CDK12 Activity-Dependent Phosphorylation Events in Human Cells

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Growth
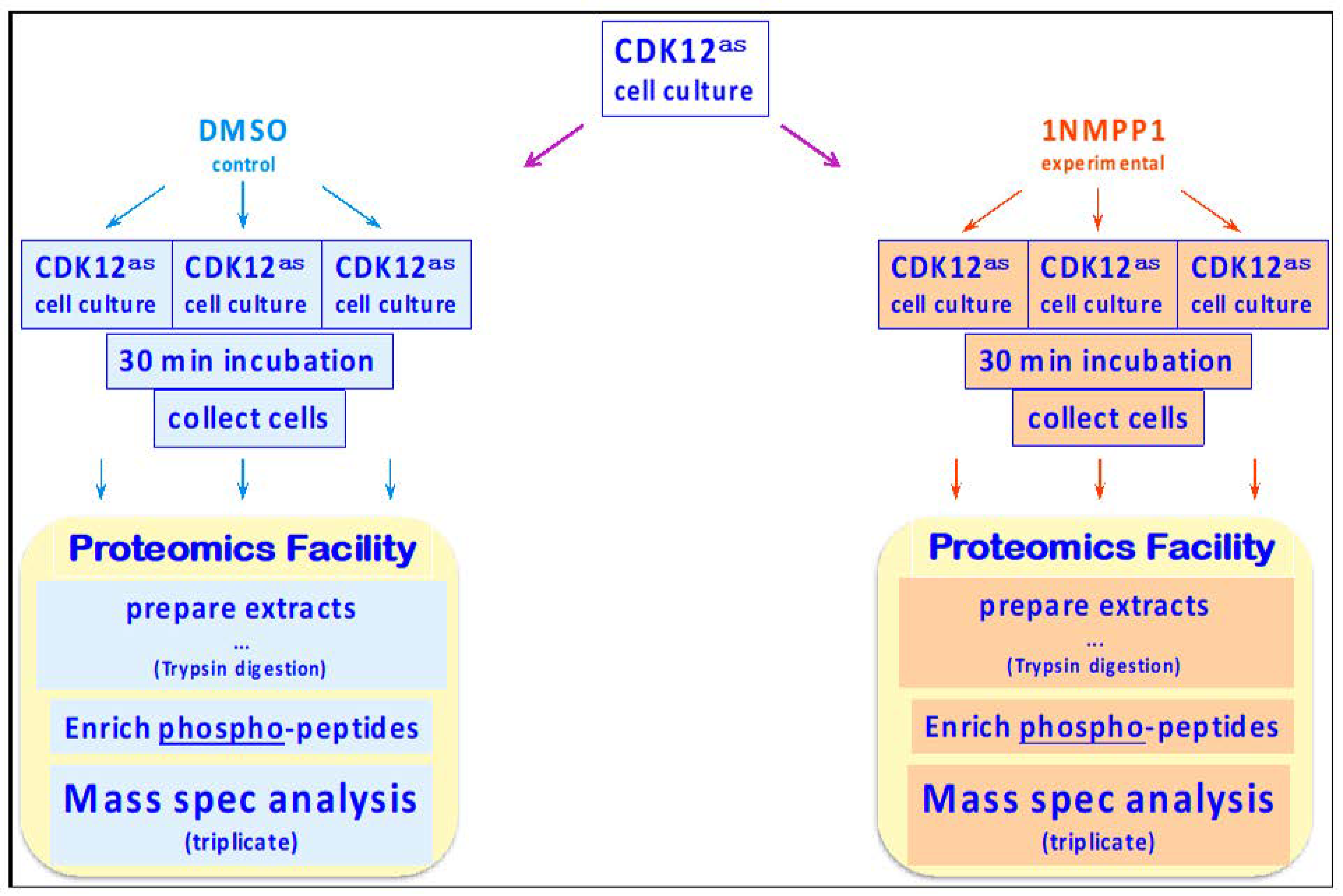
2.2. Proteomic Analyses
2.2.1. Sample Preparation
2.2.2. Phospho-Peptide Enrichment
2.2.3. Quantitative Analysis of HeLa Phosphoproteome
2.2.4. Assessment of Variability
3. Results
3.1. Phosphorylation Events Altered by Inhibitory Analog in CDK12as
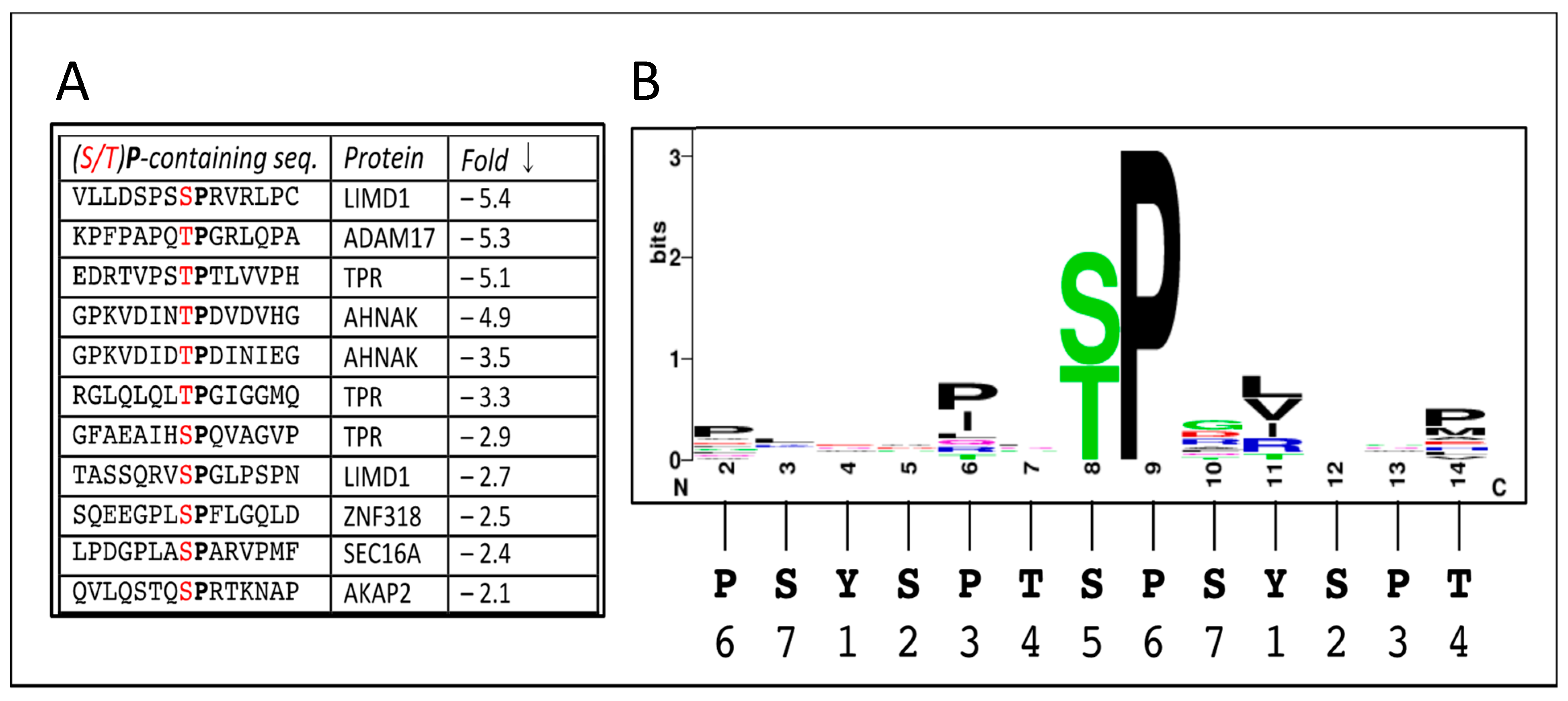
3.2. Sites Directly Phosphorylated by CDK12
3.3. Control for Off-Target Effects of Inhibitory Analog 1-NM-PP1
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- The Cancer Genome Atlas Research Network. The Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar]
- Carter, S.L.; Cibulskis, K.; Helman, E.; McKenna, A.; Shen, H.; Zack, T.; Laird, P.W.; Onofrio, R.C.; Winckler, W.; Weir, B.A.; et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol 2012, 30, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Bartkowiak, B.; Greenleaf, A.L. Expression, purification, and identification of associated proteins of the full-length hCDK12/CyclinK complex. J. Biol. Chem. 2015, 290, 1786–1795. [Google Scholar] [CrossRef]
- Blazek, D.; Kohoutek, J.; Bartholomeeusen, K.; Johansen, E.; Hulinkova, P.; Luo, Z.; Cimermancic, P.; Ule, J.; Peterlin, B.M. The Cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev 2011, 25, 2158–2172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartkowiak, B.; Yan, C.; Greenleaf, A.L. Engineering an analog-sensitive CDK12 cell line using CRISPR/Cas. Biochim Biophys Acta 2015, 1849, 1179–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bøsken, C.A.; Farnung, L.; Hintermair, C.; Schachter, M.M.; Vogel-Bachmayr, K.; Blazek, D.; Anand, K.; Fisher, R.P.; Eick, D.; Geyer, M. The structure and substrate specificity of human Cdk12/Cyclin K. Nat. Commun. 2014, 5, 1–14. [Google Scholar] [CrossRef]
