A Perspective on the Development of TGF-β Inhibitors for Cancer Treatment


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
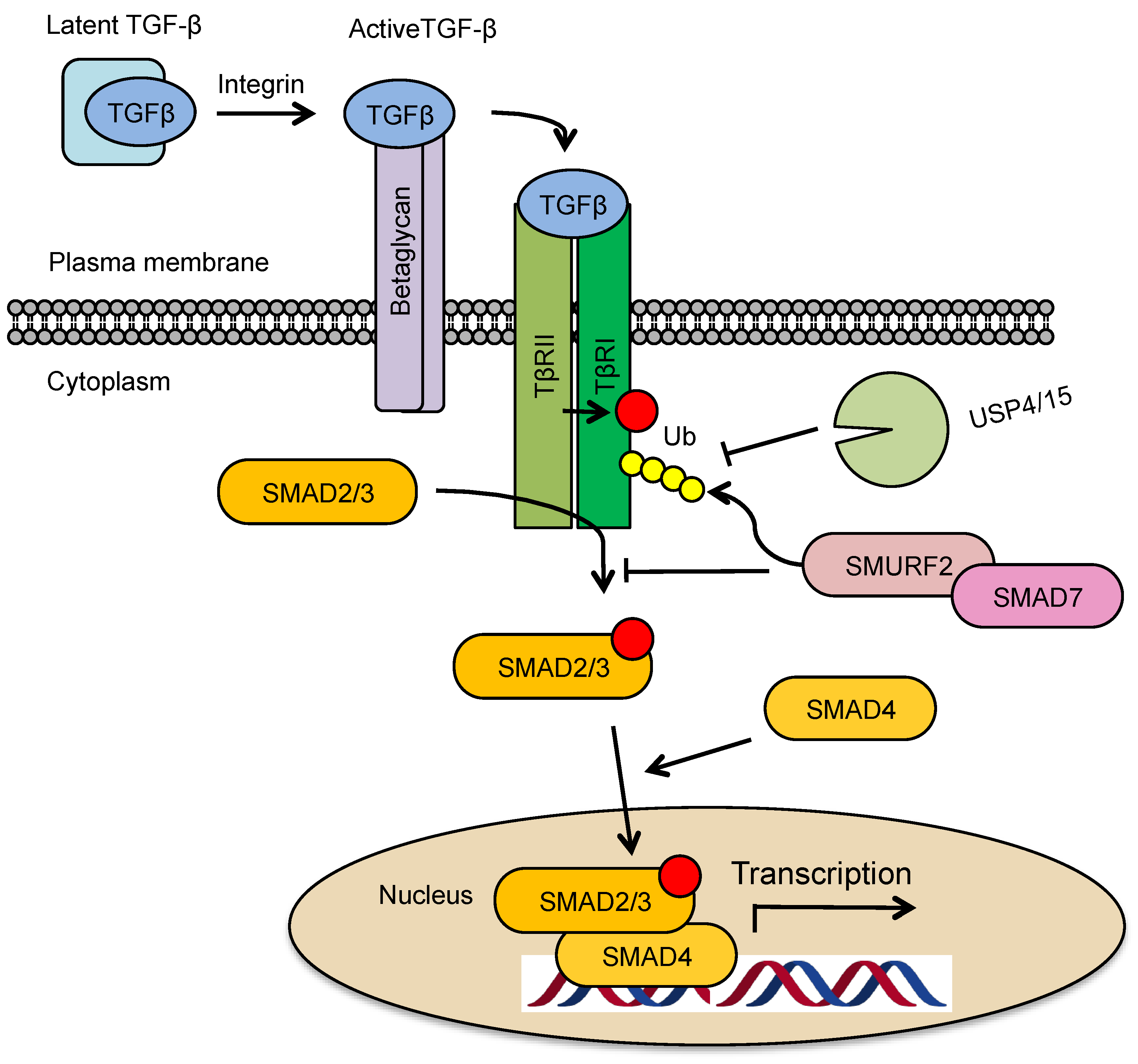
2. TGF-β Signaling Pathway
3. TGF-β Signaling in Cancer Progression
3.1. Biphasic Role in Cancer Progression
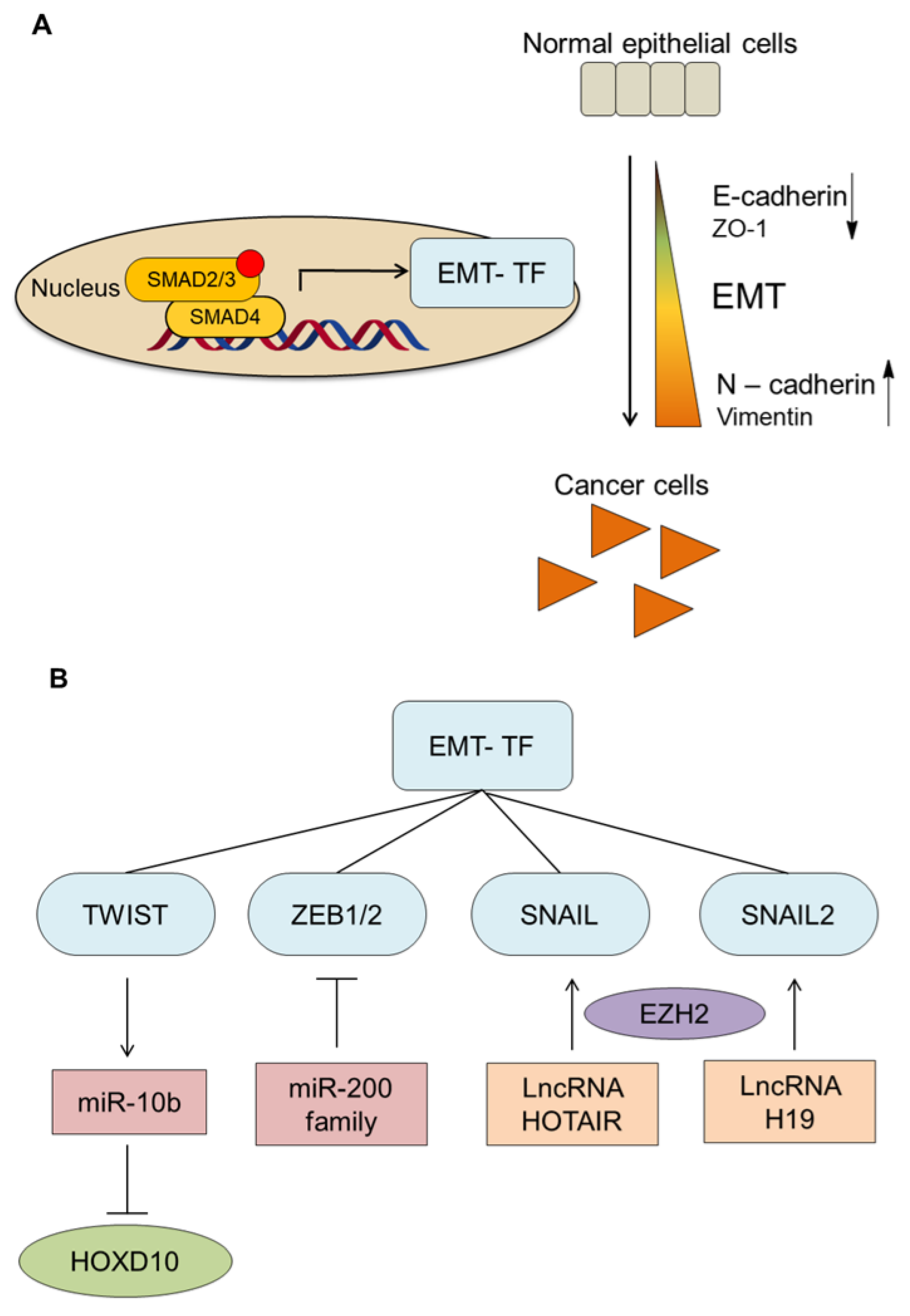
3.2. TGF-β and EMT
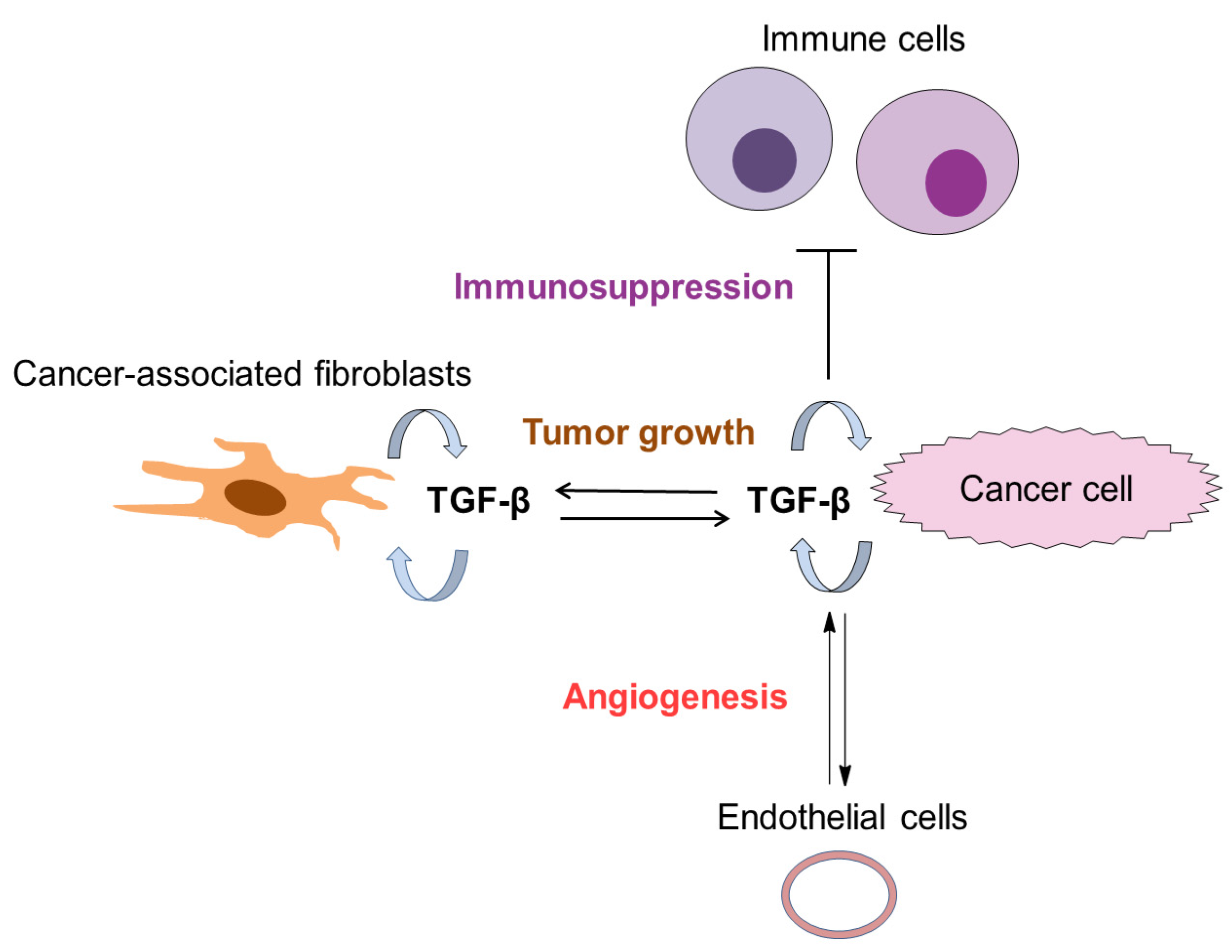
4. The Role of TGF-β in Tumor Microenvironment
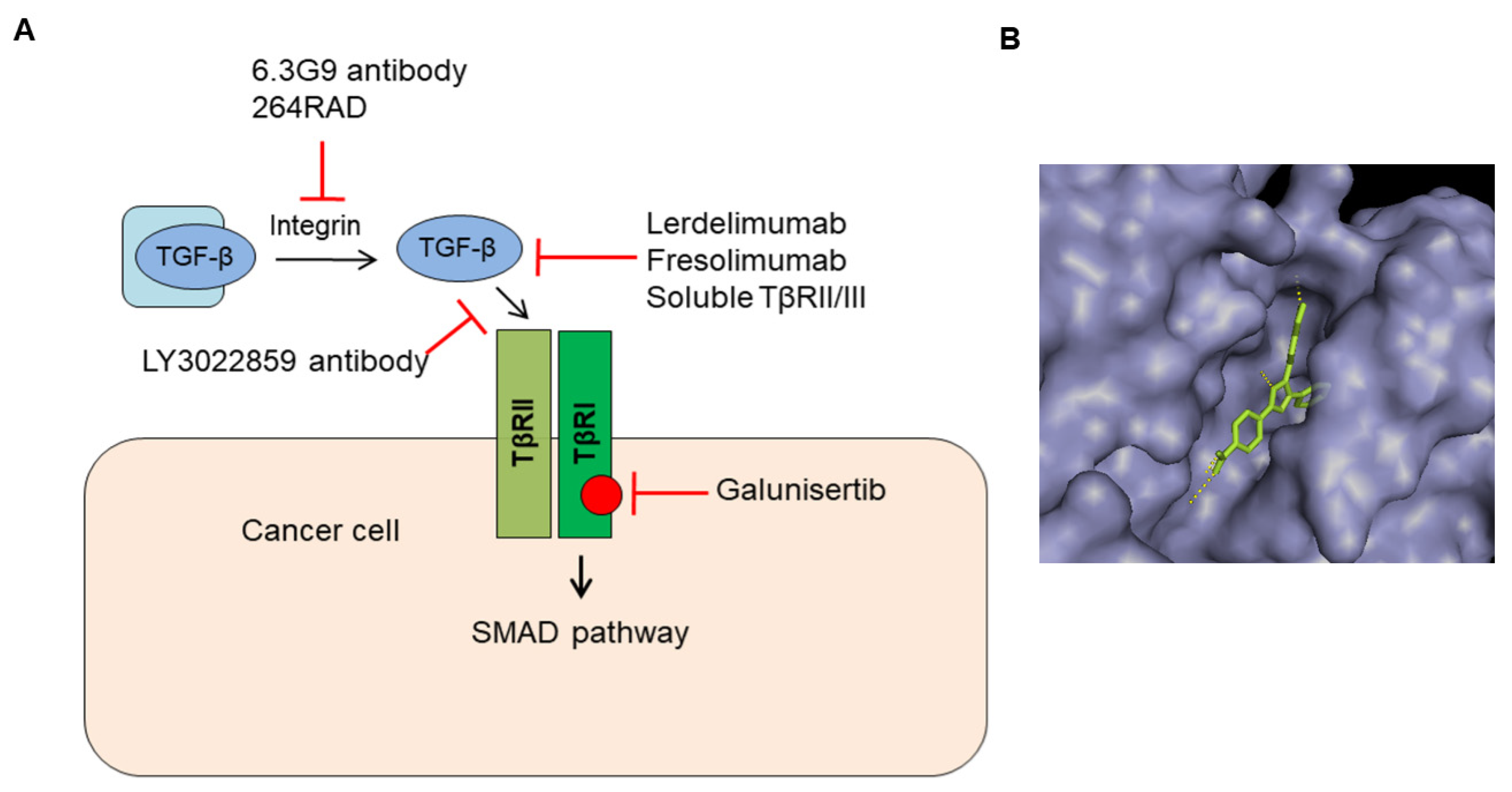
5. Inhibitors that Target TGF-β or TGF-β Receptor Function
5.1. Antibodies That Target TGF-β Activation and Ligand–Receptor Interaction
5.2. Ligand Traps That Sequester Ligands from Receptor Binding
5.3. Engineered Mutant TGF-β Ligands
5.4. Small Molecules
5.5. Future Perspective of TGF-β Signaling Inhibitors
6. Targeting TGF-β Signaling Effectors and Modulators
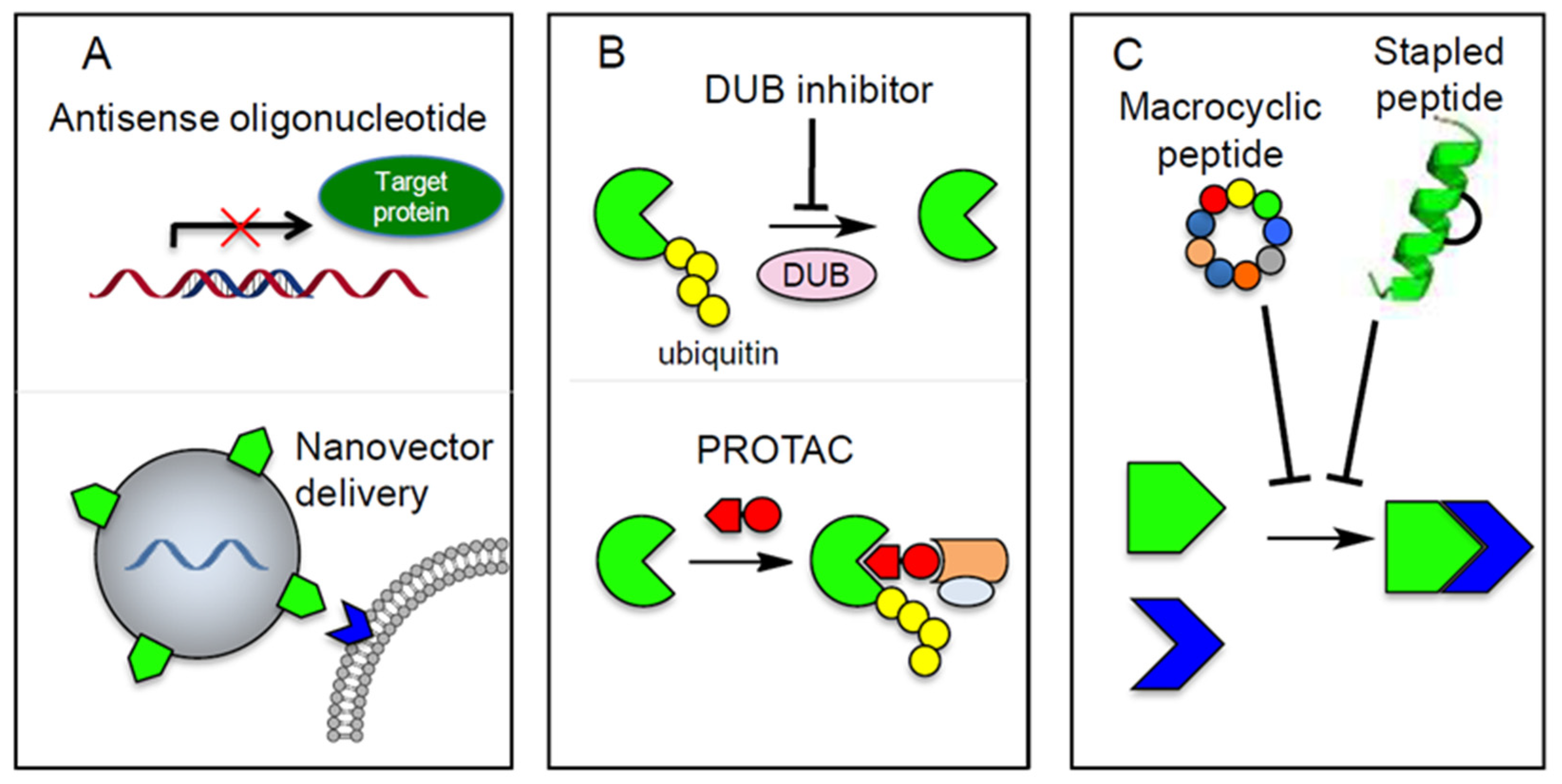
6.1. Targeting specific TGF-β Signaling Components by Using Anti-Sense Oligonucleotides
6.2. Targeting miRNA, lncRNA
6.3. Targeting of Deubiquitinating Enzymes (DUBs)
6.4. Interfering with Intracellular Protein-Protein Interactions
7. Cyclic Peptides and PROTAC as Potential Technologies for Targeting TGF-β signaling
8. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- David, C.J.; Massagué, J. Contextual determinants of TGFβ. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-β family signaling. Sci. Signal. 2019, 12, eaav5183. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-β signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Akhurst, R.J.; Derynck, R. TGF-β signaling in cancer—A double-edged sword. Trends Cell Biol. 2001, 11, S44–S51. [Google Scholar] [CrossRef] [PubMed]
- Colak, S.; ten Dijke, P. Targeting TGF-β Signaling in Cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef]
- Akhurst, R.J. Targeting TGF-β signaling for therapeutic gain. Cold Spring Harb. Perspect. Biol. 2017, 9, a022301. [Google Scholar] [CrossRef]
- Yingling, J.M.; Blanchard, K.L.; Sawyer, J.S. Development of TGFβ signalling inhibitors for cancer therapy. Nat. Rev. Drug Discov. 2004, 3, 1011–1022. [Google Scholar] [CrossRef]
- Flanders, K.C.; Wakefield, L.M. Transforming growth factor-βs and mammary gland involution; functional roles and implications for cancer progression. J. Mammary Gland. Biol. Neoplasia 2009, 14, 131–144. [Google Scholar] [CrossRef]
- Pelton, R.W.; Hogan, B.L.; Miller, D.A.; Moses, H.L. Differential expression of genes encoding TGFs β1, β2, and β3 during murine palate formation. Dev. Biol. 1990, 141, 456–460. [Google Scholar] [CrossRef]
- Robertson, I.B.; Rifkin, D.B. Regulation of the bioavailability of TGF-β and TGF-β-related proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a021907. [Google Scholar] [CrossRef]
- Nickel, J.; ten Dijke, P.; Mueller, T.D. TGF-β family co-receptor function and signaling. Acta Biochim. Biophys. Sin. 2018, 50, 12–36. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.S. Transcriptional control by the SMADs. Cold Spring Harb. Perspect. Biol. 2016, 8, a022079. [Google Scholar] [CrossRef] [PubMed]
- Dennler, S.; Itoh, S.; Vivien, D.; ten Dijke, P.; Huet, S.; Gauthier, J.M. Direct binding of Smad3 and Smad4 to critical TGFβ-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998, 17, 3091–3100. [Google Scholar] [CrossRef] [PubMed]
- Dennler, S.; Huet, S.; Gauthier, J.M. A short amino-acid sequence in MH1 domain is responsible for functional differences between Smad2 and Smad3. Oncogene 1999, 18, 1643–1648. [Google Scholar] [CrossRef]
