Pharmacophore Directed Screening of Agonistic Natural Molecules Showing Affinity to 5HT2C Receptor

 , and
, and
Abstract
:
1. Introduction
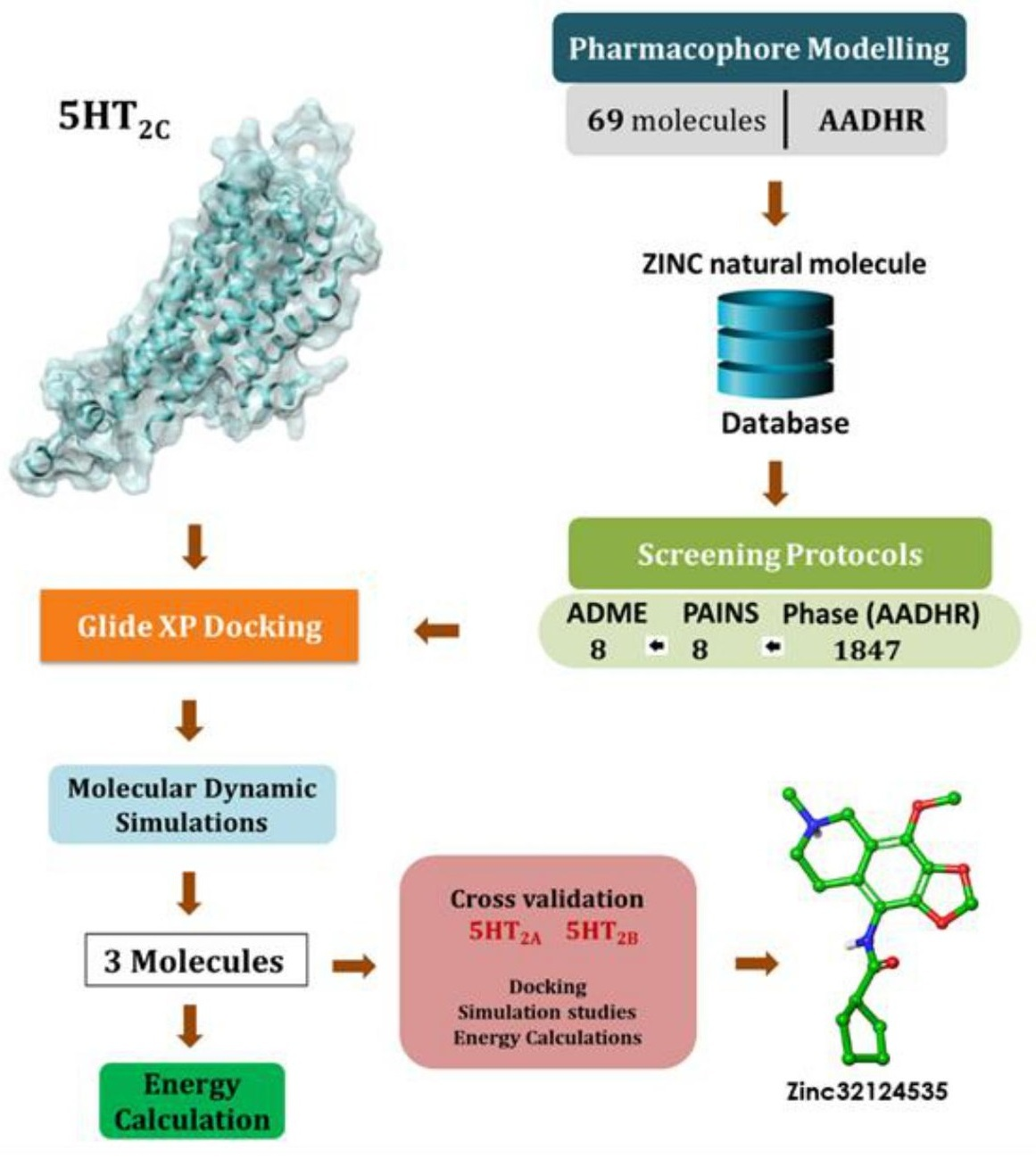
2. Materials and Methods
2.1. Pharmacophore Hypotheses Generation and Screening
2.1.1. Dataset Preparation
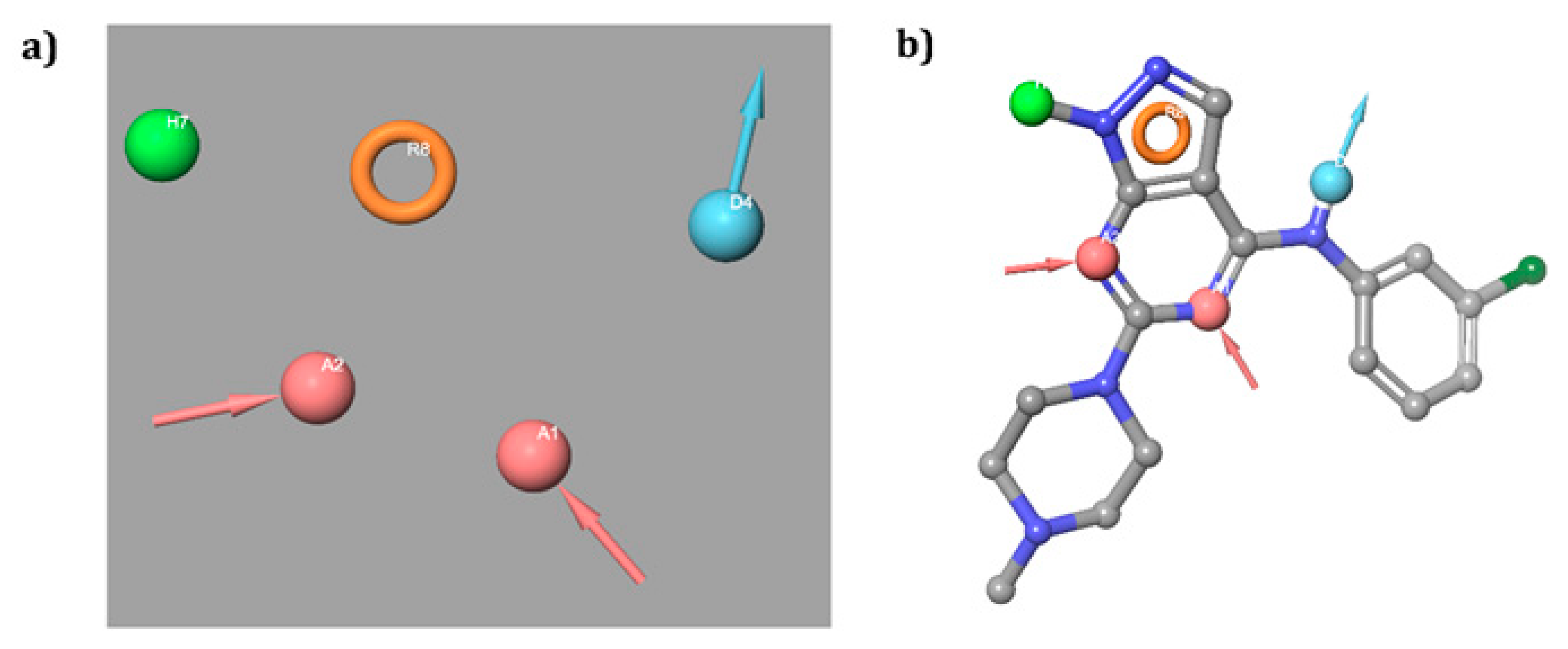
2.1.2. Pharmacophore Hypotheses Generation and Validation
2.1.3. Screening of the ZINC Database
2.2. ADME Screening and PAINS
2.3. Receptors Preparation
2.4. Ligand Docking Studies
2.5. Binding Free Energy Calculations Using Prime/MM-GBSA
2.6. Molecular Dynamics Simulations
3. Results and Discussion
3.1. Pharmacophore Modeling and Validations
3.2. Screening of the ZINC Natural Molecule Database
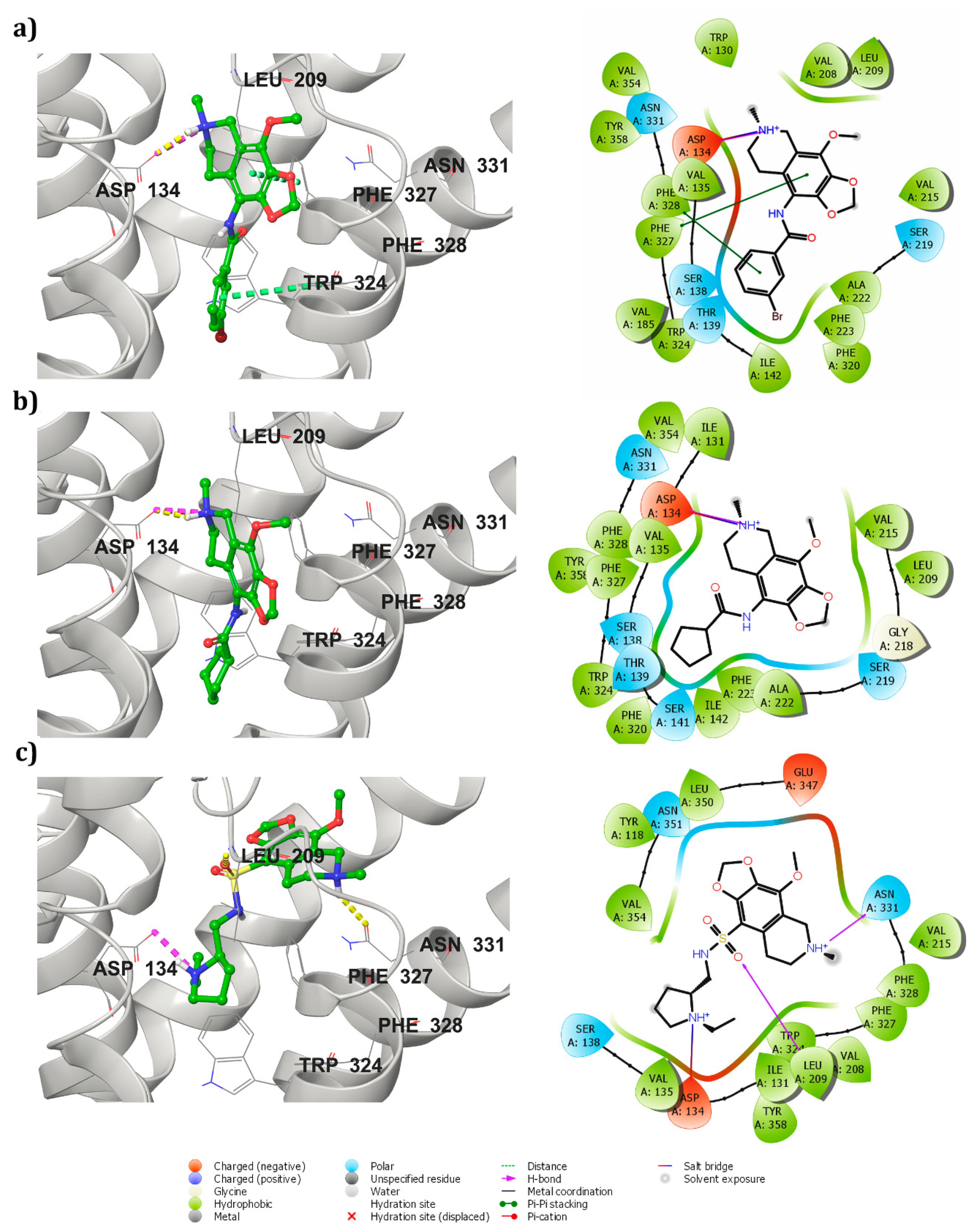
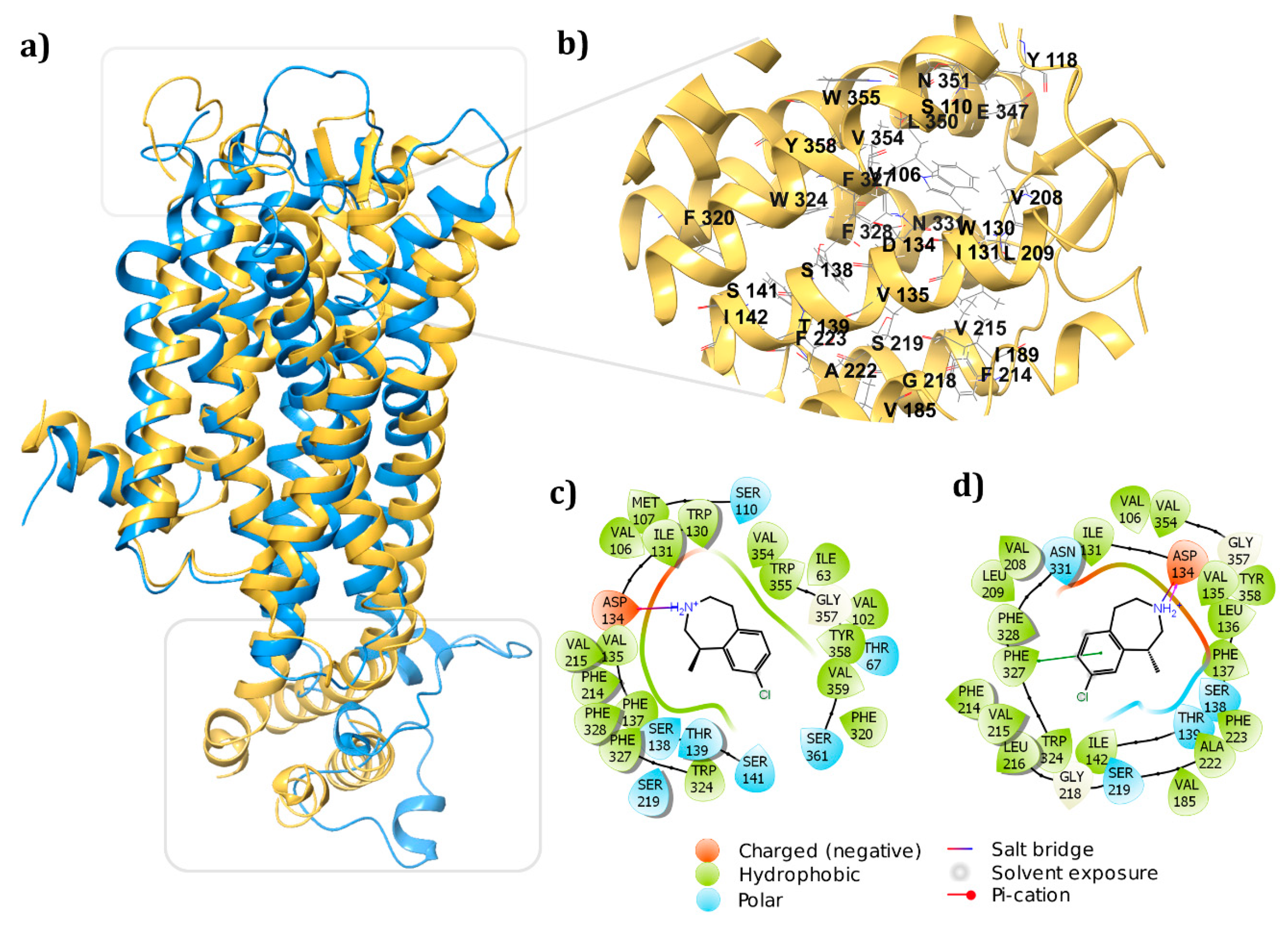
3.3. Receptor-Ligand Docking Studies (5HT2C)
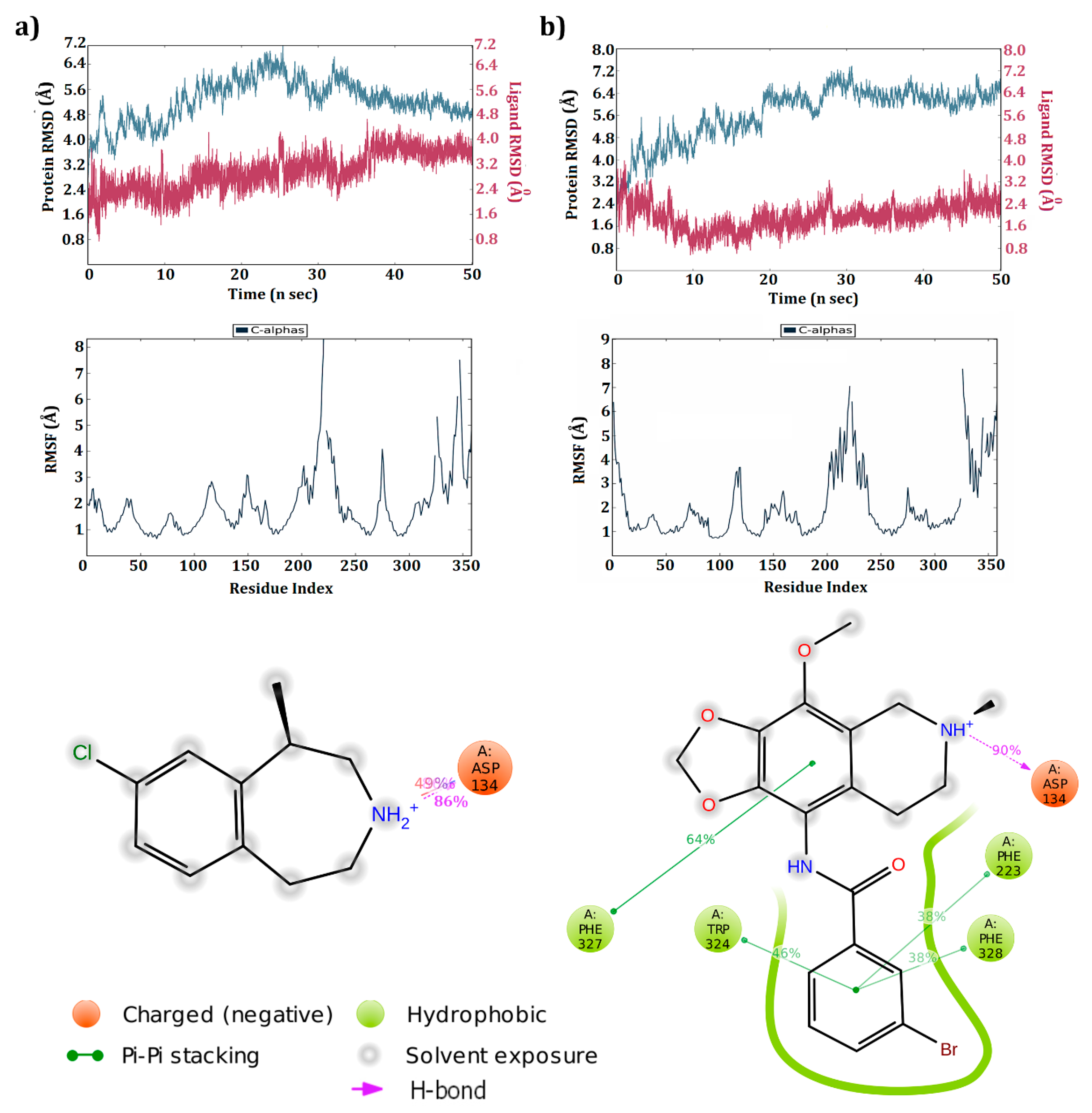
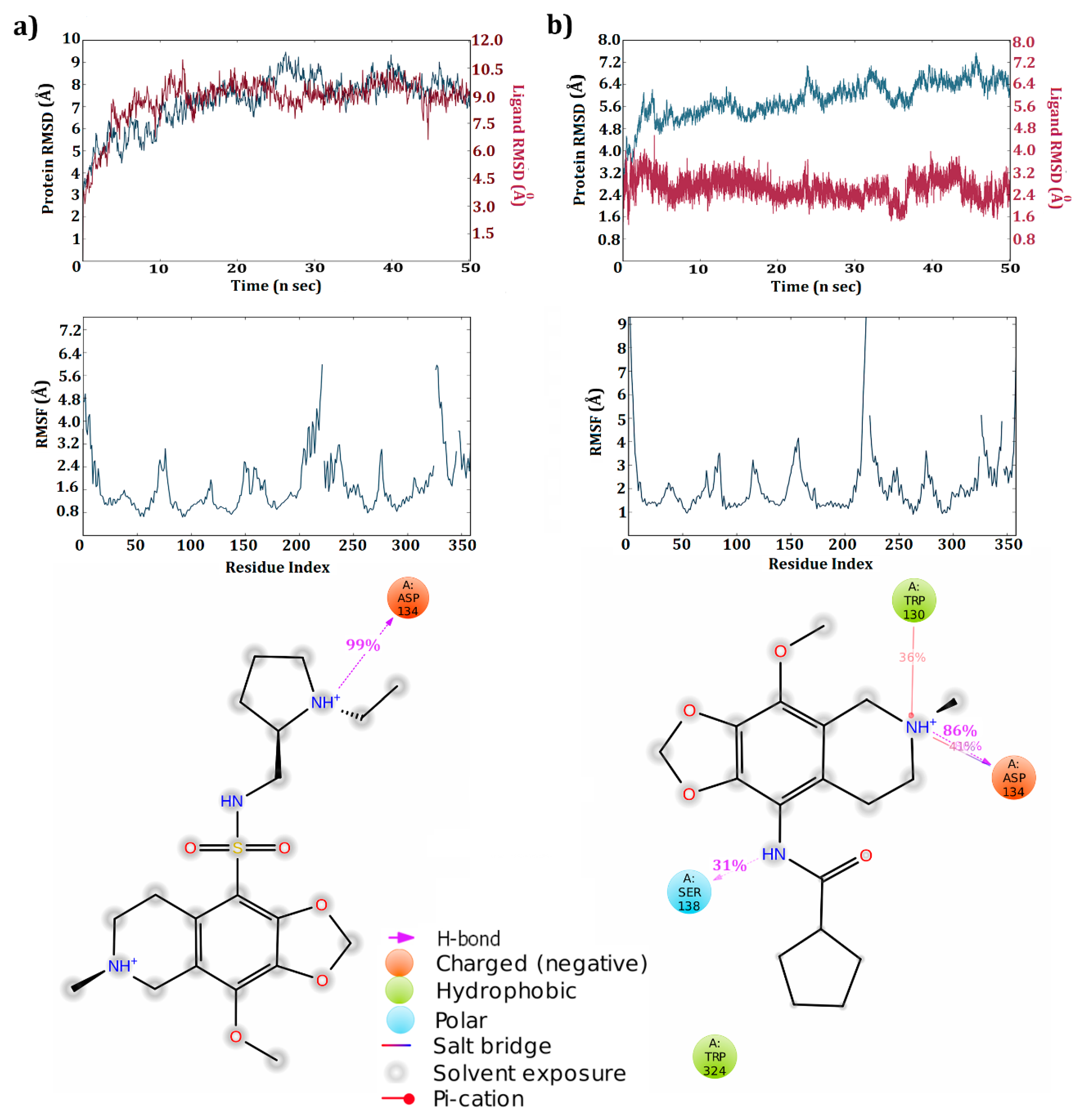
3.4. Dynamic Simulations of the 5HT2C-Ligand Complexes
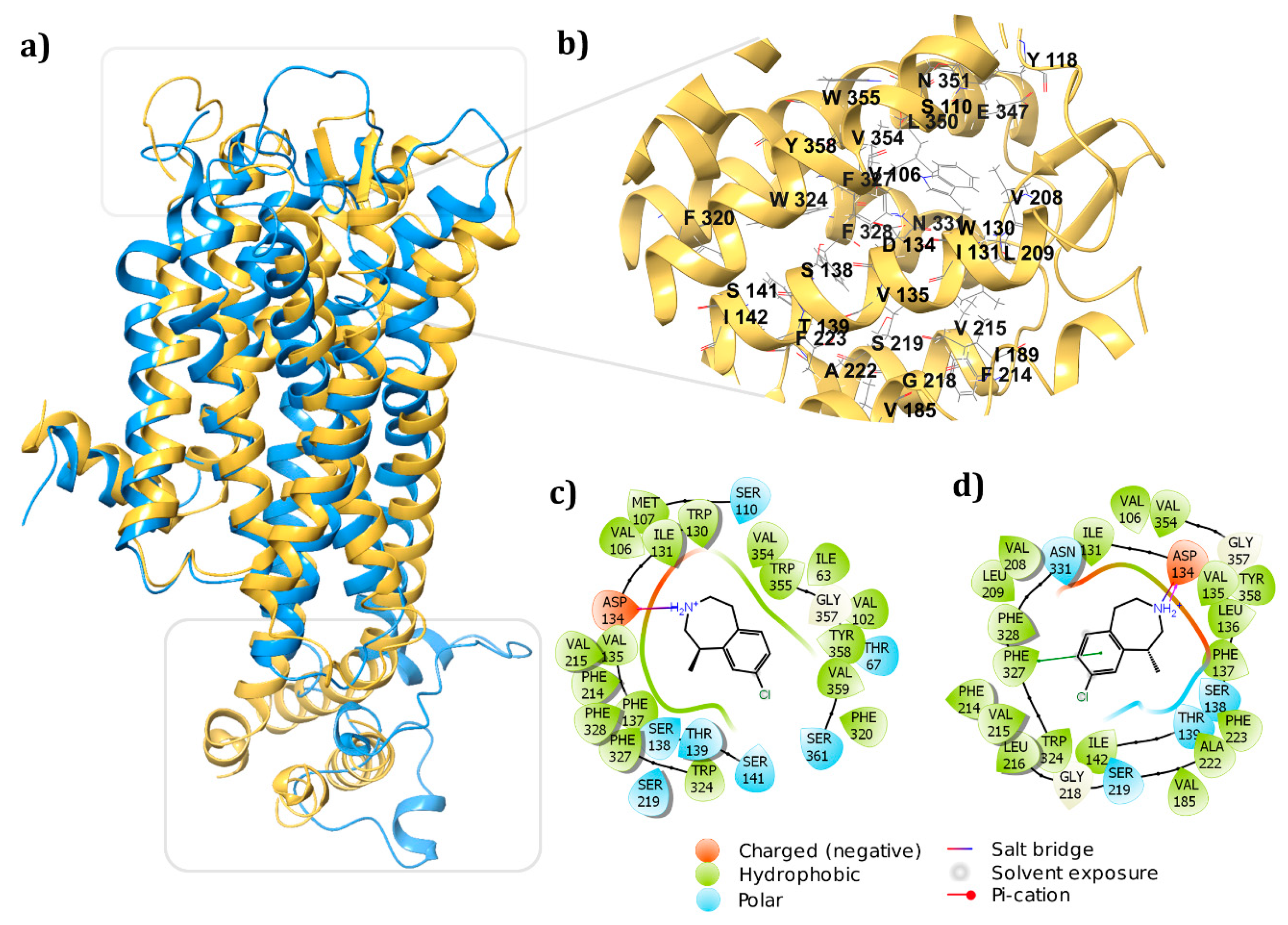
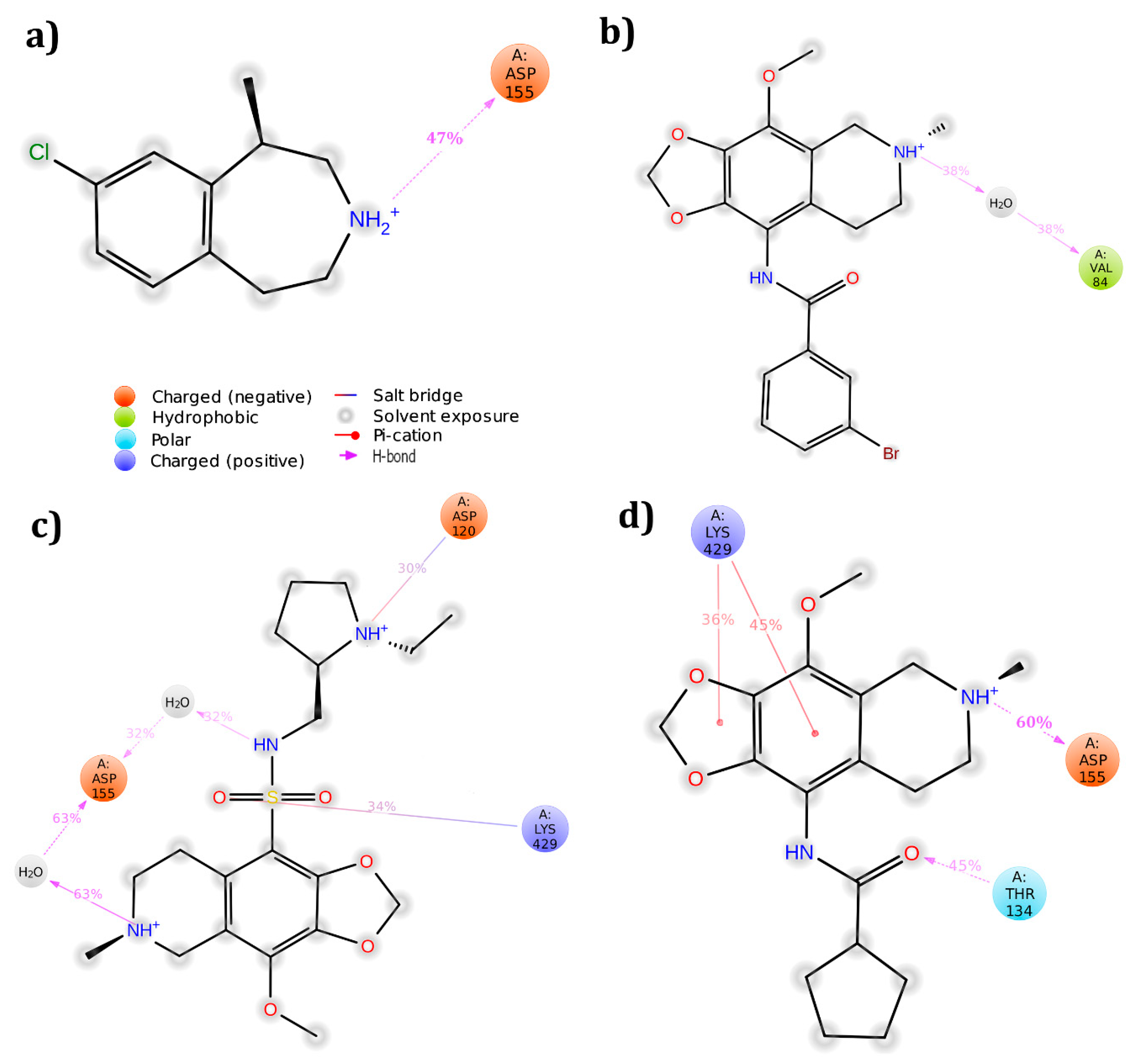
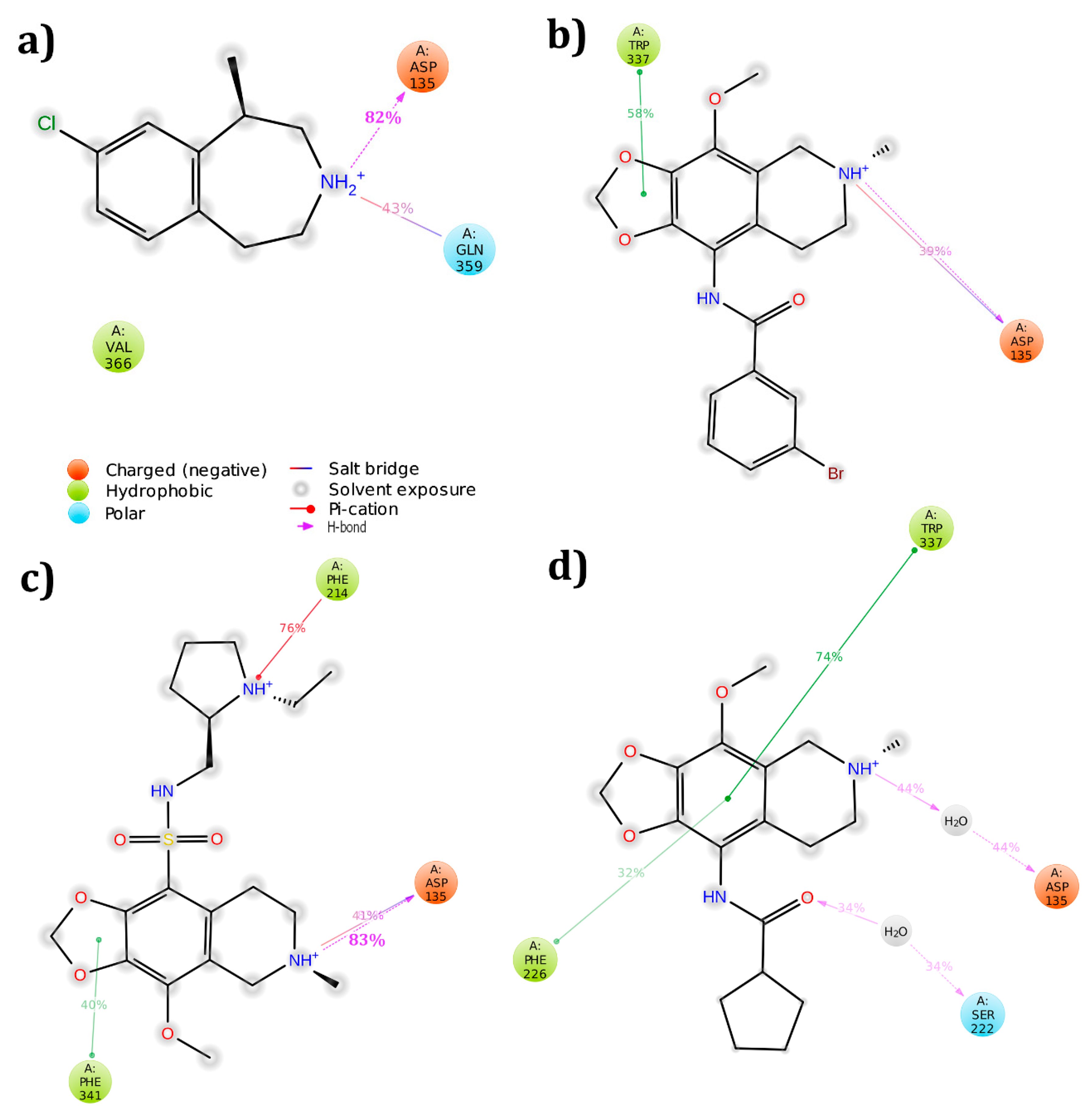
3.5. Cross Validation of the Hits
3.5.1. 5HT2A Receptor
3.5.2. 5HT2B Receptor
3.6. Homology Model vs. Crystal Structure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kopelman, P.G. Obesity as a medical problem. Nature 2000, 404, 635. [Google Scholar] [CrossRef] [PubMed]
- Jebb, S. Obesity: Causes and consequences. Women’s Health Med. 2004, 1, 38–41. [Google Scholar] [CrossRef]
- Finer, N. Medical consequences of obesity. Medicine 2006, 34, 510–514. [Google Scholar] [CrossRef]