- Lee, J.M.; Greenleaf, A.L. A protein kinase that phosphorylates the C-terminal repeat domain of the largest subunit of RNA polymerase II. Proc. Natl. Acad. Sci. USA. 1989, 86, 3624–3628. [Google Scholar] [CrossRef]
- Sterner, D.E.; Lee, J.M.; Hardin, S.E.; Greenleaf, A.L. The yeast carboxyl-terminal repeat domain kinase CTDK-I is a divergent cyclin-cyclin-dependent kinase complex. Mol. Cell. Biol. 1995, 15, 5716–5724. [Google Scholar] [CrossRef] [Green Version]
- Skaar, D.A.; Greenleaf, A.L. The RNA polymerase II CTD kinase CTDK-I affects pre-mRNA 3’ cleavage/polyadenylation through the processing component Pti1p. Mol. Cell 2002, 10, 1429–1439. [Google Scholar] [CrossRef]
- Ahn, S.H.; Kim, M.; Buratowski, S. Phosphorylation of serine 2 within the RNA polymerase II C-terminal domain couples transcription and 3’ end processing. Mol. Cell 2004, 13, 67–76. [Google Scholar] [CrossRef]
- Phatnani, H.P.; Jones, J.C.; Greenleaf, A.L. Expanding the functional repertoire of CTD kinase I and RNA polymerase II: Novel phosphoCTD-associating proteins in the yeast proteome. Biochemistry 2004, 43, 15702–15719. [Google Scholar] [CrossRef] [PubMed]
- Ostapenko, D.; Solomon, M.J. Budding yeast CTDK-I is required for DNA damage-induced transcription. Eukaryotic Cell 2003, 2, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Phatnani, H.P.; Guan, Z.; Sage, H.; Greenleaf, A.L.; Zhou, P. Solution structure of the Set2–Rpb1 interacting domain of human Set2 and its interaction with the hyperphosphorylated C-terminal domain of Rpb1. Proc. Natl. Acad. Sci. USA 2005, 102, 17636–17641. [Google Scholar] [CrossRef] [PubMed]
- Youdell, M.L.; Kizer, K.O.; Kisseleva-Romanova, E.; Fuchs, S.M.; Duro, E.; Strahl, B.D.; Mellor, J. Roles for Ctk1 and Spt6 in regulating the different methylation states of histone H3 lysine 36. Mol. Cell. Biol. 2008, 28, 4915–4926. [Google Scholar] [CrossRef]
- MacKellar, A.L.; Greenleaf, A.L. Cotranscriptional association of mRNA export factor Yra1 with C-terminal domain of RNA polymerase II. J. Biol. Chem. 2011, 286, 36385–36395. [Google Scholar] [CrossRef]
