Post-Translational Modifications in NETosis and NETs-Mediated Diseases



Abstract
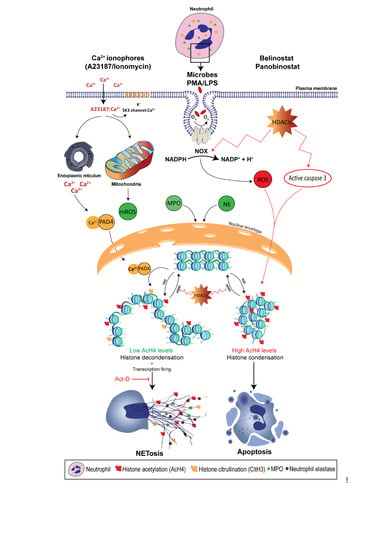
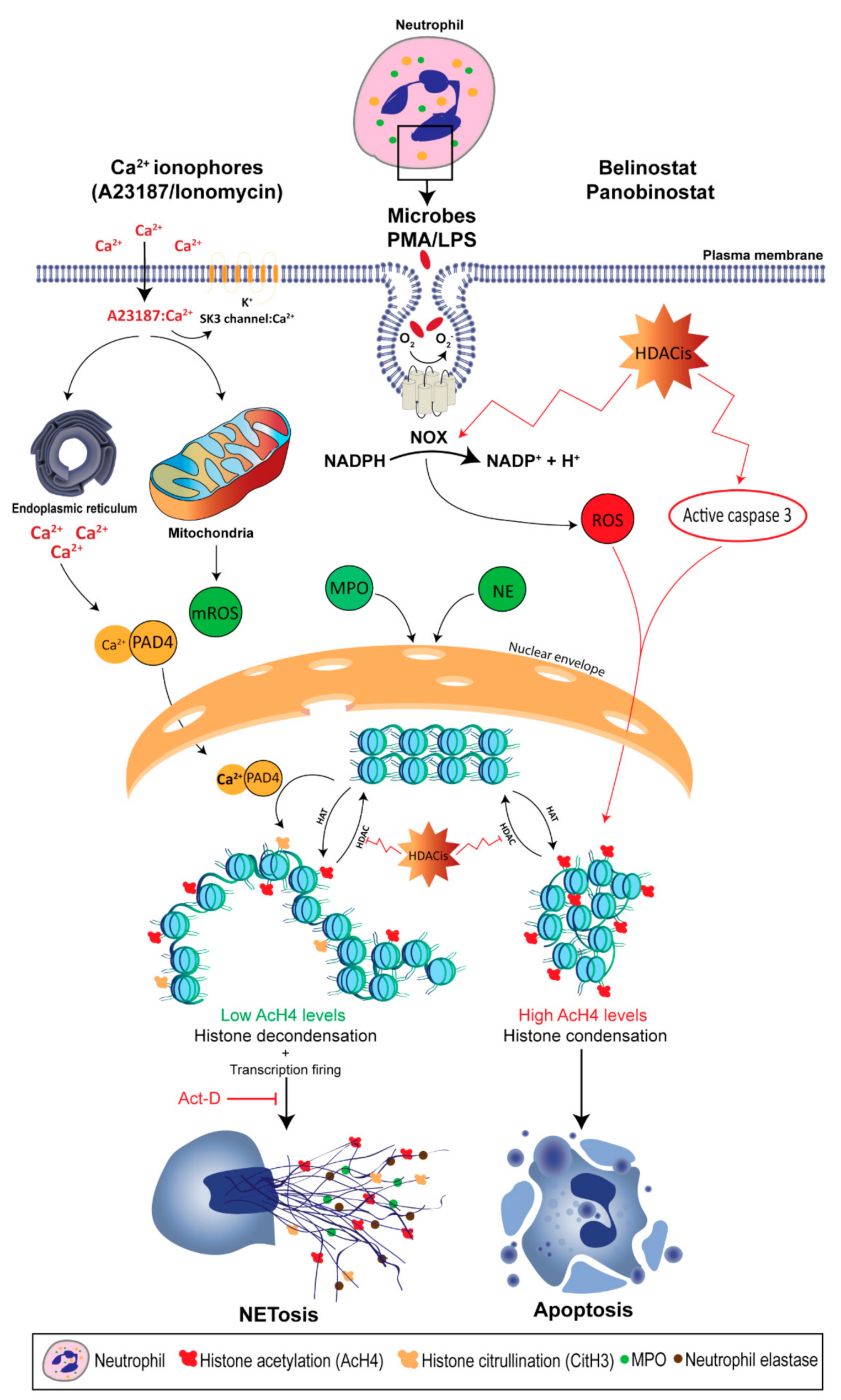
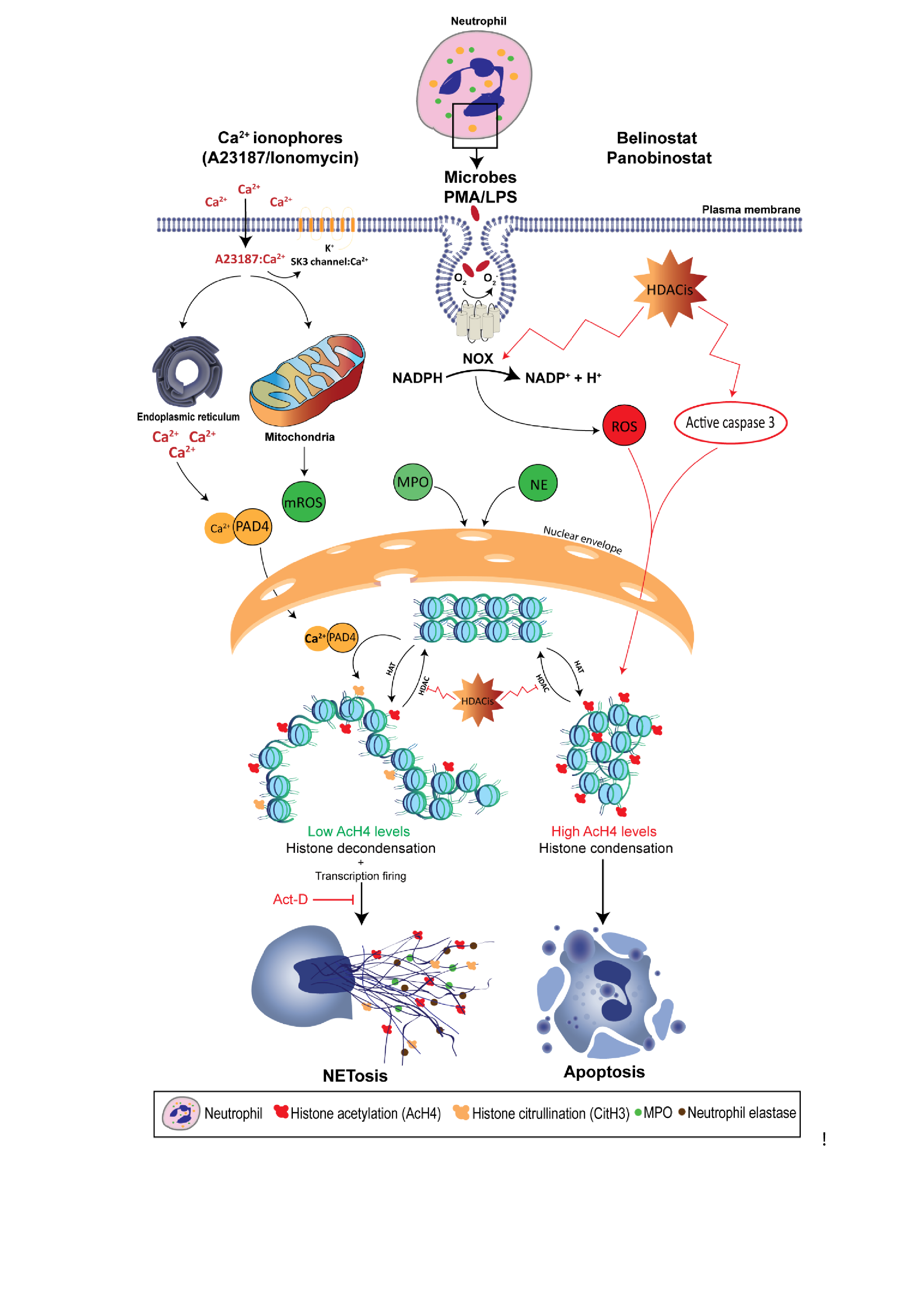{kind=link}
{kind=link}
1. Introduction
2. Neutrophils in Health and Disease
2.1. Neutrophils
2.2. Antimicrobial Functions of Neutrophils
3. Neutrophil Death
3.1. Neutrophil Apoptosis
3.2. Neutrophil Extracellular Trap Formation (NETosis)
3.3. NOX-Dependent NETosis
3.4. Other Types of NETosis
4. Histone Modification in Neutrophils
4.1. Histone Citrullination
4.2. Histone Methylation
4.3. Histone Acetylation
4.4. Role of Post-Translational Modifications in NET-Mediated Diseases
4.5. Sepsis
4.6. Systemic Lupus Erythematosus (SLE)
4.7. Cancer
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Mayadas, T.N.; Cullere, X.; Lowell, C.A. The Multifaceted Functions of Neutrophils. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 181–218. [Google Scholar] [CrossRef] [PubMed]
- Downey, D.G.; Bell, S.C.; Elborn, J.S. Neutrophils in cystic fibrosis. Thorax 2009, 64, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Kronbichler, A.; Park, D.D.Y.; Park, Y.M.; Moon, H.; Kim, H.; Choi, J.H.; Choi, Y.S.; Shim, S.; Lyu, I.S.; et al. Neutrophil extracellular traps (NETs) in autoimmune diseases: A comprehensive review. Autoimmun. Rev. 2017, 16, 1160–1173. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, S.; Stojkov, D.; Germic, N.; Simon, D.; Wang, X.; Benarafa, C.; Simon, H.U. Untangling “NETosis” from NETs. Eur. J. Immunol. 2019, 49, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Sollberger, G.; Tilley, D.O.; Zychlinsky, A. Neutrophil Extracellular Traps: The Biology of Chromatin Externalization. Dev. Cell 2018, 44, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Hamam, H.J.; Khan, M.A.; Palaniyar, N. Histone Acetylation Promotes Neutrophil Extracellular Trap Formation. Biomolecules 2019, 9, 32. [Google Scholar] [CrossRef]
- Hamam, H.J.; Palaniyar, N. Histone Deacetylase Inhibitors Dose-Dependently Switch Neutrophil Death from NETosis to Apoptosis. Biomolecules 2019, 9, 184. [Google Scholar] [CrossRef]
