Studies on the Interaction of Alyteserin 1c Peptide and Its Cationic Analogue with Model Membranes Imitating Mammalian and Bacterial Membranes

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Peptide Synthesis and Purification
2.3. Helical Wheel Projection of the Peptides
2.4. Model Membranes Preparation
2.5. Differential Scanning Calorimetry (DSC)
2.6. Attenuated Total Reflectance-Fourier Transform Infrared (ATR-FTIR)
2.7. Molecular Modelling and Molecular Dynamics Simulation of the Complex Peptide-Membrane
3. Results and Discussion
3.1. Peptide Sequence
3.2. Thermotropic Behavior of Model Membranes
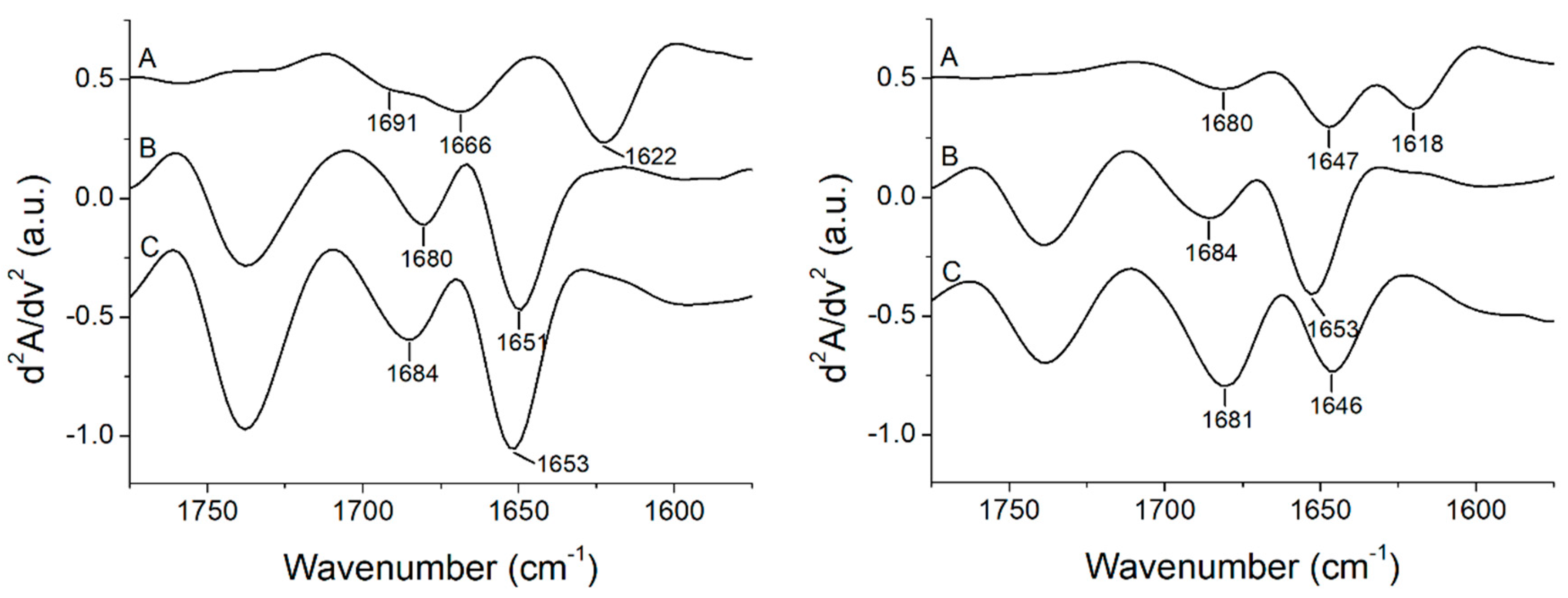
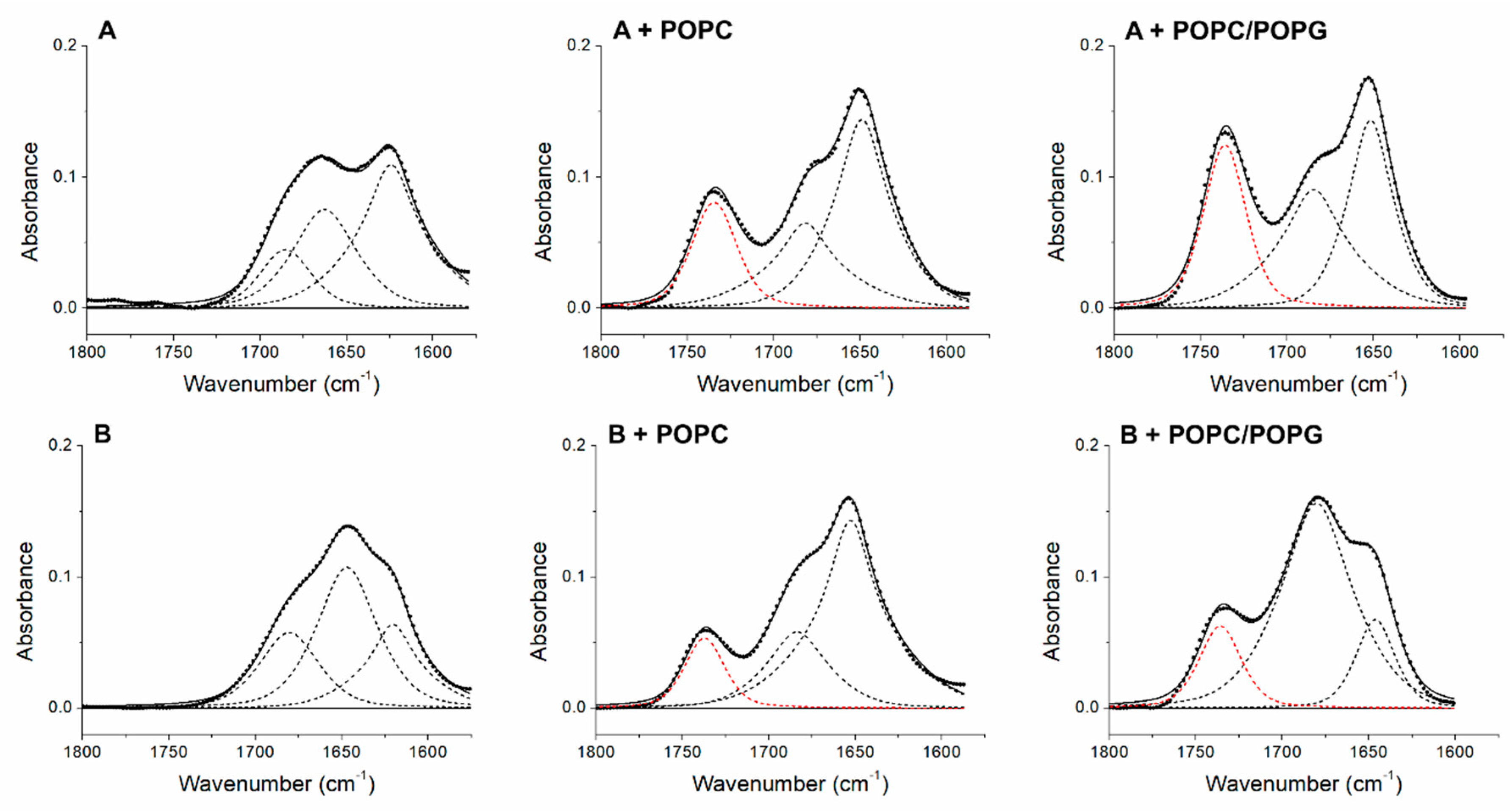
3.3. Peptide +2 and +5 Secondary Structure before and after Interacting with Membrane Models
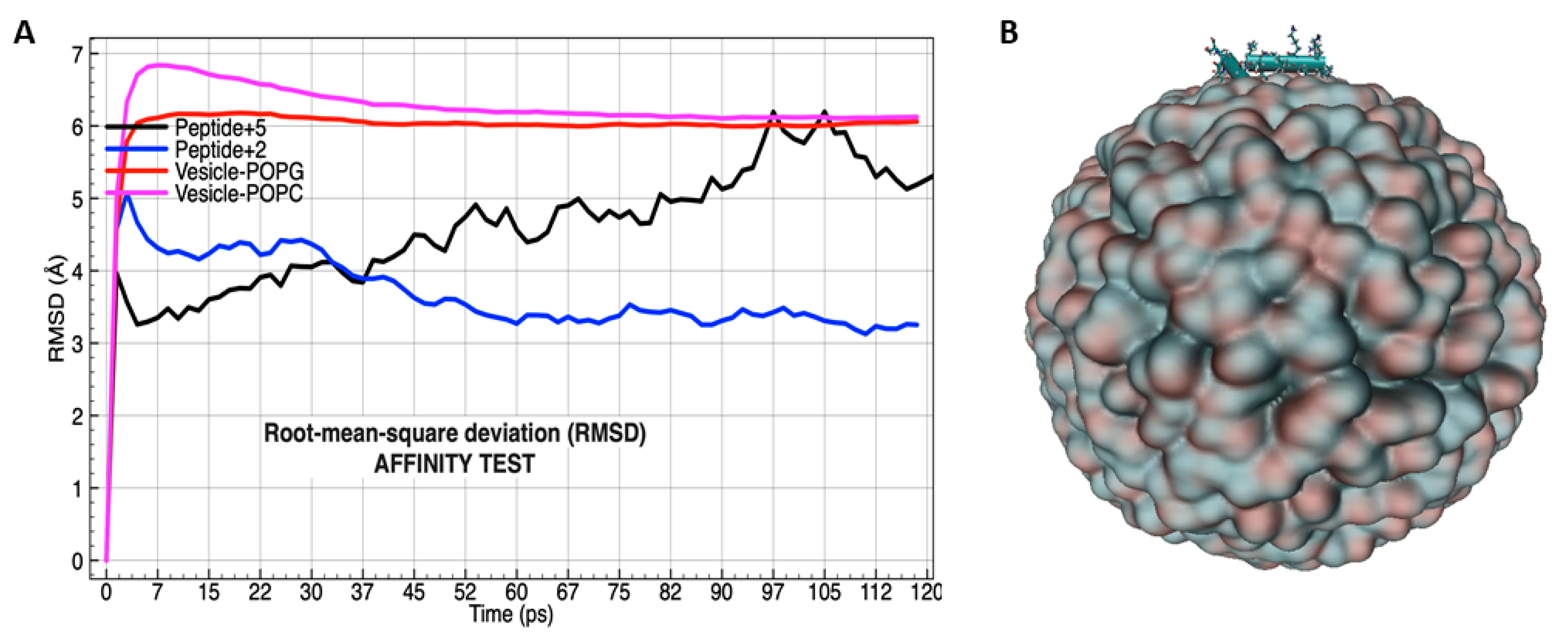
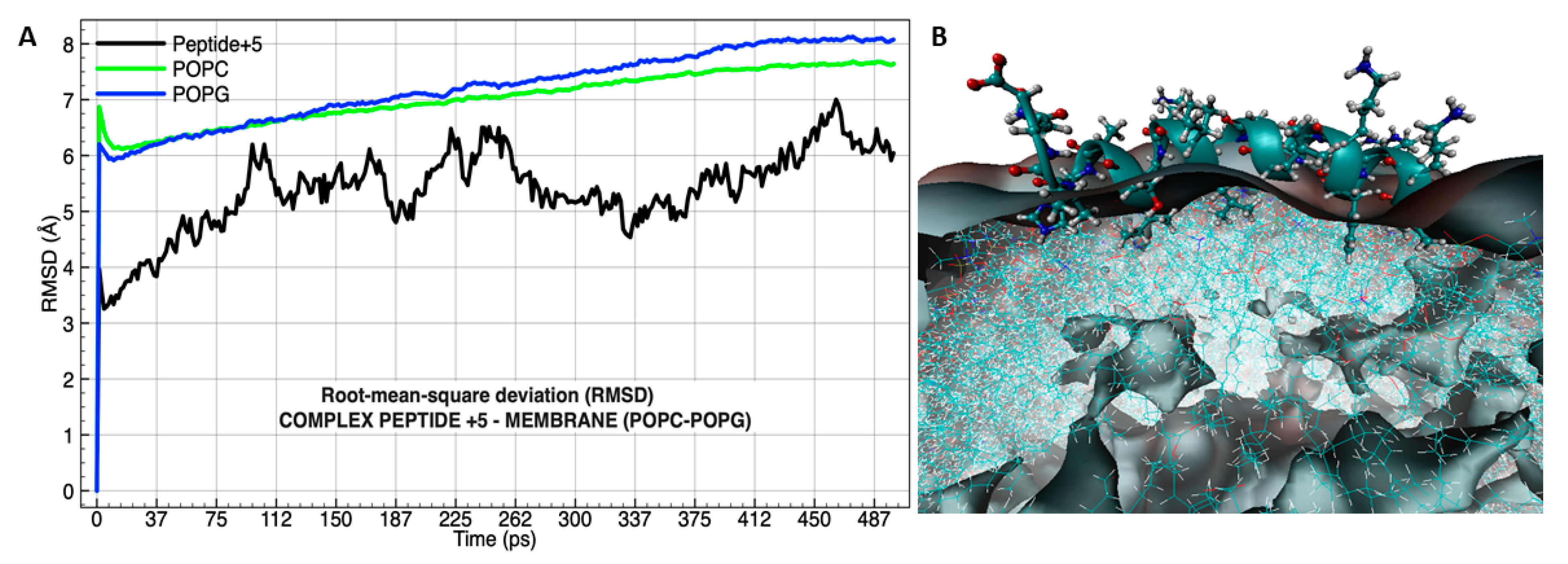
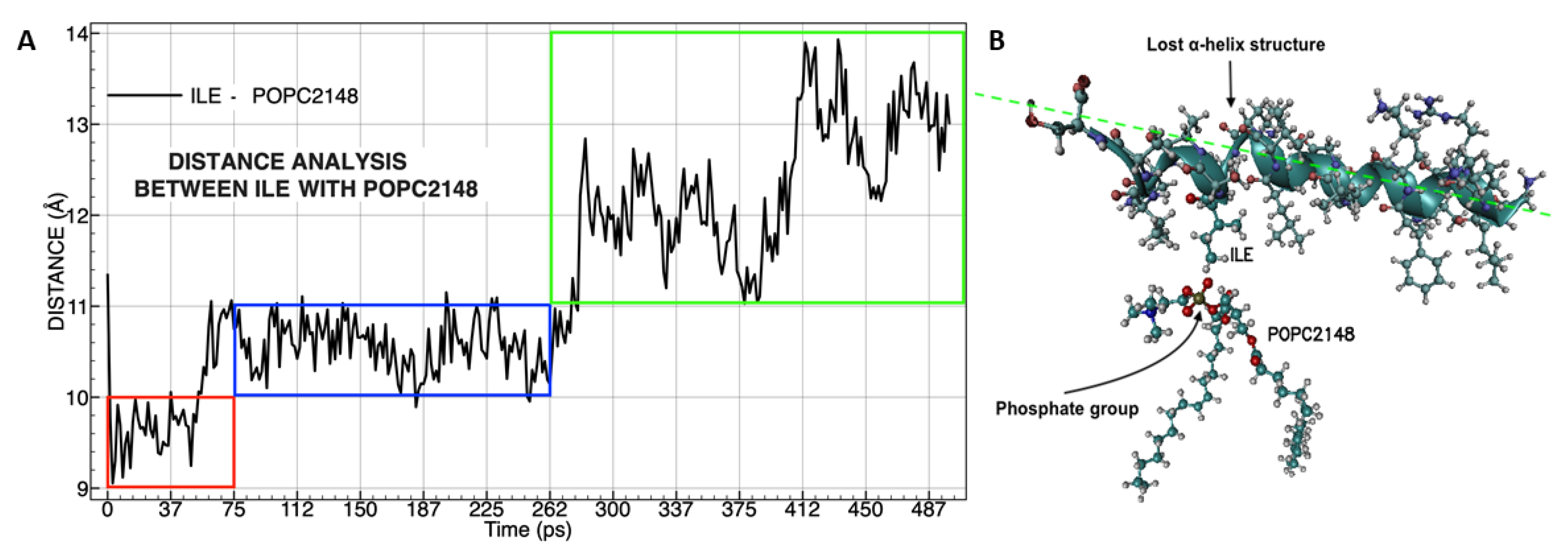
3.4. Molecular Dynamics Analysis of the Peptide-Membrane Complex
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zhang, L.J.; Gallo, R.L. Antimicrobial peptides. Curr. Biol. 2016, 26, R14–R19. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, V.; Feio, M.J.; Bastos, M. Role of lipids in the interaction of antimicrobial peptides with membranes. Prog. Lipid Res. 2012, 51, 149–177. [Google Scholar] [CrossRef] [PubMed]
- Nacif-Marçal, L.; Pereira, G.R.; Abranches, M.V.; Costa, N.C.; Cardoso, S.A.; Honda, E.R.; De Paula, S.O.; Feio, R.N.; Oliveira, L.L. Identification and characterization of an antimicrobial peptide of Hypsiboas semilineatus (Spix, 1824) (Amphibia, Hylidae). Toxicon 2015, 99, 16–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conlon, J.M. Structural diversity and species distribution of host-defense peptides in frog skin secretions. Cell. Mol. Life Sci. 2011, 68, 2303–2315. [Google Scholar] [CrossRef] [PubMed]
- Waterhous, D.V.; Johnson, W.C. Importance of Environment in Determining Secondary Structure in Proteins. Biochemistry 1994, 33, 2121–2128. [Google Scholar] [CrossRef] [PubMed]