- Meinel, D.M.; Burkert-Kautzsch, C.; Kieser, A.; O’Duibhir, E.; Siebert, M.; Mayer, A.; Cramer, P.; Söding, J.; Holstege, F.C.P.; Strässer, K. Recruitment of TREX to the Transcription Machinery by Its Direct Binding to the Phospho-CTD of RNA Polymerase II. PLoS Genetics 2013, 9, e1003914. [Google Scholar] [CrossRef]
- Winsor, T.S.; Bartkowiak, B.; Bennett, C.B.; Greenleaf, A.L. A DNA damage response system associated with the phosphoCTD of elongating RNA polymerase II. PLoS ONE 2013, 8, e60909. [Google Scholar] [CrossRef]
- Coordes, B.; Brünger, K.M.; Burger, K.; Soufi, B.; Horenk, J.; Eick, D.; Olsen, J.V.; Strässer, K. Ctk1 function is necessary for full translation initiation activity in Saccharomyces cerevisiae. Eukaryotic Cell 2015, 14, 86–95. [Google Scholar] [CrossRef]
- Liang, K.; Gao, X.; Gilmore, J.M.; Florens, L.; Washburn, M.P.; Smith, E.; Shilatifard, A. Characterization of human cyclin-dependent kinase 12 (CDK12) and CDK13 complexes in C-terminal domain phosphorylation, gene transcription, and RNA processing. Mol. Cell. Biol. 2015, 35, 928–938. [Google Scholar] [CrossRef]
- Davidson, L.; Muniz, L.; West, S. 3’ end formation of pre-mRNA and phosphorylation of Ser2 on the RNA polymerase II CTD are reciprocally coupled in human cells. Genes Dev. 2014. [Google Scholar] [CrossRef]
- Eifler, T.T.; Shao, W.; Bartholomeeusen, K.; Fujinaga, K.; Jäger, S.; Johnson, J.R.; Luo, Z.; Krogan, N.J.; Peterlin, B.M. Cyclin-dependent kinase 12 increases 3’ end processing of growth factor-induced c-FOS transcripts. Mol. Cell. Biol. 2015, 35, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Dai, Q.; Lei, T.; Zhao, C.; Zhong, J.; Tang, Y.Z.; Chen, B.; Yang, J.; Li, C.; Wang, S.; Song, X.; et al. Cyclin K-containing Kinase Complexes Maintain Self-renewal in Murine Embryonic Stem Cells. J. Biol. Chem. 2012, 287, 25344–25352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juan, H.