- Schwartz, M.W.; Seeley, R.J.; Zeltser, L.M.; Drewnowski, A.; Ravussin, E.; Redman, L.M.; Leibel, R.L. Obesity pathogenesis: An Endocrine Society scientific statement. Endocr. Rev. 2017, 38, 267–296. [Google Scholar] [CrossRef] [PubMed]
- Neeland, I.J.; Poirier, P.; Després, J.-P. Cardiovascular and metabolic heterogeneity of obesity: Clinical challenges and implications for management. Circulation 2018, 137, 1391–1406. [Google Scholar] [CrossRef]
- Gadde, K.M.; Apolzan, J.W.; Berthoud, H.-R. Pharmacotherapy for patients with obesity. Clin. Chem. 2018, 64, 118–129. [Google Scholar] [CrossRef]
- Cohen, J.B.; Gadde, K.M. Weight Loss Medications in the Treatment of Obesity and Hypertension. Curr. Hypertens. Rep. 2019, 21, 16. [Google Scholar] [CrossRef]
- Tecott, L.H.; Sun, L.M.; Akana, S.F.; Strack, A.M.; Lowenstein, D.H.; Dallman, M.F.; Julius, D. Eating disorder and epilepsy in mice lacking 5-HT2c serotonin receptors. Nature 1995, 374, 542. [Google Scholar] [CrossRef]
- Millan, M.J.; Peglion, J.-L.; Lavielle, G.; Perrin-Monneyron, S. 5-HT2C receptors mediate penile erections in rats: Actions of novel and selective agonists and antagonists. Eur. J. Pharmacol. 1997, 325, 9–12. [Google Scholar] [CrossRef]
- Delgado, P.L.; Moreno, F.A. Hallucinogens, serotonin and obsessive-compulsive disorder. J. Psychoact. Drugs 1998, 30, 359–366. [Google Scholar] [CrossRef]
- Martin, J.; Bös, M.; Jenck, F.; Moreau, J.-L.; Mutel, V.; Sleight, A.; Wichmann, J.; Andrews, J.; Berendsen, H.; Broekkamp, C. 5-HT2C receptor agonists: Pharmacological characteristics and therapeutic potential. J. Pharmacol. Exp. Ther. 1998, 286, 913–924. [Google Scholar] [PubMed]