- Yeaman, M.R.; Yount, N.Y.; Hauger, R.L.; Grigoriadis, D.E.; Dallman, M.F.; Plotsky, P.M.; Vale, W.W.; Dautzenberg, F.M. Mechanisms of Antimicrobial Peptide Action and Resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.H.; Hall, K.N.; Aguilar, M.I. Antimicrobial Peptide Structure and Mechanism of Action: A Focus on the Role of Membrane Structure. Curr. Top. Med. Chem. 2016, 16, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Oñate-Garzón, J.F.; Manrique-Moreno, M.; González, E.P. Actividad antimicrobiana de péptidos catiónicos diseñados a partir de un péptido neutro. Acta Biológica Colomb. 2017, 22, 35. [Google Scholar] [CrossRef]
- Hädicke, A.; Blume, A. Binding of the Cationic Peptide (KL)4K to Lipid Monolayers at the Air–Water Interface: Effect of Lipid Headgroup Charge, Acyl Chain Length, and Acyl Chain Saturation. J. Phys. Chem. B 2016, 120, 3880–3887. [Google Scholar] [CrossRef]
- Schmidtchen, A.; Malmsten, M. (Lipo)polysaccharide interactions of antimicrobial peptides. J. Colloid Interface Sci. 2015, 449, 136–142. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.W. Action of Antimicrobial Peptides: Two-State Model†. Biochemistry 2000, 39, 8347–8352. [Google Scholar] [CrossRef] [PubMed]
- Conlon, J.M.; Demandt, A.; Nielsen, P.F.; Leprince, J.; Vaudry, H.; Woodhams, D.C. The alyteserins: Two families of antimicrobial peptides from the skin secretions of the midwife toad Alytes obstetricans (Alytidae). Peptides 2009, 30, 1069–1073. [Google Scholar] [CrossRef] [PubMed]
- Cantor, S.; Vargas, L.; Rojas A, O.E.; Yarce, C.J.; Salamanca, C.H.; Oñate-Garzón, J. Evaluation of the Antimicrobial Activity of Cationic Peptides Loaded in Surface-Modified Nanoliposomes against Foodborne Bacteria. Int. J. Mol. Sci. 2019, 20, 680. [Google Scholar] [CrossRef] [PubMed]
- Subasinghage, A.P.; O’Flynn, D.; Conlon, J.M.; Hewage, C.M. Conformational and membrane interaction studies of the antimicrobial peptide alyteserin-1c and its analogue [E4K]alyteserin-1c. Biochim. Biophys. Acta (BBA) Biomembr. 2011, 1808, 1975–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.-H.; Heng, C.; Separovic, F.; Aguilar, M.-I. Comparison of reversible membrane destabilisation induced by antimicrobial peptides derived from Australian frogs. Biochim. Biophys. Acta (BBA) Biomembr. 