-C.; Lin, Y.; Chen, H.-R.; Fann, M.-J. Cdk12 is essential for embryonic development and the maintenance of genomic stability. Cell Death Differ. 2015, 23, 1038. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, F.; Thuma, L.; Klämbt, C. The regulation of glial-specific splicing of Neurexin IV requires HOW and Cdk12 activity. Development 2012, 139, 1765–1776. [Google Scholar] [CrossRef]
- Joshi, P.M.; Sutor, S.L.; Huntoon, C.J.; Karnitz, L.M. Ovarian Cancer-associated Mutations Disable Catalytic Activity of CDK12, a Kinase That Promotes Homologous Recombination Repair and Resistance to Cisplatin and Poly(ADP-ribose) Polymerase Inhibitors. J. Biol. Chem. 2014, 289, 9247–9253. [Google Scholar] [CrossRef] [Green Version]
- Chilà, R.; Guffanti, F.; Damia, G. Role and therapeutic potential of CDK12 in human cancers. Cancer Treat. Rev. 2016, 50, 83–88. [Google Scholar] [CrossRef]
- Popova, T.; Manié, E.; Boeva, V.; Battistella, A.; Goundiam, O.; Smith, N.K.; Mueller, C.R.; Raynal, V.; Mariani, O.; Sastre-Garau, X.; et al. Ovarian cancers harboring inactivating mutations in CDK12 display a distinct genomic instability pattern characterized by large tandem duplications. Cancer Res. 2016. [Google Scholar] [CrossRef]
- Wu, Y.-M.; Cieślik, M.; Lonigro, R.J.; Vats, P.; Reimers, M.A.; Cao, X.; Ning, Y.; Wang, L.; Kunju, L.P.; de Sarkar, N.; et al. Inactivation of CDK12 Delineates a Distinct Immunogenic Class of Advanced Prostate Cancer. Cell 2018, 173, 1770–1782.e14. [Google Scholar] [CrossRef] [Green Version]
- Yamada, T.; Yamaguchi, Y.; Inukai, N.; Okamoto, S.; Mura, T.; Handa, H. P-TEFb-mediated phosphorylation of hSpt5 C-terminal repeats is critical for processive transcription elongation. Mol. Cell 2006, 21, 227–237. [Google Scholar] [CrossRef]
- Lei, T.; Zhang, P.; Zhang, X.; Xiao, X.; Zhang, J.; Qiu, T.; Dai, Q.; Zhang, Y.; Min, L.; Li, Q.; et al. Cyclin K regulates prereplicative complex assembly to promote mammalian cell proliferation. Nat. Commun. 2018, 1–15. [Google Scholar] [CrossRef]