- Nakao, A.; Afrakhte, M.; Morén, A.; Nakayama, T.; Christian, J.L.; Heuchel, R.; Itoh, S.; Kawabata, M.; Heldin, N.E.; Heldin, C.H.; et al. Identification of Smad7, a TGFβ-inducible antagonist of TGF-β signalling. Nature 1997, 389, 631–635. [Google Scholar] [CrossRef]
- Kavsak, P.; Rasmussen, R.K.; Causing, C.G.; Bonni, S.; Zhu, H.; Thomsen, G.H.; Wrana, J.L. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGFβ receptor for degradation. Mol. Cell 2000, 6, 1365–1375. [Google Scholar] [CrossRef]
- Miyazawa, K.; Miyazono, K. Regulation of TGF-β family signaling by inhibitory Smads. Cold Spring Harb. Perspect. Biol. 2017, 9, a022095. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, X.; Xie, F.; Zhang, Z.; van Dam, H.; Zhang, L.; Zhou, F. The regulation of TGF-β/SMAD signaling by protein deubiquitination. Protein Cell 2014, 5, 503–517. [Google Scholar] [CrossRef]
- Zhang, Y.; Alexander, P.B.; Wang, X.F. TGF-β family signaling in the control of cell proliferation and survival. Cold Spring Harb. Perspect. Biol. 2017, 9, a022145. [Google Scholar] [CrossRef]
- Iavarone, A.; Massagué, J. Repression of the CDK activator Cdc25A and cell-cycle arrest by cytokine TGF-β in cells lacking the CDK inhibitor p15. Nature 1997, 387, 417–422. [Google Scholar] [CrossRef]
- Bose, P.; Simmons, G.L.; Grant, S. Cyclin-dependent kinase inhibitor therapy for hematologic malignancies. Expert Opin. Investig. Drugs 2013, 22, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, I.; Hamajima, H.; Matsuhashi, S.; Mizuta, T. Regulation of TGF-β1-induced pro-apoptotic signaling by growth factor receptors and extracellular matrix receptor integrins in the liver. Front. Physiol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Wiener, Z.; Band, A.M.; Kallio, P.; Högström, J.; Hyvönen, V.; Kaijalainen, S.; Ritvos, O.; Haglund, C.; Kruuna, O.; Robine, S.; et al. Oncogenic mutations in intestinal adenomas regulate Bim-mediated apoptosis induced by TGF-β. Proc. Natl. Acad. Sci. USA 2014, 111, E2229–E2236. [Google Scholar] [CrossRef] [PubMed]
- Spender, L.C.; O’Brien, D.I.; Simpson, D.; Dutt, D.; Gregory, C.D.; Allday, M.J.; Clark, L.J.; Inman, G.J. TGF-β induces apoptosis in human B cells by transcriptional regulation of BIK and BCL-XL. Cell Death Differ. 2009, 16, 593–602. [Google Scholar] [CrossRef]
- Korkut, A.; Zaidi, S.; Kanchi, R.S.; Rao, S.; Gough, N.R.; Schultz, A.; Li, X.; Lorenzi, P.L.; Berger, A.C.; Robertson, G.; et al. A Pan-Cancer Analysis Reveals High-Frequency Genetic Alterations in Mediators of Signaling by the TGF-β Superfamily. Cell Syst. 2018, 7, 422–437. [Google Scholar] [CrossRef]
- Fearon, E.R. PM06CH20-Fearon Molecular Genetics of Colorectal Cancer. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 479–507. [Google Scholar] [CrossRef]
- Markowitz, S.; Wang, J.; Myeroff, L.; Parsons, R.; Sun, L.; Lutterbaugh, J.; Fan, R.S.; Zborowska, E.; Kinzler, K.W.; Vogelstein, B. Inactivation of the type II TGF-β receptor in colon cancer cells with microsatellite instability. Science 1995, 268, 1336–1338. [Google Scholar] [CrossRef]
- Gryfe, R.; Kim, H.; Hsieh, E.T.; Aronson, M.D.; Holowaty, E.J.; Bull, S.B.; Redston, M.; Gallinger, S. Tumor Microsatellite Instability and Clinical Outcome in Young Patients with Colorectal Cancer. N. Engl. J. Med. 2000, 342, 69–77. [Google Scholar] [CrossRef]
