M.; Conte, M.; Tnimov, Z.; Alexandrov, K.; Capon, R.J. Nocardiopsins: New FKBP12-binding macrolide polyketides from an Australian marine-derived actinomycete, Nocardiopsis sp. Chemistry 2010, 16, 3194–3200. [Google Scholar] [CrossRef] [PubMed]
- Fehr, T.; Sanglier, J.J.; Schuler, W.; Gschwind, L.; Ponelle, M.; Schilling, W.; Wioland, C. Antascomicins A, B, C, D and E. Novel FKBP12 binding compounds from a Micromonospora strain. J. Antibiot. (Tokyo) 1996, 49, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Haltli, B.; Feng, X.; Cai, P.; Summers, M.; Lotvin, J.; He, M. Investigation of the biosynthesis of the pipecolate moiety of neuroprotective polyketide meridamycin. J. Antibiot. (Tokyo) 2011, 64, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Graziani, E.T.; Pong, K. Meridamycin Analogues for the Treatment of Neurodegenerative Disorders. U.S. Patent 7745457 B2, 29 June 2010. [Google Scholar]
- Zenke, G.; Strittmatter, U.; Fuchs, S.; Quesniaux, V.F.; Brinkmann, V.; Schuler, W.; Zurini, M.; Enz, A.; Billich, A.; Sanglier, J.J.; et al. Sanglifehrin A, a novel cyclophilin-binding compound showing immunosuppressive activity with a new mechanism of action. J. Immunol. 2001, 166, 7165–7171. [Google Scholar] [CrossRef] [PubMed]
- Sedrani, R.; Kallen, J.; Martin Cabrejas, L.M.; Papageorgiou, C.D.; Senia, F.; Rohrbach, S.; Wagner, D.; Thai, B.; Jutzi Eme, A.M.; France, J.; et al. Sanglifehrin-cyclophilin interaction: Degradation work, synthetic macrocyclic analogues, X-ray crystal structure, and binding data. J. Am. Chem. Soc. 2003, 125, 3849–3859. [Google Scholar] [CrossRef] [PubMed]
- Fischer, G.; Bang, H.; Mech, C. Determination of enzymatic catalysis for the cis-trans-isomerization of peptide binding in proline-containing peptides. Biomed. Biochim. Acta 1984, 43, 1101–1111. [Google Scholar] [PubMed]
- Handschumacher, R.E.; Harding, M.W.; Rice, J.; Drugge, R.J.; Speicher, D.W. Cyclophilin, a specific cytosolic binding protein for cyclosporin A. Science 1984, 226, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Harding, M.W.; Galat, A.; Uehling, D.E.; Schreiber, S.L. A receptor for the immunosuppressant FK-506 is a cis-trans peptidyl-prolyl isomerase. Nature 1989, 341, 761–763. [Google Scholar] [CrossRef] [PubMed]
- Galat, A.; Riviere, S. Peptidyl-Prolyl cis/trans Isomeases; Oxford University Press: Oxford, UK, 1998. [Google Scholar]
- Galat, A.; Bouet, F. Cyclophilin-B is an abundant protein whose conformation is similar to cyclophilin-A. FEBS Lett. 1994, 347, 31–36. [Google Scholar] [CrossRef]
- Kieffer, L.J.; Thalhammer, T.; Handschumacher, R.E. Isolation and characterization of a 40-kDa cyclophilin-related protein. J. Biol. Chem. 1992, 267, 5503–5507. [Google Scholar] [PubMed]
- Galat, A.; Lane, W.S.; Standaert, R.F.; Schreiber, S.L. A rapamycin-selective 25-kDa immunophilin. Biochemistry 1992, 31, 2427–2434. [Google Scholar] [CrossRef] [PubMed]
- Dunyak, B.M.; Gestwicki, J.E. Peptidyl-proline Isomerases (PPIases): Targets for natural products and natural product-inspired compounds. J. Med. Chem. 2016, 59, 9622–9644. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Farmer, J.D.; Lane, W.S.; Friedman, J.; Weissman, I.; Schreiber, S.L. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 1991, 66, 807–815. [Google Scholar] [CrossRef]