- Choi, S.H.; Martinez, T.F.; Kim, S.; Donaldson, C.; Shokhirev, M.N.; Saghatelian, A.; Jones, K.A. CDK12 phosphorylates 4E-BP1 to enable mTORC1-dependent translation and mitotic genome stability. Genes Dev. 2019, 33, 418–435. [Google Scholar] [CrossRef] [PubMed]
- Krajewska, M.; Dries, R.; Grassetti, A.V.; Dust, S.; Gao, Y.; Huang, H.; Sharma, B.; Day, D.S.; Kwiatkowski, N.; Pomaville, M.; et al. CDK12 loss in cancer cells affects DNA damage response genes through premature cleavage and polyadenylation. Nat. Commun. 2019, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Schüller, R.; Forné, I.; Straub, T.; Schreieck, A.; Texier, Y.; Shah, N.; Decker, T.-M.; Cramer, P.; Imhof, A.; Eick, D. Heptad-Specific Phosphorylation of RNA Polymerase II CTD. Mol. Cell 2016, 61, 305–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suh, H.; Ficarro, S.B.; Kang, U.-B.; Chun, Y.; Marto, J.A.; Buratowski, S. Direct Analysis of Phosphorylation Sites on the Rpb1 C-Terminal Domain of RNA Polymerase II. Mol. Cell 2016, 61, 297–304. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.C.; Phatnani, H.P.; Haystead, T.A.; MacDonald, J.A.; Alam, S.M.; Greenleaf, A.L. C-terminal repeat domain kinase I phosphorylates Ser2 and Ser5 of RNA polymerase II C-terminal domain repeats. J. Biol. Chem. 2004, 279, 24957–24964. [Google Scholar] [CrossRef]
- Krull, S.; Thyberg, J.; Björkroth, B.; Rackwitz, H.-R.; Cordes, V.C. Nucleoporins as components of the nuclear pore complex core structure and Tpr as the architectural element of the nuclear basket. Mol. Biol. Cell 2004, 15, 4261–4277. [Google Scholar] [CrossRef]
- Galy, V.; Gadal, O.; Fromont-Racine, M.; Romano, A.; Jacquier, A.; Nehrbass, U. Nuclear retention of unspliced mRNAs in yeast is mediated by perinuclear Mlp1. Cell 2004, 116, 63–73. [Google Scholar] [CrossRef]