- Hahn, S.A.; Schutte, M.; Hoque, A.S.; Moskaluk, C.A.; Da Costa, L.T.; Rozenblum, E.; Weinstein, C.L.; Fischer, A.; Yeo, C.J.; Hruban, R.H.; et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996, 271, 350–353. [Google Scholar] [CrossRef]
- Riggins, G.J.; Kinzler, K.W.; Vogelstein, B.; Thiagalingam, S. Frequency of Smad gene mutations in human cancers. Cancer Res. 1997, 57, 2578–2580. [Google Scholar]
- Zhang, L.; Zhou, F.; ten Dijke, P. Signaling interplay between transforming growth factor-β receptor and PI3K/AKT pathways in cancer. Trends Biochem. Sci. 2013, 38, 612–620. [Google Scholar] [CrossRef] [PubMed]
- Padua, D.; Massagué, J. Roles of TGFβ in metastasis. Cell Res. 2009, 19, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Weinberg, R.A. EMT and Cancer: More Than Meets the Eye. Dev. Cell 2019, 49, 313–316. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Cano, A.; Pérez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor Snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef]
- Haslehurst, A.M.; Koti, M.; Dharsee, M.; Nuin, P.; Evans, K.; Geraci, J.; Childs, T.; Chen, J.; Li, J.; Weberpals, J.; et al. EMT transcription factors snail and slug directly contribute to cisplatin resistance in ovarian cancer. BMC Cancer 2012, 12, 91. [Google Scholar] [CrossRef]
- Kurrey, N.K.; Amit, K.; Bapat, S.A. Snail and Slug are major determinants of ovarian cancer invasiveness at the transcription level. Gynecol. Oncol. 2005, 97, 155–165. [Google Scholar] [CrossRef]
- Kudo-Saito, C.; Shirako, H.; Takeuchi, T.; Kawakami, Y. Cancer Metastasis Is Accelerated through Immunosuppression during Snail-Induced EMT of Cancer Cells. Cancer Cell 2009, 15, 195–206. [Google Scholar] [CrossRef]
- Ansieau, S.; Morel, A.P.; Hinkal, G.; Bastid, J.; Puisieux, A. TWISTing an embryonic transcription factor into an oncoprotein. Oncogene 2010, 29, 3173–3184. [Google Scholar] [CrossRef]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Vandewalle, C.; Van Roy, F.; Berx, G. The role of the ZEB family of transcription factors in development and disease. Cell. Mol. Life Sci. 2009, 66, 773–787. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.-S.; Gao, W.; Chan, J.Y.-W. Transcription Regulation of E-Cadherin by Zinc Finger E-Box Binding Homeobox Proteins in Solid Tumors. BioMed Res. Int. 2014, 2014, 921564. [Google Scholar] [CrossRef] [PubMed]
- Eger, A.; Aigner, K.; Sonderegger, S.; Dampier, B.; Oehler, S.; Schreiber, M.; Berx, G.; Cano, A.; Beug, H.; Foisner, R. δEF1 is a transcriptional repressor of E-cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene 2005, 24, 2375–2385. [Google Scholar] [CrossRef] [PubMed]
- Aigner, K.; Dampier, B.; Descovich, L.; Mikula, M.; Sultan, A.; Schreiber, M.; Mikulits, W.; Brabletz, T.; Strand, D.; Obrist, P.; et al. The transcription factor ZEB1 (δEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene 2007, 26, 6979–6988. [Google Scholar] [CrossRef] [PubMed]
- Dohadwala, M.; Yang, S.C.; Luo, J.; Sharma, S.; Batra, R.K.; Huang, M.; Lin, Y.; Goodglick, L.; Krysan, K.; Fishbein, M.C.; et al. Cyclooxygenase-2-dependent regulation of E-cadherin: Prostaglandin E 2 induces transcriptional repressors ZEB1 and snail in non-small cell lung cancer. Cancer Res. 2006, 66, 5338–5345. [Google Scholar] [CrossRef]
- Spaderna, S.; Schmalhofer, O.; Hlubek, F.; Berx, G.; Eger, A.; Merkel, S.; Jung, A.; Kirchner, T.; Brabletz, T. A Transient, EMT-Linked Loss of Basement Membranes Indicates Metastasis and Poor Survival in Colorectal Cancer. Gastroenterology 2006, 131, 830–840. [Google Scholar] [CrossRef]