2014, 1838, 2205–2215. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Kasetty, G.; Schmidtchen, A.; Malmsten, M. Membrane and lipopolysaccharide interactions of C-terminal peptides from S1 peptidases. Biochim. Biophys. Acta (BBA) Biomembr. 2012, 1818, 2244–2251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, S.B.; Otzen, D.E. Impact of the antimicrobial peptide Novicidin on membrane structure and integrity. J. Colloid Interface Sci. 2010, 345, 248–256. [Google Scholar] [CrossRef]
- Oñate-Garzón, J.; Ausili, A.; Manrique-Moreno, M.; Torrecillas, A.; Aranda, F.J.; Patiño, E.; Gomez-Fernández, J.C. The increase in positively charged residues in cecropin D-like Galleria mellonella favors its interaction with membrane models that imitate bacterial membranes. Arch. Biochem. Biophys. 2017, 629, 54–62. [Google Scholar] [CrossRef]
- Oñate-Garzón, J.; Manrique-Moreno, M.; Trier, S.; Leidy, C.; Torres, R.; Patiño, E. Antimicrobial activity and interactions of cationic peptides derived from Galleria mellonella cecropin D-like peptide with model membranes. J. Antibiot 2017, 70, 238–245. [Google Scholar] [CrossRef]
- Correa, W.; Manrique-Moreno, M.; Patino, E.; Peláez-Jaramillo, C.; Kaconis, Y.; Gutsmann, T.; Garidel, P.; Heinbockel, L.; Brandenburg, K. Galleria mellonella native and analogue peptides Gm1 and ΔGm1. I) Biophysical characterization of the interaction mechanisms with bacterial model membranes. Biochim. Biophys. Acta (BBA) Biomembr. 2014, 1838, 2728–2738. [Google Scholar] [CrossRef]
- Abraham, T.; Prenner, E.J.; Lewis, R.N.; Mant, C.T.; Keller, S.; Hodges, R.S.; McElhaney, R.N. Structure–activity relationships of the antimicrobial peptide gramicidin S and its analogs: Aqueous solubility, self-association, conformation, antimicrobial activity and interaction with model lipid membranes. Biochim. Biophys. Acta (BBA) Biomembr. 2014, 1838, 1420–1429. [Google Scholar] [CrossRef] [PubMed]