- Khan, M.A.; Palaniyar, N. Transcriptional firing helps to drive NETosis. Sci. Rep. 2017, 7, 41749. [Google Scholar] [CrossRef]
- Douda, D.N.; Khan, M.A.; Grasemann, H.; Palaniyar, N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc. Natl. Acad. Sci. USA 2015, 112, 2817–2822. [Google Scholar] [CrossRef]
- Yuen, J.; Pluthero, F.G.; Douda, D.N.; Riedl, M.; Cherry, A.; Ulanova, M.; Kahr, W.H.A.; Palaniyar, N.; Licht, C. NETosing neutrophils activate complement both on their own NETs and bacteria via alternative and non-alternative pathways. Front. Immunol. 2016, 7, 137. [Google Scholar] [CrossRef]
- Liew, P.X.; Kubes, P. The Neutrophil’s Role During Health and Disease. Physiol. Rev. 2019, 99, 1223–1248. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Wang, L.; Yu, C.; Yu, D.; Yu, G. Histone Acetylation Modifiers in the Pathogenesis of Alzheimer’s Disease. Front. Cell. Neurosci. 2015, 9, 226. [Google Scholar] [CrossRef] [PubMed]
- Grigoryev, S.A.; Bulynko, Y.A.; Popova, E.Y. The end adjusts the means: Heterochromatin remodeling during terminal cell differentiation. Chromosom. Res. 2006, 14, 53–69. [Google Scholar] [CrossRef] [PubMed]
- Kosak, S.T.; Groudine, M. Form follows function: The genomic organization of cellular differentiation. Genes Dev. 2004, 18, 1371–1384. [Google Scholar] [CrossRef]
- Lukášová, E.; Kořistek, Z.; Falk, M.; Kozubek, S.; Grigoryev, S.; Kozubek, M.; Ondřej, V.; Kroupová, I. Methylation of histones in myeloid leukemias as a potential marker of granulocyte abnormalities. J. Leukoc. Biol. 2005, 77, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Urdinguio, R.G.; Lopez, V.; Bayón, G.F.; Diaz de la Guardia, R.; Sierra, M.I.; García-Toraño, E.; Perez, R.F.; García, M.G.; Carella, A.; Pruneda, P.C.; et al. Chromatin regulation by Histone H4 acetylation at Lysine 16 during cell death and differentiation in the myeloid compartment. Nucleic Acids Res. 2019, 47, 5016–5037. [Google Scholar] [CrossRef] [PubMed]
- Lukášová, E.; Kořistek, Z.; Klabusay, M.; Ondřej, V.; Grigoryev, S.; Bačíková, A.; Řezáčová, M.; Falk, M.; Vávrová, J.; Kohútová, V.; et al. Granulocyte maturation determines ability to release chromatin NETs and loss of DNA damage response; these properties are absent in immature AML granulocytes. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 767–779. [Google Scholar] [CrossRef]