- Calon, A.; Lonardo, E.; Berenguer-Llergo, A.; Espinet, E.; Hernando-Momblona, X.; Iglesias, M.; Sevillano, M.; Palomo-Ponce, S.; Tauriello, D.V.; Byrom, D.; et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat. Genet. 2015, 47, 320–329. [Google Scholar] [CrossRef]
- Calon, A.; Espinet, E.; Palomo-Ponce, S.; Tauriello, D.V.F.; Iglesias, M.; Céspedes, M.V.; Sevillano, M.; Nadal, C.; Jung, P.; Zhang, X.H.-F.; et al. Dependency of colorectal cancer on a TGF-β-driven programme in stromal cells for metastasis initiation. Cancer Cell 2012, 22, 571–584. [Google Scholar] [CrossRef]
- Petridout, S. Myofibroblasts differentiate from fibroblasts when plated at low density. Proc. Natl. Acad. Sci. USA 1996, 93, 4219–4223. [Google Scholar]
- Mishra, P.J.; Mishra, P.J.; Humeniuk, R.; Medina, D.J.; Alexe, G.; Mesirov, J.P.; Ganesan, S.; Glod, J.W.; Banerjee, D. Carcinoma-Associated Fibroblast—Like Differentiation of Human Mesenchymal Stem Cells. Cancer Res. 2008, 68, 4331–4339. [Google Scholar] [CrossRef] [PubMed]
- Quante, M.; Tu, S.P.; Tomita, H.; Gonda, T.; Wang, S.S.; Takashi, S.; Baik, G.H.; Shibata, W.; DiPrete, B.; Betz, K.S.; et al. Bone Marrow-Derived Myofibroblasts Contribute to the Mesenchymal Stem Cell Niche and Promote Tumor Growth. Cancer Cell 2011, 19, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; ten Dijke, P. Immunoregulation by members of the TGFβ superfamily. Nat. Rev. Immunol. 2016, 16, 723–740. [Google Scholar] [CrossRef] [PubMed]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.A.; Massagué, J. TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGFβ in the tumour microenvironment. Nat Rev Cancer. 2013, 13, 788–799. [Google Scholar] [CrossRef]
- Gorelink, L.; Flavell, R.A. Immune-mediated eradication of tumors through the blockade of transforming growth factor-β signaling in T cells. Nat. Med. 2001, 7, 1118–1122. [Google Scholar] [CrossRef]
- Rouce, R.H.; Shaim, H.; Sekine, T.; Weber, G.; Ballard, B.; Ku, S.; Barese, C.; Murali, V.; Wu, M.F.; Liu, H.; et al. The TGF-β/SMAD pathway is an important mechanism for NK cell immune evasion in childhood B-acute lymphoblastic leukemia. Leukemia 2016, 30, 800–811. [Google Scholar] [CrossRef]
- Nam, J.; Terabe, M.; Kang, M.; Chae, H.; Voong, N.; Yang, Y.A.; Laurence, A.; Michalowska, A.; Mamura, M.; Lonning, S.; et al. Transforming growth factor β subverts the immune system into directly promoting tumor growth through interleukin-17. Cancer Res. 2008, 68, 3915–3923. [Google Scholar] [CrossRef]
- Vanpouille-Box, C.; Diamond, J.M.; Pilones, K.A.; Zavadil, J.; Babb, J.S.; Formenti, S.C.; Barcellos-Hoff, M.H.; Demaria, S. TGFβ is a master regulator of radiation therapy-induced antitumor immunity. Cancer Res. 2015, 75, 2232–2242. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, I.I.I.E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, K.; Massague, J. Previews TGF-β Inhibition and Immunotherapy: Checkmate. Immunity 2018, 48, 626–628. [Google Scholar] [CrossRef] [PubMed]
- Chang, Z.L.; Lorenzini, M.H.; Chen, X.; Tran, U.; Bangayan, N.J.; Chen, Y.Y. Rewiring T-cell responses to soluble factors with chimeric antigen receptors. Nat. Chem. Biol. 2018, 14, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Fjellbirkeland, L.; Cambier, S.; Broaddus, V.C.; Hill, A.; Brunetta, P.; Dolganov, G.; Jablons, D.; Nishimura, S.L. Integrin αvβ8-mediated activation of transforming growth factor-β inhibits human airway epithelial proliferation in intact bronchial tissue. Am. J. Pathol. 2003, 163, 533–542. [Google Scholar] [CrossRef]