- Berglund, N.A.; Piggot, T.J.; Jefferies, D.; Sessions, R.B.; Bond, P.J.; Khalid, S. Interaction of the Antimicrobial Peptide Polymyxin B1 with Both Membranes of E. coli: A Molecular Dynamics Study. PLoS Comput. Boil. 2015, 11, e1004180. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Schlamadinger, D.E.; Kim, J.E.; McCammon, J.A. Comparative Molecular Dynamics Simulations of the Antimicrobial Peptide CM15 in Model Lipid Bilayers. Biochim. Biophys. Acta (BBA) Bioenerg. 2012, 1818, 1402–1409. [Google Scholar] [CrossRef] [PubMed]
- Arrondo, J.L.R.; Muga, A.; Castresana, J.; Goñi, F.M. Quantitative studies of the structure of proteins in solution by fourier-transform infrared spectroscopy. Prog. Biophys. Mol. Boil. 1993, 59, 23–56. [Google Scholar] [CrossRef]
- Sánchez, M.; Scirè, A.; Tanfani, F.; Ausili, A. The thermal unfolding of the ribosome-inactivating protein saporin-S6 characterized by infrared spectroscopy. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2015, 1854, 1357–1364. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; Beer, T.A.P.D.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Martinez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekhar, J.; Impey, R.W.; Jorgensen, W.L.; Madura, J.D.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. [Google Scholar]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Feig, M.; Brooks, C.L. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D.; Pastor, R.W. Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef] [PubMed]
- MacKerell, A.D.; Feig, M.; Brooks, C.L. Improved Treatment of the Protein Backbone in Empirical Force Fields. J. Am. Chem. Soc. 2004, 126, 698–699. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordo, D.; Argos, P. Suggestions for “safe” residue substitutions in site-directed mutagenesis. J. Mol. Boil. 1991, 217, 721–729. [Google Scholar] [CrossRef]
- Lohner, K.; Latal, A.; Lehrer, R.I.; Ganz, T. Differential Scanning Microcalorimetry Indicates That Human Defensin, HNP-2, Interacts Specifically with Biomembrane Mimetic Systems. Biochemistry 1997, 36, 1525–1531. [Google Scholar] [CrossRef] [PubMed]
- Jing, W.; Demcoe, A.R.; Vogel, H.J. Conformation of a Bactericidal Domain of Puroindoline a: Structure and Mechanism of Action of a 13-Residue Antimicrobial Peptide. J. Bacteriol. 2003, 185, 4938–4947. [Google Scholar] [CrossRef] [Green Version]