- Schreiber, S.L. Chemistry and biology of the immunophilins and their immunosuppressive ligands. Science 1991, 251, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, G.R.; Schreiber, S.L. SnapShot: Ca2+-calcineurin-NFAT signaling. Cell 2009, 138, 210. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.J.; Albers, M.W.; Shin, T.B.; Ichikawa, K.; Keith, C.T.; Lane, W.S.; Schreiber, S.L. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 1994, 369, 756–758. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Chen, J.; Schreiber, S.L.; Clardy, J. Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science 1996, 273, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Harrison, S.C. Crystal structure of human calcineurin complexed with cyclosporin A and human cyclophilin. Proc. Natl. Acad. Sci. USA 2002, 99, 13522–13526. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Stroud, J.C.; Chen, L. Crystal Structure of Human NFAT1 and Fos-JUN on the IL-2 ARRE1 Site; 14 June 2005; 1S9K.pdb; RCSB PDB: Piscataway, NJ, USA. [Google Scholar] [CrossRef]
- Hein, M.Y.; Hubner, N.C.; Poser, I.; Cox, J.; Nagaraj, N.; Toyoda, Y.; Gak, I.A.; Weisswange, I.; Mansfeld, J.; Buchholz, F.; et al. Human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 2015, 163, 712–723. [Google Scholar] [CrossRef] [PubMed]
- Cung, T.T.; Morel, O.; Cayla, G.; Rioufol, G.; Garcia-Dorado, D.; Angoulvant, D.; Bonnefoy-Cudraz, E.; Guérin, P.; Elbaz, M.; Delarche, N.; et al. Cyclosporine before PCI in patients with acute myocardial infarction. N. Engl. J. Med. 2015, 373, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Murphy, M.; Kratowicz, S.; Wang, A.; Levine, A.J.; George, D.L. Down-regulation of the stathmin/Op18 and FKBP25 genes following p53 induction. Oncogene 1999, 18, 5954–5958. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.M.; Yao, Y.L.; Seto, E. The FK506-binding protein 25 functionally associates with histone deacetylases and with transcription factor YY1. EMBO J. 2001, 20, 4814–4825. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.L.; Yang, W.M. The metastasis-associated proteins 1 and 2 form distinct protein complexes with histone deacetylase activity. J. Biol. Chem. 2003, 278, 42560–42568. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, M.; Vinci, F.; Galat, A. Mammalian FKBP-25 and its associated proteins. Arch. Biochem. Biophys. 2000, 380, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Mas, C.; Guimiot-Maloum, I.; Guimiot, F.; Khelfaoui, M.; Nepote, V.; Bourgeois, F.; Boda, B.; Levacher, B.; Galat, A.; Moalic, J.M.; et al. Molecular cloning and expression pattern of the Fkbp25 gene during cerebral cortical neurogenesis. Gene Expr. Patterns 2005, 5, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Troiani, S.; Lupi, R.; Perego, R.; Depaolini, S.R.; Thieffine, S.; Bosotti, R.; Rusconi, L. Identification of candidate substrates for poly(ADP-ribose) polymerase-2 (PARP2) in the absence of DNA damage using high-density protein microarrays. FEBS J. 2011, 278, 3676–3687. [Google Scholar] [CrossRef] [PubMed]
- Ochocka, A.M.; Kampanis, P.; Nicol, S.; Allende-Vega, N.; Cox, M.; Marcar, L.; Milne, D.; Fuller-Pace, F.; Meek, D. FKBP25, a novel regulator of the p53 pathway, induces the degradation of MDM2 and activation of p53. FEBS Lett. 2009, 583, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Yuchi, Z.; Yuen, S.M.; Lau, K.; Underhill, A.Q.; Cornea, R.L.; Fessenden, J.D.; Van Petegem, F. Crystal structures of ryanodine receptor SPRY1 and tandem-repeat domains reveal a critical FKBP12 binding determinant. Nat. Commun. 2015, 6, 7947. [Google Scholar] [CrossRef] [PubMed]
- Lopez, E.; Berna-Erro, A.; Salido, G.M.; Rosado, J.A.; Redondo, P.C. FKBP25 and FKBP38 regulate non-capacitative calcium entry through TRPC6. Biochim. Biophys. Acta 2015, 1853, 2684–2696. [Google Scholar] [CrossRef] [PubMed]