- Eberlein, C.; Kendrew, J.; McDaid, K.; Alfred, A.; Kang, J.S.; Jacobs, V.N.; Ross, S.J.; Rooney, C.; Smith, N.R.; Rinkenberger, J.; et al. A human monoclonal antibody 264RAD targeting αvβ6 integrin reduces tumour growth and metastasis, and modulates key biomarkers in vivo. Oncogene 2013, 32, 4406–4416. [Google Scholar] [CrossRef]
- Moore, K.M.; Thomas, G.J.; Duffy, S.W.; Warwick, J.; Gabe, R.; Chou, P.; Ellis, I.O.; Green, A.R.; Haider, S.; Brouilette, K.; et al. Therapeutic targeting of integrin αvβ6 in breast cancer. J. Natl. Cancer Inst. 2014, 106, dju169. [Google Scholar] [CrossRef]
- Morris, J.C.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Reiss, M.; Hsu, F.J.; Berzofsky, J.A.; Lawrence, D.P. Phase I study of GC1008 (fresolimumab): A human anti-transforming growth factor-beta (TGFβ) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS ONE 2014, 9, e90353. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Berlin, J.D.; Cosaert, J.; Kauh, J.; Chan, E.; Piha-Paul, S.A.; Amaya, A.; Tang, S.; Driscoll, K.; Kimbung, R.; et al. A phase 1 study of anti-TGFβ receptor type-II monoclonal antibody LY3022859 in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2017, 79, 673–680. [Google Scholar] [CrossRef]
- Muraoka, R.S.; Dumont, N.; Ritter, C.A.; Dugger, T.C.; Brantley, D.M.; Chen, J.; Easterly, E.; Roebuck, L.R.; Ryan, S.; Gotwals, P.J.; et al. Blockade of TGF-β inhibits mammary tumor cell viability, migration, and metastases. J. Clin. Investig. 2002, 109, 1551–1559. [Google Scholar] [CrossRef]
- Bandyopadhyay, A.; López-Casillas, F.; Malik, S.N.; Montiel, J.L.; Mendoza, V.; Yang, J.; Sun, L.Z. Antitumor activity of a recombinant soluble betaglycan in human breast cancer xenograft. Cancer Res. 2002, 62, 4690–4695. [Google Scholar] [PubMed]
- Qin, T.; Barron, L.; Xia, L.; Huang, H.; Villarreal, M.M.; Zwaagstra, J.; Collins, C.; Yang, J.; Zwieb, C.; Kodali, R.; et al. A novel highly potent trivalent TGF-β receptor trap inhibits early-stage tumorigenesis and tumor cell invasion in murine Pten-deficient prostate glands. Oncotarget 2016, 7, 86087–86102. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Gu, X.; Xia, L.; Zhou, Y.; Bouamar, H.; Yang, J.; Ding, X.; Zwieb, C.; Zhang, J.; Hinck, A.P.; et al. A novel TGFβ trap blocks chemotherapeutics-induced TGFβ1 signaling and enhances their anticancer activity in gynecologic cancers. Clin. Cancer Res. 2018, 24, 2780–2793. [Google Scholar] [CrossRef] [PubMed]
- Hinck, A.P. Structure-guided engineering of TGF-βs for the development of novel inhibitors and probing mechanism. Bioorg. Med. Chem. 2018, 26, 5239–5246. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Garcia-Carbonero, R.; Macarulla, T.; Pezet, D.; Deplanque, G.; Fuchs, M.; Trojan, J.; Oettle, H.; Kozloff, M.; Cleverly, A.; et al. A phase II, double-blind study of galunisertib+gemcitabine (GG) vs gemcitabine+placebo (GP) in patients (pts) with unresectable pancreatic cancer (PC). J. Clin. Oncol. 2017, 34, 4019. [Google Scholar] [CrossRef]
- Faivre, S.J.; Santoro, A.; Gane, E.; Kelley, R.K.; Hourmand, I.O.; Assenat, E.; Gueorguieva, I.; Cleverly, A.; Desaiah, D.; Lahn, M.M.F.; et al. A phase 2 study of galunisertib, a novel transforming growth factor-beta (TGF-β) receptor I kinase inhibitor, in patients with advanced hepatocellular carcinoma (HCC) and low serum alpha fetoprotein (AFP). J. Clin. Oncol. 2017, 34, 4070. [Google Scholar] [CrossRef]