- Sánchez, M.; Teruel, J.A.; Espuny, M.J.; Marqués, A.M.; Aranda, F.J.; Manresa, A.; Ortiz, A. Modulation of the physical properties of dielaidoylphosphatidylethanolamine membranes by a dirhamnolipid biosurfactant produced by Pseudomonas aeruginosa. Chem. Phys. Lipids 2006, 142, 118–127. [Google Scholar] [CrossRef]
- Conlon, J.M.; Ahmed, E.; Pal, T.; Sonnevend, A. Potent and rapid bactericidal action of alyteserin-1c and its [E4K] analog against multidrug-resistant strains of Acinetobacter baumannii. Peptides 2010, 31, 1806–1810. [Google Scholar] [CrossRef]
- Wieprecht, T.; Dathe, M.; Krause, E.; Beyermann, M.; Maloy, W.; Macdonald, D.L.; Bienert, M. Modulation of membrane activity of amphipathic, antibacterial peptides by slight modifications of the hydrophobic moment. FEBS Lett. 1997, 417, 135–140. [Google Scholar] [CrossRef] [Green Version]
- Ausili, A.; Cobucci-Ponzano, B.; Di Lauro, B.; D’Avino, R.; Scirè, A.; Rossi, M.; Tanfani, F.; Moracci, M. Structural basis of the destabilization produced by an amino-terminal tag in the β-glycosidase from the hyperthermophilic archeon Sulfolobus solfataricus. Biochimie 2006, 88, 807–817. [Google Scholar] [CrossRef]
- Susi, H.; Timasheff, S.N.; Stevens, L. Infrared spectra and protein conformations in aqueous solutions. I. The amide I band in H2O and D2O solutions. J. Biol. Chem. 1967, 242, 5460–5466. [Google Scholar] [PubMed]
- Mannini, R.; Rivieccio, V.; D’Auria, S.; Tanfani, F.; Ausili, A.; Facchiano, A.; Pedone, C.; Grimaldi, G. Structure/function of KRAB repression domains: Structural properties of KRAB modules inferred from hydrodynamic, circular dichroism, and FTIR spectroscopic analyses. Proteins: Struct. Funct. Bioinform. 2006, 65, 524. [Google Scholar] [CrossRef]
- Yassine, W.; Taib, N.; Federman, S.; Milochau, A.; Castano, S.; Sbi, W.; Manigand, C.; Laguerre, M.; Desbat, B.; Oda, R.; et al. Reversible transition between α-helix and β-sheet conformation of a transmembrane domain. Biochim. Biophys. Acta (BBA) Biomembr. 2009, 1788, 1722–1730. [Google Scholar] [CrossRef] [PubMed]
- Barth, A. The infrared absorption of amino acid side chains. Prog. Biophys. Mol. Boil. 2000, 74, 141–173. [Google Scholar] [CrossRef]