- Elvira, G.; Wasiak, S.; Blandford, V.; Tong, X.K.; Serrano, A.; Fan, X.; del Rayo Sanchez-Carbente, M.; Servant, F.; Bell, A.W.; Boismenu, D.; et al. Characterization of an RNA granule from developing brain. Mol. Cell. Proteom. 2006, 5, 635–651. [Google Scholar] [CrossRef] [PubMed]
- Kovalev, N.; Nagy, P.D. Cyclophilin A binds to the viral RNA and replication proteins resulting in inhibition of tombusviral replicase assembly. J. Virol. 2013, 87, 13330–13342. [Google Scholar] [CrossRef] [PubMed]
- Arevalo-Rodriguez, M.; Heitman, J. Cyclophilin A is localized to the nucleus and controls meiosis in Saccharomyces cerevisiae. Eukaryot. Cell 2005, 4, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Wang, X.; Deinum, J.; Huang, Z.; Gao, J.; Modjtahedi, N.; Neagu, M.R.; Nilsson, M.; Eriksson, P.S.; Hagberg, H.; et al. Cyclophilin A participates in the nuclear translocation of apoptosis-inducing factor in neurons after cerebral hypoxia-ischemia. J. Exp. Med. 2007, 204, 1741–1748. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Song, J.; Milne, T.A.; Wang, G.G.; Li, H.; Allis, C.D.; Patel, D.J. Pro Isomerization in MLL1 PHD3-Bromo cassette connects H3K4me readout to CyP33 and HDAC-mediated repression. Cell 2010, 141, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Galat, A.; Thai, R. Rapamycin-binding FKBP25 associates with diverse proteins that form large intracellular entities. Biochem. Biophys. Res. Commun. 2014, 450, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- Will, C.L.; Luhrmann, R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Galat, A.; Thai, R.; Stura, E.A. Diversified targets of FKBP25 and its complex with rapamycin. Int. J. Biol. Macromol. 2014, 69, 344–532. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.; Shin, J.; Rajan, S.; Yoon, H.S. Structural basis of nucleic acid recognition by FK506-binding protein 25 FKBP25, a nuclear immunophilin. Nucleic Acids Res. 2016, 44, 2909–2925. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Stefanovic, B. LARP6 meets collagen mRNA: Specific regulation of type I collagen expression. Int. J. Mol. Sci. 2016, 17, 419. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Wootton, J.C.; Gertz, E.M.; Agarwala, R.; Morgulis, A.; Schäffer, A.A.; Yu, Y.K. Protein database searches using compositionally adjusted substitution matrices. FEBS J. 2005, 272, 5101–5109. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.L.; Barrett, T.; Benson, D.A.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; Dicuccio, M.; Edgar, R.; Federhen, S.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2008, 36, D13–D21. [Google Scholar] [CrossRef] [PubMed]
- Galat, A. Multidimensional drift of sequence attributes and functional profiles in the superfamily of the three-finger proteins and their structural homologues. J. Chem. Inf. Model. 2015, 55, 2026–2041. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X Version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Galat, A. Functional drift of sequence attributes in the FK506-binding proteins FKBPs. J. Chem. Inf. Model. 2008, 48, 1118–1130. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Henrick, K.; Nakamura, H.; Markley, J.L. The worldwide Protein Data Bank (wwPDB): Ensuring a single, uniform archive of PDB data. Nucleic Acids Res. 2007, 35, D301–D303. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. The PyMOL Molecular Graphics System; DeLano Scientific: San Carlos, CA, USA, 2002; Available online: www://pymol.sourceforge.net/ (accessed on 5 January 2017).