- Fasken, M.B.; Stewart, M.; Corbett, A.H. Functional significance of the interaction between the mRNA-binding protein, Nab2, and the nuclear pore-associated protein, Mlp1, in mRNA export. J. Biol. Chem. 2008, 283, 27130–27143. [Google Scholar] [CrossRef]
- Coyle, J.H.; Bor, Y.-C.; Rekosh, D.; Hammarskjold, M.-L. The Tpr protein regulates export of mRNAs with retained introns that traffic through the Nxf1 pathway. RNA 2011, 17, 1344–1356. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Lopez, M.S.; Dar, A.C.; Ladow, E.; Finkbeiner, S.; Yun, C.-H.; Eck, M.J.; Shokat, K.M. Structure-guided inhibitor design expands the scope of analog-sensitive kinase technology. ACS Chem. Biol. 2013, 8, 1931–1938. [Google Scholar] [CrossRef]
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, C.J.; McLauchlan, H.; Klevernic, I.; Arthur, J.S.C.; Alessi, D.R.; Cohen, P. The selectivity of protein kinase inhibitors: A further update. Biochem. J. 2007, 408, 297–315. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.B.; Westmoreland, T.J.; Verrier, C.S.; Blanchette, C.A.B.; Sabin, T.L.; Phatnani, H.P.; Mishina, Y.V.; Huper, G.; Selim, A.L.; Madison, E.R.; et al. Yeast screens identify the RNA polymerase II CTD and SPT5 as relevant targets of BRCA1 interaction. PLoS ONE 2008, 3, e1448. [Google Scholar] [CrossRef] [PubMed]
- Cattoglio, C.; Zhang, E.T.; Grubisic, I.; Chiba, K.; Fong, Y.W.; Tjian, R. Functional and mechanistic studies of XPC DNA-repair complex as transcriptional coactivator in embryonic stem cells. PNAS 2015, 112, 2317–2326. [Google Scholar] [CrossRef] [PubMed]

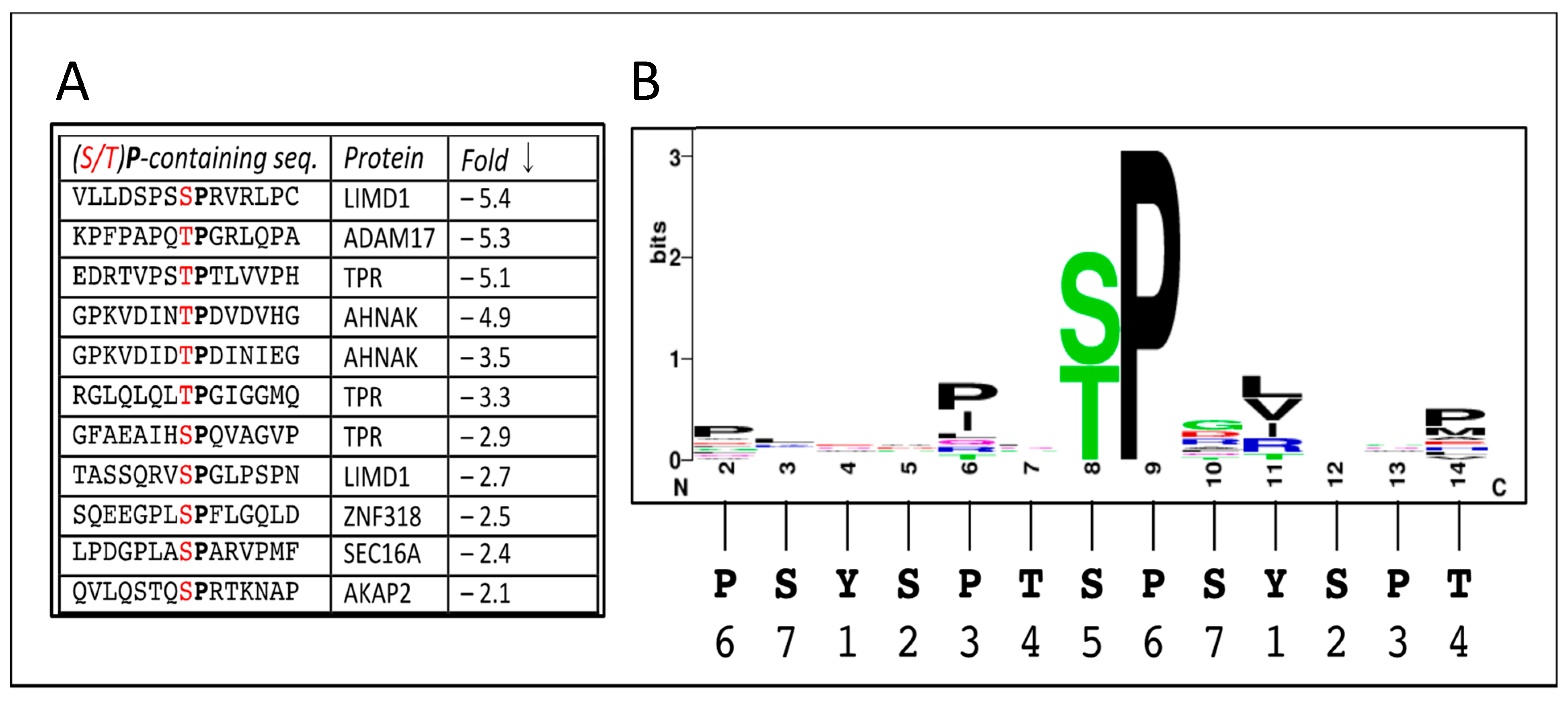

{kind=link}
{kind=link}
| Protein Name | Protein Description | Gene Name | Fold Change | Functional “Keywords” | Comments |
|---|---|---|---|---|---|
| MK03_HUMAN | Mitogen-activated protein kinase 3 | MAPK3 | −6.3 | signaling; Txn-regulation | |
| SOS1_HUMAN | Son of sevenless homolog 1 | SOS1 | −6.1 | signaling | |
| RHG35_HUMAN | Rho GTPase-activating protein 35 | ARHGAP35 | −5.6 | Txn-regulation; signaling | |