- White, S.H.; Wimley, W.C. Membrane Protein Folding And Stability: Physical Principles. Annu. Rev. Biophys. Biomol. Struct. 2002, 28, 319–365. [Google Scholar] [CrossRef] [PubMed]
- Krimm, S.; Bandekar, J. Vibrational Spectroscopy and Conformation of Peptides, Polypeptides, and Proteins. Membr. Proteins 1986, 38, 181–364. [Google Scholar]
- Rankin, S.E.; Watts, A.; Pinheiro, T.J.T. Electrostatic and hydrophobic contributions to the folding mechanism of apocytochrome c driven by the interaction with lipid. Biochemistry 1998, 37, 12588–12595. [Google Scholar] [CrossRef] [PubMed]
- Ausili, A.; De Godos, A.; Torrecillas, A.; Corbalán-García, S.; Gómez-Fernández, J.C.; Torrecillas-Sanchez, A. The interaction of the Bax C-terminal domain with membranes is influenced by the presence of negatively charged phospholipids. Biochim. Biophys. Acta (BBA) Biomembr. 2009, 1788, 1924–1932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafalski, M.; Lear, J.D.; DeGrado, W.F. Phospholipid interactions of synthetic peptides representing the N-terminus of HIV gp41. Biochemistry 1990, 29, 7917–7922. [Google Scholar] [CrossRef]
- Nieva, J.L.; Nir, S.; Muga, A.; Goñi, F.M.; Wilschut, J. Interaction of the HIV-1 Fusion Peptide with Phospholipid Vesicles: Different Structural Requirements for Fusion and Leakage. Biochemistry 1994, 33, 3201–3209. [Google Scholar] [CrossRef] [PubMed]
- Mobley, P.W.; Waring, A.J.; Sherman, M.A.; Gordon, L.M. Membrane interactions of the synthetic N-terminal peptide of HIV-1 gp41 and its structural analogs. Biochim. Biophys. Acta (BBA) Biomembr. 1999, 1418, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Heller, W.T.; Zolnierczuk, P.A. The helix-to-sheet transition of an HIV-1 fusion peptide derivative changes the mechanical properties of lipid bilayer membranes. Biochim. Biophys. Acta (BBA) Biomembr. 2019, 1861, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Krone, M.; Stone, J.; Ertl, T.; Schulten, K. Fast Visualization of Gaussian Density Surfaces for Molecular Dynamics and Particle System Trajectories. Presented at EuroVis, Austria, 5–8 June 2012. Available online: https://www.cg.tuwien.ac.at/eurovis2012/ (accessed on 27 August 2019).