- Weissman, N. Appetite suppressant valvulopathy: A review of current data. Cardiovasc. Rev. Rep. 1999, 20, 146–155. [Google Scholar]
- Roth, B.L.; Lopez, E.; Patel, S.; Kroeze, W.K. The multiplicity of serotonin receptors: Uselessly diverse molecules or an embarrassment of riches? Neuroscience 2000, 6, 252–262. [Google Scholar] [CrossRef]
- Shapiro, D.A.; Roth, B.L. Insights into the structure and function of 5-HT2 familyserotonin receptors reveal novel strategies for therapeutic target development. Expert Opin. Ther. Targets 2001, 5, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Rocha, B.A.; Goulding, E.H.; O’Dell, L.E.; Mead, A.N.; Coufal, N.G.; Parsons, L.H.; Tecott, L.H. Enhanced locomotor, reinforcing, and neurochemical effects of cocaine in serotonin 5-hydroxytryptamine 2C receptor mutant mice. J. Neurosci. 2002, 22, 10039–10045. [Google Scholar] [CrossRef]
- Chou-Green, J.M.; Holscher, T.D.; Dallman, M.F.; Akana, S.F. Compulsive behavior in the 5-HT2C receptor knockout mouse. Physiol. Behav. 2003, 78, 641–649. [Google Scholar] [CrossRef]
- Isaac, M. Serotonergic 5-HT2C receptors as a potential therapeutic target for the design antiepileptic drugs. Curr. Top. Med. Chem. 2005, 5, 59–67. [Google Scholar] [CrossRef]
- Miller, K.J. Serotonin 5-ht2c receptor agonists: Potential for the treatment of obesity. Mol. Interv. 2005, 5, 282–291. [Google Scholar] [CrossRef]
- Rashid, M.; Manivet, P.; Nishio, H.; Pratuangdejkul, J.; Rajab, M.; Ishiguro, M.; Launay, J.-M.; Nagatomo, T. Identification of the binding sites and selectivity of sarpogrelate, a novel 5-HT2 antagonist, to human 5-HT2A, 5-HT2B and 5-HT2C receptor subtypes by molecular modeling. Life Sci. 2003, 73, 193–207. [Google Scholar] [CrossRef]