| LIMD1_HUMAN | LIM domain-containing protein 1 | LIMD1 | −5.4 | Txn-regulation; scaffold prot; cell fate | |
| ANS1A_HUMAN | Ankyrin repeat and SAM domain-containing protein 1A | ANKS1A | −4.2 | signaling; cell migration | |
| KANK2_HUMAN | KN motif and ankyrin repeat domain-containing protein 2 | KANK2 | −4.0 | Txn-regulation; steroid co-activators | |
| MK01_HUMAN | Mitogen-activated protein kinase 1 | MAPK1 | −3.7 | signaling; Txn-regulation | |
| BCAR3_HUMAN | Breast cancer anti-estrogen resistance protein 3 | BCAR3 | −3.1 | signaling; reg. DNA synthesis | |
| NU214_HUMAN | Nuclear pore complex protein Nup214 | NUP214 | −5.3 | mRNA maturation: nuclear export | |
| TPR_HUMAN | Nucleoprotein TPR | TPR | −5.1 | mRNA maturation: nuclear export | TPR, pep 1 |
| AHNK_HUMAN | Neuroblast differentiation-associated protein AHNAK | AHNAK | −4.9 | mRNA maturation: poly A binding; splicing | AHNK, pep 1 |
| DDX20 / Gemin 3 | Probable ATP-dependent RNA helicase DDX20 | DDX20 | −4.2 | mRNA maturation: splicing; snRNP | |
| AHNK_HUMAN | Neuroblast differentiation-associated protein AHNAK | AHNAK | −3.6 | mRNA maturation: poly A binding; splicing | AHNK, pep 2 |
| AHNK_HUMAN | Neuroblast differentiation-associated protein AHNAK | AHNAK | −3.5 | mRNA maturation: poly A binding; splicing | AHNK, pep 3 |
| TPR_HUMAN | Nucleoprotein TPR | TPR | −3.2 | mRNA maturation: nuclear export | TPR, pep 2 |
| PARD3_HUMAN | Partitioning defective 3 homolog | PARD3 | −11.1 | cell division; signaling | |
| SEPT7_HUMAN | Septin-7 | SEPT7 | −6.3 | mitosis; kinetochore; ciliogenesis | |
| ADA17_HUMAN | ADAM 17 | ADAM17 | −5.3 | mitosis; signaling; spindle | |
| CLAP2_HUMAN | CLIP-associating protein 2 | CLASP2 | −3.4 | microtubule-binding; kinetochore | |
| XPC_HUMAN | DNA repair protein complementing XP-C | XPC | >−12 | DNA repair; Txn | |
| DPOLA_HUMAN | DNA polymerase alpha catalytic subunit | POLA1 | −3.0 | DNA synthesis | |