- Agopian, A.; Castano, S. Structure and orientation study of Ebola fusion peptide inserted in lipid membrane models. Biochim. Biophys. Acta (BBA) Biomembr. 2014, 1838, 117–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]

 ); 1:100 (
); 1:100 (  ); 1:50 (
); 1:50 (  ); and 1:25 (
); and 1:25 (  ).
); 1:100 ( ); 1:50 ( ); and 1:25 ( ).
).
); 1:100 ( ); 1:50 ( ); and 1:25 ( ).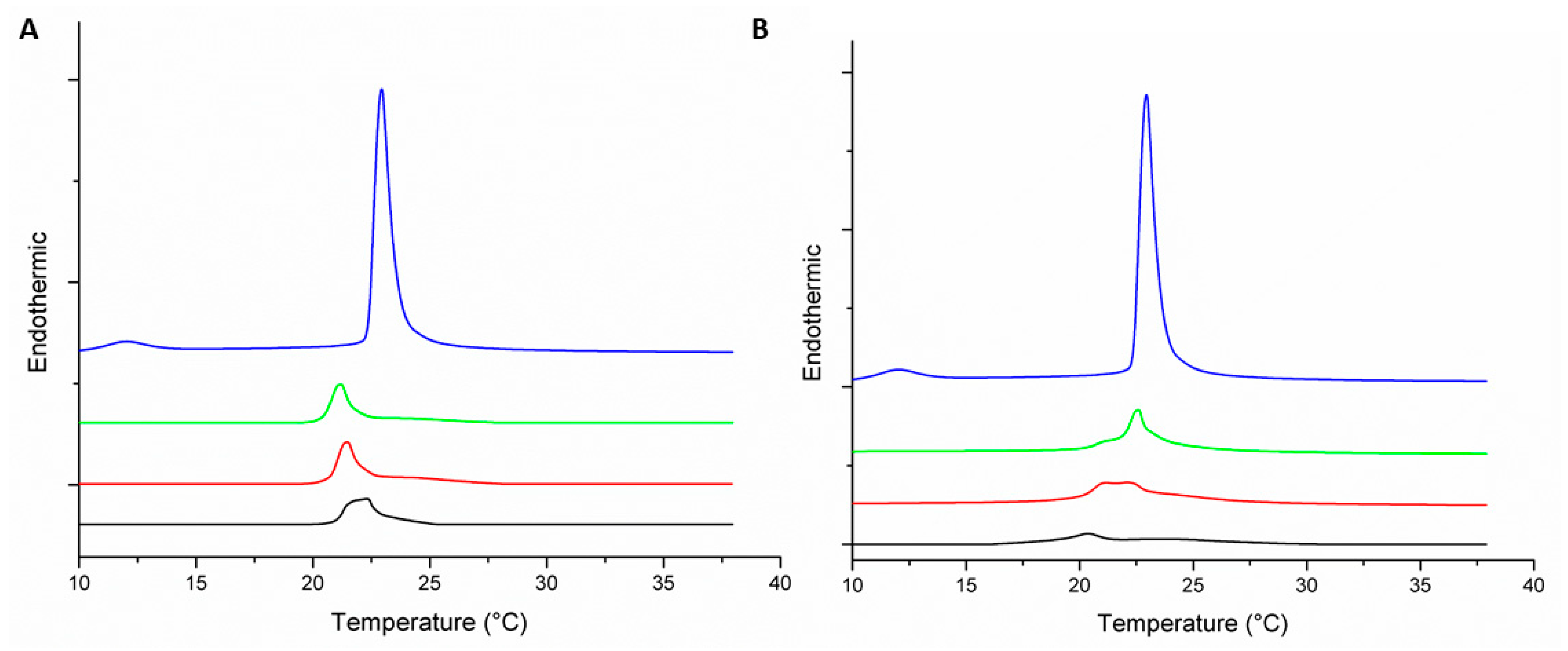 ); 1:100 ( ); 1:50 ( ); and 1:25 ( ).
); 1:100 ( ); 1:50 ( ); and 1:25 ( ).
); 1:100 ( ); 1:50 ( ); and 1:25 ( ).