- Girardet, C.; Butler, A.A. Neural melanocortin receptors in obesity and related metabolic disorders. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2014, 1842, 482–494. [Google Scholar] [CrossRef]
- Martin, C.K.; Redman, L.M.; Zhang, J.; Sanchez, M.; Anderson, C.M.; Smith, S.R.; Ravussin, E. Lorcaserin, a 5-HT2C receptor agonist, reduces body weight by decreasing energy intake without influencing energy expenditure. J. Clin. Endocrinol. Metab. 2011, 96, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Blundell, J.E.; Halford, J.C. Serotonin and appetite regulation. CNS Drugs 1998, 9, 473–495. [Google Scholar] [CrossRef]
- Fitzgerald, L.W.; Ennis, M.D. 5-HT2C receptor modulators: Progress in development of new CNS medicines. Annu. Rep. Med. Chem. 2002, 37, 21–30. [Google Scholar]
- Smith, S.R.; Prosser, W.A.; Donahue, D.J.; Morgan, M.E.; Anderson, C.M.; Shanahan, W.R.; Group, A.S. Lorcaserin (APD356), a selective 5-HT2C agonist, reduces body weight in obese men and women. Obesity 2009, 17, 494–503. [Google Scholar] [CrossRef]
- Bohula, E.A.; Wiviott, S.D.; McGuire, D.K.; Inzucchi, S.E.; Kuder, J.; Im, K.; Fanola, C.L.; Qamar, A.; Brown, C.; Budaj, A. Cardiovascular safety of lorcaserin in overweight or obese patients. N. Engl. J. Med. 2018, 379, 1107–1117. [Google Scholar] [CrossRef]
- DiNicolantonio, J.J.; Chatterjee, S.; O’Keefe, J.H.; Meier, P. Lorcaserin for the treatment of obesity? A closer look at its side effects. Arch. Dis. Child. 2014. [Google Scholar] [CrossRef]