- Park, A.; Kingdom, U.; Park, A. Induction of Heart Valve Lesions by Small-Molecule. Toxicol. Pathol. 2011, 39, 916–924. [Google Scholar] [CrossRef]
- Herbertz, S.; Sawyer, J.S.; Stauber, A.J.; Gueorguieva, I.; Driscoll, K.E.; Estrem, S.T.; Cleverly, A.L.; Desaiah, D.; Guba, S.C.; Benhadji, K.A.; et al. Clinical development of galunisertib (Ly2157299 monohydrate), a small molecule inhibitor of transforming growth factor-β signaling pathway. Drug Des. Dev. Ther. 2015, 9, 4479–4499. [Google Scholar] [CrossRef]
- Gueorguieva, I.; Cleverly, A.L.; Stauber, A.; Pillay, N.S.; Rodon, J.A.; Miles, C.P.; Yingling, J.M.; Lahn, M.M. Defining a therapeutic window for the novel TGF-β inhibitor LY2157299 monohydrate based on a pharmacokinetic/pharmacodynamic model. Br. J. Clin. Pharmacol. 2014, 77, 796–807. [Google Scholar] [CrossRef]
- Kovacs, R.J.; Maldonado, G.; Azaro, A.; Fernández, M.S.; Romero, F.L.; Sepulveda-Sánchez, J.M.; Corretti, M.; Carducci, M.; Dolan, M.; Gueorguieva, I.; et al. Cardiac Safety of TGF-β Receptor I Kinase Inhibitor LY2157299 Monohydrate in Cancer Patients in a First-in-Human Dose Study. Cardiovasc. Toxicol. 2015, 15, 309–323. [Google Scholar] [CrossRef]
- Rodon, J.; Carducci, M.A.; Sepulveda-Sánchez, J.M.; Azaro, A.; Calvo, E.; Seoane, J.; Brana, I.; Sicart, E.; Gueorguieva, I.; Cleverly, A.L.; et al. First-in-human dose study of the novel transforming growth factor-β receptor I kinase inhibitor LY2157299 monohydrate in patients with advanced cancer and glioma. Clin. Cancer Res. 2015, 21, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, J.S.; Beight, D.W.; Britt, K.S.; Anderson, B.D.; Campbell, R.M.; Goodson, T., Jr.; Herron, D.K.; Li, H.Y.; McMillen, W.T.; Mort, N.; et al. Synthesis and activity of new aryl- and heteroaryl-substituted 5,6-dihydro-4H-pyrrolo[1,2-b]pyrazole inhibitors of the transforming growth factor-β type I receptor kinase domain. Bioorg Med Chem Lett. 2004, 14, 3581–3584. [Google Scholar] [CrossRef] [PubMed]
- Dias, N.; Stein, C.A. Antisense oligonucleotides: Basic concepts and mechanisms. Mol. Cancer Ther. 2002, 1, 347–355. [Google Scholar] [PubMed]
- Tibbitt, M.W.; Dahlman, J.E.; Langer, R. Emerging Frontiers in Drug Delivery. J. Am. Chem. Soc. 2016, 138, 704–717. [Google Scholar] [CrossRef] [PubMed]
- Seystahl, K.; Papachristodoulou, A.; Burghardt, I.; Schneider, H.; Hasenbach, K.; Janicot, M.; Roth, P.; Weller, M. Biological Role and Therapeutic Targeting of TGF-β3 in Glioblastoma. Mol. Cancer Ther. 2017, 16, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Kemaladewi, D.U.; Pasteuning, S.; van der Meulen, J.W.; van Heiningen, S.H.; van Ommen, G.-J.; ten Dijke, P.; Aartsma-Rus, A.; AC’t Hoen, P.; Hoogaars, W.M. Targeting TGF-β Signaling by Antisense Oligonucleotide-mediated Knockdown of TGF-β Type I Receptor. Mol. Ther. Nucleic Acids 2014, 3, e156. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Straub, V.; Hemmings, R.; Haas, M.; Schlosser-Weber, G.; Stoyanova-Beninska, V.; Mercuri, E.; Muntoni, F.; Sepodes, B.; Vroom, E.; et al. Development of Exon Skipping Therapies for Duchenne Muscular Dystrophy: A Critical Review and a Perspective on the Outstanding Issues. Nucleic Acid Ther. 2017, 27, 251–259. [Google Scholar] [CrossRef]