- Borregaard, N. Neutrophils, from Marrow to Microbes. Immunity 2010, 33, 657–670. [Google Scholar] [CrossRef]
- Edwards, S.W. The development and structure of mature neutrophils. In Biochemistry and Physiology of the Neutrophil; Cambridge University Press: Cambridge, UK, 2010. [Google Scholar]
- Basu, S.; Hodgson, G.; Katz, M.; Dunn, A.R. Evaluation of role of G-CSF in the production, survival, and release of neutrophils from bone marrow into circulation. Blood 2002, 100, 854–861. [Google Scholar] [CrossRef]
- Galli, S.J.; Borregaard, N.; Wynn, T.A. Phenotypic and functional plasticity of cells of innate immunity: Macrophages, mast cells and neutrophils. Nat. Immunol. 2011, 12, 1035. [Google Scholar] [CrossRef]
- Pillay, J.; Den Braber, I.; Vrisekoop, N.; Kwast, L.M.; De Boer, R.J.; Borghans, J.A.M.; Tesselaar, K.; Koenderman, L. In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood 2010, 116, 625–627. [Google Scholar] [CrossRef] [PubMed]
- Summers, C.; Rankin, S.M.; Condliffe, A.M.; Singh, N.; Peters, A.M.; Chilvers, E.R. Neutrophil kinetics in health and disease. Trends Immunol. 2010, 31, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Colotta, F.; Re, F.; Polentarutti, N.; Sozzani, S.; Mantovani, A. Modulation of granulocyte survival and programmed cell death by cytokines and bacterial products. Blood 1992, 80, 2012–2020. [Google Scholar] [PubMed]
- Wang, J.; Hossain, M.; Thanabalasuriar, A.; Gunzer, M.; Meininger, C.; Kubes, P. Visualizing the function and fate of neutrophils in sterile injury and repair. Science 2017, 358, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.M.; Everhart, J.E.; Byrd-Holt, D.D.; Tisdale, J.F.; Rodgers, G.P. Prevalence of neutropenia in the U.S. population: Age, sex, smoking status, and ethnic differences. Ann. Intern. Med. 2007, 146, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Biron, B.M.; Chung, C.-S.; Chen, Y.; Wilson, Z.; Fallon, E.A.; Reichner, J.S.; Ayala, A. PADI4 Deficiency Leads to Decreased Organ Dysfunction and Improved Survival in a Dual Insult Model of Hemorrhagic Shock and Sepsis. J. Immunol. 2018, 200, 1817–1828. [Google Scholar] [PubMed]