- Ahmed, A.; Choo, H.; Cho, Y.S.; Park, W.-K.; Pae, A.N. Identification of novel serotonin 2C receptor ligands by sequential virtual screening. Bioorg. Med. Chem. 2009, 17, 4559–4568. [Google Scholar] [CrossRef]
- Dixon, S.L.; Smondyrev, A.M.; Knoll, E.H.; Rao, S.N.; Shaw, D.E.; Friesner, R.A. PHASE: A new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput. Aided Mol. Des. 2006, 20, 647–671. [Google Scholar] [CrossRef]
- Veeramachaneni, G.K.; Raj, K.K.; Chalasani, L.M.; Annamraju, S.K.; JS, B.; Talluri, V.R. Shape based virtual screening and molecular docking towards designing novel pancreatic lipase inhibitors. Bioinformation 2015, 11, 535. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Berman, H.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.; Weissig, H.; Shindyalov, I.; Bourne, P. The protein data Bank nucleic acids research. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein—Ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Guo, Z.; Mohanty, U.; Noehre, J.; Sawyer, T.K.; Sherman, W.; Krilov, G. Probing the α-helical structural stability of stapled p53 peptides: Molecular dynamics simulations and analysis. Chem. Biol. Drug Des. 2010, 75, 348–359. [Google Scholar] [CrossRef]
- Veeramachaneni, G.K.; Raj, K.K.; Chalasani, L.M.; Bondili, J.S.; Talluri, V.R. High-throughput virtual screening with e-pharmacophore and molecular simulations study in the designing of pancreatic lipase inhibitors. Drug Des. Dev. Ther. 2015, 9, 4397. [Google Scholar] [CrossRef]