- Ma, L.; Reinhardt, F.; Pan, E.; Soutschek, J.; Bhat, B.; Marcusson, E.G.; Teruya-Feldstein, J.; Bell, G.W.; Weinberg, R.A. Therapeutic silencing of miR-10b inhibits metastasis in a mouse mammary tumor model. Nat. Biotechnol. 2010, 28, 341–347. [Google Scholar] [CrossRef]
- Raveh, E.; Matouk, I.J.; Gilon, M.; Hochberg, A. The H19 Long non-coding RNA in cancer initiation, progression and metastasis—A proposed unifying theory. Mol. Cancer 2015, 14, 184. [Google Scholar] [CrossRef]
- Battistelli, C.; Cicchini, C.; Santangelo, L.; Tramontano, A.; Grassi, L.; Gonzalez, F.J.; De Nonno, V.; Grassi, G.; Amicone, L.; Tripodi, M. The Snail repressor recruits EZH2 to specific genomic sites through the enrollment of the lncRNA HOTAIR in epithelial-to-mesenchymal transition. Oncogene 2017, 36, 942–955. [Google Scholar] [CrossRef]
- Vega, S.; Morales, A.V.; Ocaña, O.H.; Valdés, F.; Fabregat, I.; Nieto, M.A. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 2004, 18, 1131–1143. [Google Scholar] [CrossRef] [PubMed]
- Van Staalduinen, J.; Baker, D.; Ten Dijke, P.; van Dam, H. Epithelial-mesenchymal-transition-inducing transcription factors: New targets for tackling chemoresistance in cancer? Oncogene 2018, 37, 6195–6211. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.M.; Shahin, S.A.; Wen, W.; Finlay, J.B.; Dong, J.; Wang, R.; Dellinger, T.H.; Zink, J.I.; Tamanoi, F.; Glackin, C.A. Nanoparticle delivery of siRNA against TWIST to reduce drug resistance and tumor growth in ovarian cancer models. Nanomedicine 2017, 13, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; De Boeck, M.; Van Dam, H.; Ten Dijke, P. Regulation of the TGF-β pathway by deubiquitinases in cancer. Int. J. Biochem. Cell Biol. 2016, 76, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, F.; Drabsch, Y.; Gao, R.; Snaar-Jagalska, B.E.; Mickanin, C.; Huang, H.; Sheppard, K.A.; Porter, J.A.; Lu, C.X.; et al. USP4 is regulated by AKT phosphorylation and directly deubiquitylates TGF-β type i receptor. Nat. Cell Biol. 2012, 14, 717–726. [Google Scholar] [CrossRef]
- Eichhorn, P.J.A.; Rodón, L.; Gonzàlez-Juncà, A.; Dirac, A.; Gili, M.; Martínez-Sáez, E.; Aura, C.; Barba, I.; Peg, V.; Prat, A.; et al. USP15 stabilizes TGF-β2 receptor I and promotes oncogenesis through the activation of TGF-β2 signaling in glioblastoma. Nat. Med. 2012, 18, 429–435. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, J.; Chen, C.; Yuan, H.; Wen, X.; Sun, H. USP7: Target Validation and Drug Discovery for Cancer Therapy. Med. Chem. 2018, 14, 3–18. [Google Scholar] [CrossRef]
- Mullard, A. Proteing-protein interaction inhibitors get into the groove. Nat. Rev. Drug. Discov. 2012, 11, 173–175. [Google Scholar] [CrossRef]
- Cui, Q.; Lim, S.K.; Zhao, B.; Hoffmann, F.M. Selective inhibition of TGF-β responsive genes by Smad-interacting peptide aptamers from FoxH1, Lef1 and CBP. Oncogene 2005, 24, 3864–3874. [Google Scholar] [CrossRef]
- Kang, J.H.; Jung, M.Y.; Yin, X.; Andrianifahanana, M.; Hernandez, D.M.; Leof, E.B. Cell-penetrating peptides selectively targeting SMAD3 inhibit profibrotic TGF-β signaling. J. Clin. Investig. 2017, 127, 2541–2554. [Google Scholar] [CrossRef]
- Ramsey, J.D.; Flynn, N.H. Cell-penetrating peptides transport therapeutics into cells. Pharmacol. Ther. 2015, 154, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.; Pardali, E.; Van Der Horst, G.; Cheung, H.; Van Den Hoogen, C.; Van Der Pluijm, G.; Ten Dijke, P. Smad2 and Smad3 have opposing roles in breast cancer bone metastasis by differentially affecting tumor angiogenesis. Oncogene 2010, 29, 1351–1361. [Google Scholar] [CrossRef] [PubMed]