- Ke, H.M.; Zydowsky, L.D.; Liu, J.; Walsh, C.T. Crystal structure of recombinant human T-cell cyclophilin A at 2.5 Å resolution. Proc. Natl. Acad. Sci. USA 1991, 88, 9483–9487. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.L.; Walker, J.R.; Campagna-Slater, V.; Finerty, P.J.; Paramanathan, R.; Bernstein, G.; MacKenzie, F.; Tempel, W.; Ouyang, H.; Lee, W.H.; et al. Structural and biochemical characterization of the human cyclophilin family of peptidyl-prolyl isomerases. PLoS Biol. 2010, 8, e1000439. [Google Scholar] [CrossRef] [PubMed]
- Van Duyne, G.D.; Standaert, R.F.; Karplus, P.A.; Schreiber, S.L.; Clardy, J. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J. Mol. Biol. 1993, 229, 105–124. [Google Scholar] [CrossRef]
- Huai, Q.; Kim, H.Y.; Liu, Y.; Zhao, Y.; Mondragon, A.; Liu, J.O.; Ke, H. Crystal structure of calcineurin-cyclophilin-cyclosporin shows common but distinct recognition of immunophilin-drug complexes. Proc. Natl. Acad. Sci. USA 2002, 99, 12037–12042. [Google Scholar] [CrossRef] [PubMed]
- Griffith, J.P.; Kim, J.L.; Kim, E.E.; Sintchak, M.D.; Thomson, J.A.; Fitzgibbon, M.J.; Fleming, M.A.; Caron, P.R.; Hsiao, K.; Navia, M.A. X-ray structure of calcineurin inhibited by the immunophilin-immunosuppressant FKBP12-FK506 complex. Cell 1995, 82, 507–522. [Google Scholar] [CrossRef]
- Shimobayashi, M.; Hall, M.N. Making new contacts: The mTOR network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 2014, 15, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, D.; Colombi, M.; Moroni, C.; Hall, M.N. Rapamycin passes the torch: A new generation of mTOR inhibitors. Nat. Rev. Drug Discov. 2011, 10, 868–880. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. mTOR kinase structure, mechanism and regulation. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Murr, R.; Vaissière, T.; Sawan, C.; Shukla, V.; Herceg, Z. Orchestration of chromatin-based processes: Mind the TRRAP. Oncogene 2007, 26, 5358–5372. [Google Scholar] [CrossRef] [PubMed]
- Cimprich, K.A.; Shin, T.B.; Keith, C.T.; Schreiber, S.L. cDNA cloning and gene mapping of a candidate human cell cycle checkpoint protein. Proc. Natl. Acad. Sci. USA 1996, 93, 2850–2855. [Google Scholar] [CrossRef] [PubMed]
- Gatti, R.A.; Berkel, I.; Boder, E.; Braedt, G.; Charmley, P.; Concannon, P.; Ersoy, F.; Foroud, T.; Jaspers, N.G.; Kenneth, L.; et al. Localization of an ataxia-telangiectasia gene to chromosome 11q22-23. Nature 1988, 336, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, P.; Yepiskoposyan, H.; Metze, S.; Zamudio-Orozco, R.; Kleinschmidt, N.; Muhlemann, O. Nonsense-mediated mRNA decay in human cells: Mechanistic insights, functions beyond quality control and the double-life of NMD factors. Cell. Mol. Life Sci. 2010, 67, 677–700. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhong, C.; Wang, F.; Qu, F.; Ding, J. Crystal structures of S6K1 provide insights into the regulation mechanism of S6K1 by the hydrophobic motif. Biochem. J. 2013, 454, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Schmidpeter, P.A.; Schmid, F.X. Prolyl isomerization and its catalysis in protein folding and protein function. J. Mol. Biol. 2015, 427, 1609–1631. [Google Scholar] [CrossRef] [PubMed]
- Taipale, M.; Tucker, G.; Peng, J.; Krykbaeva, I.; Lin, Z.Y.; Larsen, B.; Choi, H.; Berger, B.; Gingras, A.C.; Lindquist, S. A quantitative chaperone interaction network reveals the architecture of cellular protein homeostasis pathways. Cell 2014, 158, 434–448. [Google Scholar] [CrossRef] [PubMed]
- Van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahni, N.; Yi, S.; Taipale, M.; Fuxman Bass, J.I.; Coulombe-Huntington, J.; Yang, F.; Peng, J.; Weile, J.; Karras, G.I.; Wang, Y.; et al. Widespread macromolecular interaction perturbations in human genetic disorders. Cell 2015, 161, 647–660. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C. Thermodynamic description of β-amyloid formation using physicochemical scales and fractal bioinformatic scales. ACS Chem. Neurosci. 2015, 6, 745–750. [Google Scholar] [CrossRef] [PubMed]