| CTNA1_HUMAN | Catenin alpha-1 | CTNNA1 | −6.1 | other… cell adhesion | |
| ARFP1_HUMAN | Arfaptin-1 | ARFIP1 | −5.2 | other… Golgi; vessicles; phospholipase D |
| Protein | Peptide | Fold Change | Potential Protein Kinase | |
|---|---|---|---|---|
| ‘as’ | ‘WT’ | |||
| HDAC4 | SSPLLR (S266) | −2.7 | −5.8 | PIM1; PRKG1; PIM3 |
| AHNK | VSMPDVELNLKSPK (S3426) | −3 | −2.7 | JNK1/3; P38d (MAPK13) |
| ANLN | TQSLPVTEK (S485) | −4.3 | −2.5 | ANPa; COT; ANPb; |
| RAB34 | INSDDSNLYLTASK (S241) | −2.5 | −2.4 | MAPKAPK3; ANPa; PRKG2 |
| NF1 | SFDHLISDTK (S2543) | −4.1 | −2.2 | PIM1/3; MSK1 |
| WDCP | DSFSHSPGAVSSLK (S690) | −2.6 | −2 | ERK1/2/5 |
| AFAP1 | LSSERPSSDGEGVVE… (S283) | (+)2.1 | (+) 3.4 | CK2a2 (CSNK2A2); CK2a1 (CSNK2A1); MAPKAPK3 |
| Potential Kinase | # Sites |
|---|---|
| PIM 1, 2 or 3 | 5 |
| PRKG1 or 2 | 5 |
| CDK2 or 3 | 3 |
| JNK1 or 3 | 3 |
| MAPKAPK3 or 2 | 3 |
| mTOR/FRAP | 2 |
| PLK3 | 2 |
| BARK2/1 | 1 |
| CAMK4 | 1 |
| CDK7 | 1 |
| CDK9 | 1 |
| CHK2 | 1 |
| GSK2B | 1 |
| GSK3A | 1 |
| IKKb | 1 |
| PFTAIRE2 | 1 |
| RHOK | 1 |
| ROCK1 | 1 |
| SCYL2 | 1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartkowiak, B.; Yan, C.M.; Soderblom, E.J.; Greenleaf, A.L. CDK12 Activity-Dependent Phosphorylation Events in Human Cells. Biomolecules 2019, 9, 634. https://doi.org/10.3390/biom9100634
Bartkowiak B, Yan CM, Soderblom EJ, Greenleaf AL. CDK12 Activity-Dependent Phosphorylation Events in Human Cells. Biomolecules. 2019; 9(10):634. https://doi.org/10.3390/biom9100634
Chicago/Turabian StyleBartkowiak, Bartlomiej, Christopher M. Yan, Erik J. Soderblom, and Arno L. Greenleaf. 2019. "CDK12 Activity-Dependent Phosphorylation Events in Human Cells" Biomolecules 9, no. 10: 634. https://doi.org/10.3390/biom9100634
APA StyleBartkowiak, B., Yan, C. M., Soderblom, E. J., & Greenleaf, A. L. (2019). CDK12 Activity-Dependent Phosphorylation Events in Human Cells. Biomolecules, 9(10), 634. https://doi.org/10.3390/biom9100634