- Liu, K.K.-C.; Cornelius, P.; Patterson, T.A.; Zeng, Y.; Santucci, S.; Tomlinson, E.; Gibbons, C.; Maurer, T.S.; Marala, R.; Brown, J. Design and synthesis of orally-active and selective azaindane 5HT2c agonist for the treatment of obesity. Bioorg. Med. Chem. Lett. 2010, 20, 266–271. [Google Scholar] [CrossRef]
- Peng, Y.; McCorvy, J.D.; Harpsøe, K.; Lansu, K.; Yuan, S.; Popov, P.; Qu, L.; Pu, M.; Che, T.; Nikolajsen, L.F. 5-HT2C receptor structures reveal the structural basis of GPCR polypharmacology. Cell 2018, 172, 719–730.e14. [Google Scholar] [CrossRef]
- Peng, Y.; Zhao, S.; Wu, Y.; Cao, H.; Xu, Y.; Liu, X.; Shui, W.; Cheng, J.; Zhao, S.; Shen, L. Identification of natural products as novel ligands for the human 5-HT 2C receptor. Biophys. Rep. 2018, 4, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, J.; Wang, Y.; Wu, G.; Feng, J.; Ye, X.-Y.; Morales, C.L.; Broekema, M.; Rossi, K.A.; Miller, K.J.; Murphy, B.J. Utilization of an active site mutant receptor for the identification of potent and selective atypical 5-HT2C receptor agonists. J. Med. Chem. 2017, 60, 6166–6190. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Z.; Chen, G.; Luo, X.; Puah, C.; Zhu, W.; Chen, K.; Jiang, H. Pharmacophore-directed Homology Modeling and Molecular Dynamics Simulation of G Protein-coupled Receptor: Study of Possible Binding Modes of 5-HT2C Receptor Agonists. Acta Biochim. Biophys. Sin. 2007, 39, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Nagarajan, S.; Doddareddy, M.R.; Cho, Y.-S.; Pae, A.-N. Binding mode prediction of 5-hydroxytryptamine 2C receptor ligands by homology modeling and molecular docking analysis. Bull. Korean Chem. Soc. 2011, 32, 2008–2014. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hypothesis ID | No. of Sites | Survival Score | Site Score | Vector Score | Volume Score | Selectivity |