- Teng, T.-S.; Ji, A.; Ji, X.-Y.; Li, Y.-Z. Neutrophils and Immunity: From Bactericidal Action to Being Conquered. J. Immunol. Res. 2017, 2017. [Google Scholar] [CrossRef]
- Rigby, K.M.; DeLeo, F.R. Neutrophils in innate host defense against Staphylococcus aureus infections. In Seminars in Immunopathology; Springer: Berlin, Germany, 2012; pp. 237–259. [Google Scholar]
- Voisin, M.B.; Nourshargh, S. Neutrophil transmigration: Emergence of an adhesive cascade within venular walls. J. Innate Immun. 2013, 5, 336–347. [Google Scholar] [CrossRef]
- Sahakian, E.; Chen, J.; Powers, J.J.; Chen, X.; Maharaj, K.; Deng, S.L.; Achille, A.N.; Lienlaf, M.; Wang, H.W.; Cheng, F.; et al. Essential role for histone deacetylase 11 (HDAC11) in neutrophil biology. J. Leukoc. Biol. 2017, 102, 475–486. [Google Scholar] [CrossRef]
- Lee, W.L.; Harrison, R.E.; Grinstein, S. Phagocytosis by neutrophils. Microbes Infect. 2003, 5, 1299–1306. [Google Scholar] [CrossRef]
- Cheng, O.Z.; Palaniyar, N. NET balancing: A problem in inflammatory lung diseases. Front. Immunol. 2013, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Cowland, J.B.; Borregaard, N. Granulopoiesis and granules of human neutrophils. Immunol. Rev. 2016, 273, 11–28. [Google Scholar] [CrossRef] [PubMed]
- Roos, D.; Van Bruggen, R.; Meischl, C. Oxidative killing of microbes by neutrophils. Microbes Infect. 2003, 5, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Sengeløv, H.; Follin, P.; Kjeldsen, L.; Lollike, K.; Dahlgren, C.; Borregaard, N. Mobilization of granules and secretory vesicles during in vivo exudation of human neutrophils. J. Immunol. 1995, 154, 4157–4165. [Google Scholar] [PubMed]
- Masson, P.L. Lactoferrin, an iron-binbing protein Ni neutrophilic leukocytes. J. Exp. Med. 1969, 130, 643–658. [Google Scholar] [CrossRef] [PubMed]
- Rice, W.G.; Ganz, T.; Kinkade, J.M.; Selsted, M.E.; Lehrer, R.I.; Parmley, R.T. Defensin-rich dense granules of human neutrophils. Blood 1987, 70, 757–765. [Google Scholar] [PubMed]