); 1:100 ( ); 1:50 ( ); and 1:25 ( ).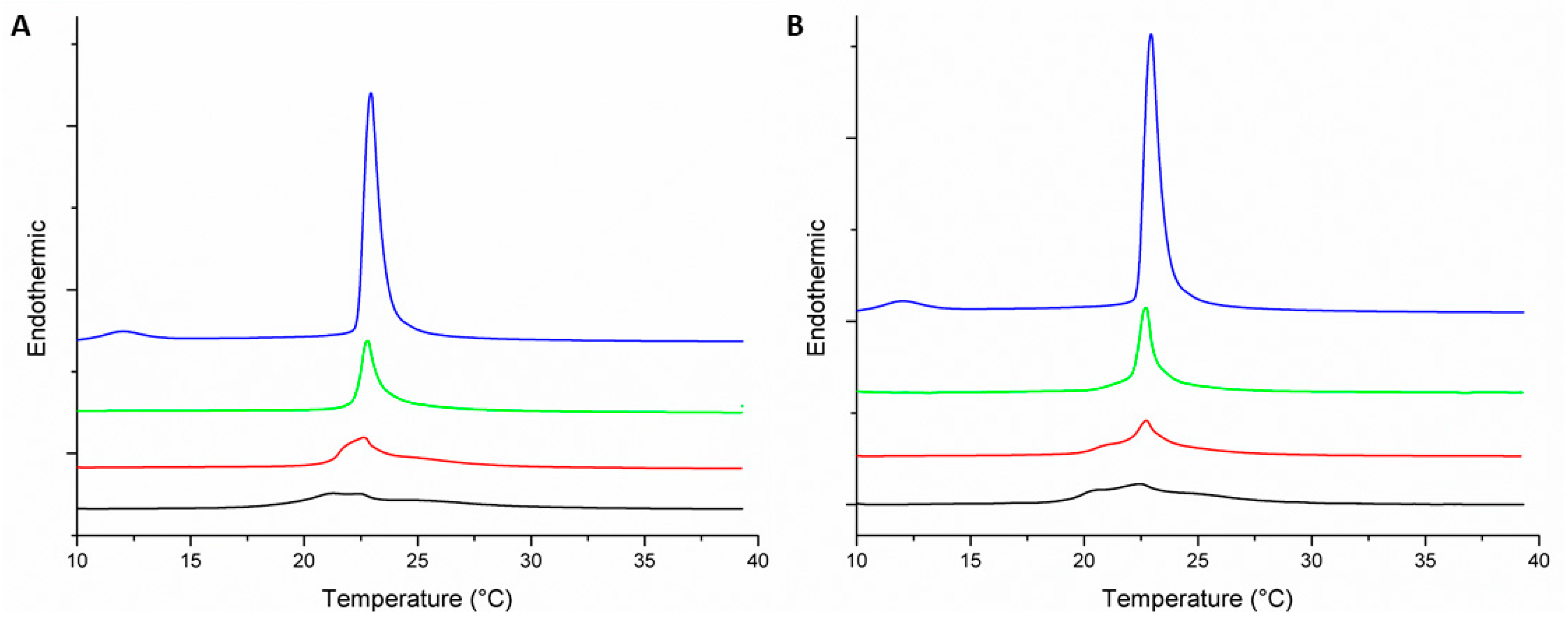


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MLV | Peptide–Lipid Molar Ratio | Pre-Transition Temperature (°C) | Tm (°C) | ΔH (J·g−1) |
|---|---|---|---|---|
| DMPC | 0:1 | 12.9 | 22.9 | 1.47 |
| DMPC-Peptide +2 | 1:100 | 21.2 | 0.63 | |
| 1:50 | 21.5 | 0.62 | ||
| 1:25 | 21.2 | 0.62 | ||
| DMPC-Peptide +5 | 1:100 | 22.7 | 0.8 | |
| 1:50 | 22.2 | 0.63 | ||
| 1:25 | 20.5 | 0.18 | ||
| DMPC/DMPG (3:1) | 0:1 | 12 | 22.9 | 1.6 |
| DMPC/DMPG (3:1)-Peptide +2 | 1:100 | 22.9 | 0.54 | |
| 1:50 | 22.8 | 0.38 | ||
| 1:25 | 21.3 | 0.30 | ||
| DMPC/DMPG (3:1)-Peptide +5 | 1:100 | 22.9 | 0.64 | |
| 1:50 | 23 | 0.45 | ||
| 1:25 | 22.6 | 0.44 |
| Peptide | α-Helix (%) | β-Sheet (%) | Turns (%) | Unordered (%) | Aggregation (%) | C=O Band Area/Total Area (%) |
|---|---|---|---|---|---|---|
| +2 | 66 | 34 | ||||
| +2 + POPC | 64 | 36 | 22 | |||
| +2 + POPC/POPG | 51 | 49 | 29 | |||
| +5 | 48 | 52 | ||||
| +5 + POPC | 72 | 28 | 14 | |||
| +5 + POPC/POPG | 81 | 19 | 16 | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aragón-Muriel, A.; Ausili, A.; Sánchez, K.; Rojas A., O.E.; Londoño Mosquera, J.; Polo-Cerón, D.; Oñate-Garzón, J. Studies on the Interaction of Alyteserin 1c Peptide and Its Cationic Analogue with Model Membranes Imitating Mammalian and Bacterial Membranes. Biomolecules 2019, 9, 527. https://doi.org/10.3390/biom9100527
Aragón-Muriel A, Ausili A, Sánchez K, Rojas A. OE, Londoño Mosquera J, Polo-Cerón D, Oñate-Garzón J. Studies on the Interaction of Alyteserin 1c Peptide and Its Cationic Analogue with Model Membranes Imitating Mammalian and Bacterial Membranes. Biomolecules. 2019; 9(10):527. https://doi.org/10.3390/biom9100527
Chicago/Turabian StyleAragón-Muriel, Alberto, Alessio Ausili, Kevin Sánchez, Oscar E. Rojas A., Juan Londoño Mosquera, Dorian Polo-Cerón, and Jose Oñate-Garzón. 2019. "Studies on the Interaction of Alyteserin 1c Peptide and Its Cationic Analogue with Model Membranes Imitating Mammalian and Bacterial Membranes" Biomolecules 9, no. 10: 527. https://doi.org/10.3390/biom9100527