|---|---|---|---|---|---|---|
| AADHR | 5 | 5.143 | 0.766 | 0.825 | 0.622 | 1.515 |
| AAADH | 4.909 | 0.688 | 0.769 | 0.517 | 1.214 | |
| AAAH | 4 | 5.064 | 0.957 | 0.994 | 0.650 | 1.015 |
| AAHR | 5.171 | 0.860 | 0.972 | 0.631 | 1.217 | |
| DHRR | 5.090 | 0.660 | 0.934 | 0.592 | 1.490 | |
| ADHR | 4.939 | 0.737 | 0.919 | 0.581 | 1.287 |
| Core Moiety |  | |||||||
|---|---|---|---|---|---|---|---|---|
| Molecule | R | G-Score | PAINS | CNS a | mol_MW b | QPlog BB c | Percent Human Oral Absorption d | Rule of Five e |
| ZINC32123898 |  | −9.58 | Passed | 2 | 304.34 | 0.475 | 92.5 | 0 |
| ZINC32123782 |  | −8.93 | Passed | 2 | 374.82 | 0.499 | 96.8 | 0 |
| ZINC32123870 |  | −8.66 | Passed | 2 | 419.27 | 0.457 | 95.7 | 0 |
| ZINC32124535 |  | −8.51 | Passed | 2 | 332.39 | 0.476 | 96.2 | 0 |
| ZINC40312983 |  | −7.79 | Passed | 2 | 411.51 | 0.539 | 74.3 | 0 |
| ZINC40309640 |  | −7.80 | Passed | 2 | 413.48 | 0.462 | 66.3 | 0 |
| ZINC32124107 |  | −6.26 | Passed | 2 | 374.45 | 0.472 | 100 | 0 |
| Lorcaserin | - | −6.55 | Passed | 2 | 181.58 | 0.483 | 100 | 0 |
| Molecule | Receptor | Before MDS | After MDS | ||
|---|---|---|---|---|---|
| Interactions Profile | PRIME Energy (Kcal/mol) | Interactions | PRIME Energy (Kcal/mol) | ||
| Lorcaserin | 5HT2C | Asp134(1HB & 1SB) Phe327(π–π) | −41.27 | Asp134(1HB) | −43.72 |
| 5HT2B | Asp135(1SB) | −43.24 | Asp135(1HB) Gln359(1SB) | −42.04 | |
| 5HT2A | Ser 432 | −32.48 | −33.01 | ||
| ZINC 32123870 | 5HT2C | Asp134(1HB & 1SB) Phe327(π–π), Phe328(π–π) | −58.30 | Asp134(1HB) Phe327(1HB) Phe223(π–π) Trp324(π–π) Phe328(π–π) | −76.03 |