- Galat, A. On transversal hydrophobicity of some proteins and their modules. J. Chem. Inf. Model. 2009, 49, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Hankey, A. A complexity basis for phenomenology: How information states at criticality offer a new approach to understanding experience of self, being and time. Prog. Biophys. Mol. Biol. 2015, 119, 288–302. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Liu, L.; Wu, Y.; Singh, K.; Su, B.; Zhang, N.; Liu, X.; Shen, Y.; Huang, S. Rapamycin inhibits mSin1 phosphorylation independently of mTORC1 and mTORC2. Oncotarget 2015, 6, 4286–4298. [Google Scholar] [CrossRef] [PubMed]
- Stepanenko, A.A.; Dmitrenko, V.V. Pitfalls of the MTT assay: Direct and off-target effects of inhibitors can result in over/underestimation of cell viability. Gene 2015, 574, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Arriola-Apelo, S.I.; Neuman, J.C.; Baar, E.L.; Syed, F.A.; Cummings, N.E.; Brar, H.K.; Pumper, C.P.; Kimple, M.E.; Lamming, D.W. Alternative rapamycin treatment regimens mitigate the impact of rapamycin on glucose homeostasis and the immune system. Aging Cell 2016, 15, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Marz, A.M.; Fabian, A.K.; Kozany, C.; Bracher, A.; Hausch, F. Large FK506-binding proteins shape the pharmacology of rapamycin. Mol. Cell. Biol. 2013, 33, 1357–1367. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Lee, H.; Lee, H.K.; Lee, S.W.; Ha, S.C.; Kwon, T.; Seo, J.K.; Lee, C.; Rhee, H.W. Proximity-directed labeling reveals a new rapamycin-induced heterodimer of FKBP25 and FRB in live cells. ACS Cent. Sci. 2016, 2, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Sundin, T.; Peffley, D.M.; Hentosh, P. Disruption of an hTERT-mTOR-RAPTOR protein complex by a phytochemical perillyl alcohol and rapamycin. Mol. Cell. Biochem. 2013, 375, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Cox, L.S. Live fast, die young: New lessons in mammalian longevity. Rejuvenation Res. 2009, 12, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Cox, L.S.; Mattison, J.A. Increasing longevity through caloric restriction or rapamycin feeding in mammals: Common mechanisms for common outcomes? Aging Cell 2009, 8, 607–1385. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, K.H.; Ortiz, D.; Academia, E.C.; Anies, A.C.; Liao, C.Y.; Kennedy, B.K. Rapamycin-mediated mTORC2 inhibition is determined by the relative expression of FK506-binding proteins. Aging Cell 2015, 14, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Rodrik-Outmezguine, V.S.; Okaniwa, M.; Yao, Z.; Novotny, C.J.; McWhirter, C.; Banaji, A.; Won, H.; Wong, W.; Berger, M.; de Stanchina, E.; et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature 2016, 534, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; Reddy, E.P.; Shokat, K.M.; Soucek, L. Drugging the ‘undruggable’ cancer targets. Nat. Rev. Cancer 2017, 17, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Gaali, S.; Feng, X.; Hahle, A.; Sippel, C.; Bracher, A.; Hausch, F. Rapid, structure-based exploration of pipecolic acid amides as novel selective antagonists of the FK506-Binding protein 51. J. Med. Chem. 2016, 59, 2410–2422. [Google Scholar] [CrossRef] [PubMed]
- Maiaru, M.; Tochiki, K.K.; Cox, M.B.; Annan, L.V.; Bell, C.G.; Feng, X.; Hausch, F.; Géranton, S.M. The stress regulator FKBP51 drives chronic pain by modulating spinal glucocorticoid signaling. Sci. Transl. Med. 2016, 8, 325ra19. [Google Scholar] [CrossRef] [PubMed]












© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galat, A. Peptidylprolyl Isomerases as In Vivo Carriers for Drugs That Target Various Intracellular Entities. Biomolecules 2017, 7, 72. https://doi.org/10.3390/biom7040072
Galat A. Peptidylprolyl Isomerases as In Vivo Carriers for Drugs That Target Various Intracellular Entities. Biomolecules. 2017; 7(4):72. https://doi.org/10.3390/biom7040072
Chicago/Turabian StyleGalat, Andrzej. 2017. "Peptidylprolyl Isomerases as In Vivo Carriers for Drugs That Target Various Intracellular Entities" Biomolecules 7, no. 4: 72. https://doi.org/10.3390/biom7040072
APA StyleGalat, A. (2017). Peptidylprolyl Isomerases as In Vivo Carriers for Drugs That Target Various Intracellular Entities. Biomolecules, 7(4), 72. https://doi.org/10.3390/biom7040072