- Fuxe, J.; Vincent, T.; De Herreros, A.G. Transcriptional crosstalk between TGFβ and stem cell pathways in tumor cell invasion: Role of EMT promoting Smad complexes. Cell Cycle 2010, 9, 2363–2374. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; He, W.; Tulley, S.; Gupta, G.P.; Serganova, I.; Chen, C.R.; Manova-Todorova, K.; Blasberg, R.; Gerald, W.L.; Massagué, J. Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. Proc. Natl. Acad. Sci. USA 2005, 102, 13909–13914. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, P.G.; Qian, Z.; Pei, D. Macrocycles as protein–protein interaction inhibitors. Biochem. J. 2017, 474, 1109–1125. [Google Scholar] [CrossRef] [PubMed]
- Cardote, T.A.F.; Ciulli, A. Cyclic and Macrocyclic Peptides as Chemical Tools to Recognise Protein Surfaces and Probe Protein-Protein Interactions. ChemMedChem 2016, 11, 787–794. [Google Scholar] [CrossRef]
- Driggers, E.M.; Hale, S.P.; Lee, J.; Terrett, N.K. The exploration of macrocycles for drug discovery—An underexploited structural class. Nat. Rev. Drug Discov. 2008, 7, 608–624. [Google Scholar] [CrossRef]
- Qian, Z.; Dougherty, P.G.; Pei, D. Targeting intracellular protein–protein interactions with cell-permeable cyclic peptides. Curr. Opin. Chem. Biol. 2017, 38, 80–86. [Google Scholar] [CrossRef]
- Zorzi, A.; Deyle, K.; Heinis, C. Cyclic peptide therapeutics: Past, present and future. Curr. Opin. Chem. Biol. 2017, 38, 24–29. [Google Scholar] [CrossRef]
- Bechara, C.; Sagan, S. Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett. 2013, 587, 1693–1702. [Google Scholar] [CrossRef]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The Future of Peptide-based Drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Walensky, L.D.; Bird, G.H. Hydrocarbon-stapled peptides: Principles, practice, and progress. J. Med. Chem. 2014, 57, 6275–6288. [Google Scholar] [CrossRef] [PubMed]
- Rezaei Araghi, R.; Bird, G.H.; Ryan, J.A.; Jenson, J.M.; Godes, M.; Pritz, J.R.; Grant, R.A.; Letai, A.; Walensky, L.D.; Keating, A.E. Iterative optimization yields Mcl-1-targeting stapled peptides with selective cytotoxicity to Mcl-1-dependent cancer cells. Proc. Natl. Acad. Sci. USA 2018, 115, E886–E895. [Google Scholar] [CrossRef] [PubMed]
- Passioura, T.; Katoh, T.; Goto, Y.; Suga, H. Selection-Based Discovery of Druglike Macrocyclic Peptides. Annu. Rev. Biochem. 2014, 83, 727–752. [Google Scholar] [CrossRef]
- Guo, J.; Liu, J.; Wei, W. Degrading proteins in animals: “PROTAC”tion goes in vivo. Cell Res. 2019, 29, 179–180. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huynh, L.K.; Hipolito, C.J.; ten Dijke, P. A Perspective on the Development of TGF-β Inhibitors for Cancer Treatment. Biomolecules 2019, 9, 743. https://doi.org/10.3390/biom9110743
Huynh LK, Hipolito CJ, ten Dijke P. A Perspective on the Development of TGF-β Inhibitors for Cancer Treatment. Biomolecules. 2019; 9(11):743. https://doi.org/10.3390/biom9110743
Chicago/Turabian StyleHuynh, Linh Khanh, Christopher John Hipolito, and Peter ten Dijke. 2019. "A Perspective on the Development of TGF-β Inhibitors for Cancer Treatment" Biomolecules 9, no. 11: 743. https://doi.org/10.3390/biom9110743
APA StyleHuynh, L. K., Hipolito, C. J., & ten Dijke, P. (2019). A Perspective on the Development of TGF-β Inhibitors for Cancer Treatment. Biomolecules, 9(11), 743. https://doi.org/10.3390/biom9110743