| 5HT2B | Asp135(1HB & 1SB) Phe340(pi-cation) | −62.85 | Asp135(1HB & 1SB),Trp337(π–π) | −64.20 | |
| 5HT2A | Ser 432(1HB) | −44.62 | No interactions | −58.27 | |
| ZINC 40312983 | 5HT2C | Asp134(1SB) Leu209(1HB) Asn331(1HB) | −52.52 | Asp134(1HB) Tyr358(pi-cation) Ser110(1HB) | −34.74 |
| 5HT2B | Asp135(1HB & 1SB) Phe340(pi-cation) | −62.56 | Asp135(1HB & 1SB) Phe214(pi-cation) Phe341(π–π) | −63.55 | |
| 5HT2A | Lys 429(1HB) | −48.20 | Asp120(pi-cation) | −31.12 | |
| ZINC 32124535 | 5HT2C | Asp134(1HB & 1SB) | −49.66 | Asp134 (1HB & 1SB) | −70.54 |
| 5HT2B | Asp135(1SB) | −55.75 | Phe226(π–π) Trp337(π–π) | −70.24 | |
| 5HT2A | Lys 429 (1HB&2pi-cation) | −38.39 | Asp155(1HB) Thr134(1HB) Lys 429(2pi-cation) | −41.34 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Veeramachaneni, G.K.; Thunuguntla, V.B.S.C.; Bhaswant, M.; Mathai, M.L.; Bondili, J.S. Pharmacophore Directed Screening of Agonistic Natural Molecules Showing Affinity to 5HT2C Receptor. Biomolecules 2019, 9, 556. https://doi.org/10.3390/biom9100556
Veeramachaneni GK, Thunuguntla VBSC, Bhaswant M, Mathai ML, Bondili JS. Pharmacophore Directed Screening of Agonistic Natural Molecules Showing Affinity to 5HT2C Receptor. Biomolecules. 2019; 9(10):556. https://doi.org/10.3390/biom9100556
Chicago/Turabian StyleVeeramachaneni, Ganesh Kumar, V B S C Thunuguntla, Maharshi Bhaswant, Michael L. Mathai, and Jayakumar Singh Bondili. 2019. "Pharmacophore Directed Screening of Agonistic Natural Molecules Showing Affinity to 5HT2C Receptor" Biomolecules 9, no. 10: 556. https://doi.org/10.3390/biom9100556