- Campbell, E.J.; Campbell, M.A.; Owen, C.A. Bioactive Proteinase 3 on the Cell Surface of Human Neutrophils: Quantification, Catalytic Activity, and Susceptibility to Inhibition. J. Immunol. 2000, 165, 3366–3374. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.J.; Peppin, G.; Ortiz, X.; Ragsdale, C.; Test, S.T. Oxidative autoactivation of latent collagenase by human neutrophils. Science 1985, 227, 747–749. [Google Scholar] [CrossRef]
- Kasama, T.; Miwa, Y.; Isozaki, T.; Odai, T.; Adachi, M.; Kunkel, S. Neutrophil-Derived Cytokines: Potential Therapeutic Targets in Inflammation. Curr. Drug Targets Inflamm. Allergy 2005, 4, 273–279. [Google Scholar] [CrossRef]
- Kato, T.; Kitagawa, S. Regulation of Neutrophil Functions by Proinflammatory Cytokines. Int. J. Hematol. 2006, 84, 205–209. [Google Scholar] [CrossRef]
- McCracken, J.M.; Allen, L.A.H. Regulation of human neutrophil apoptosis and lifespan in health and disease. J. Cell Death 2014, 7, JCD-S11038. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.D.; Deleo, F.R. Neutrophil apoptosis and the resolution of infection. Immunol. Res. 2009, 43, 25–61. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Salvesen, G.S.; Duckett, C.S. IAP proteins: Blocking the road to death’s door. Nat. Rev. Mol. Cell Biol. 2002, 3, 401. [Google Scholar] [CrossRef] [PubMed]
- Squier, M.K.T.; Sehnert, A.J.; Sellins, K.S.; Malkinson, A.M.; Takano, E.; Cohen, J.J. Calpain and calpastatin regulate neutrophil apoptosis. J. Cell. Physiol. 1999, 178, 311–319. [Google Scholar] [CrossRef]
- Cory, S.; Adams, J.M. The BCL2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Milot, E.; Filep, J.G. Regulation of Neutrophil Survival/Apoptosis by Mcl-1. Sci. World J. 2011, 11, 1948–1962. [Google Scholar] [CrossRef]
- Andina, N.; Conus, S.; Schneider, E.M.; Fey, M.F.; Simon, H.U. Induction of Bim limits cytokine-mediated prolonged survival of neutrophils. Cell Death Differ. 2009, 16, 1248. [Google Scholar] [CrossRef]
- Villunger, A.; Scott, C.; Bouillet, P.; Strasser, A. Essential role for the BH3-only protein Bim but redundant roles for Bax, Bcl-2, and Bcl-w in the control of granulocyte survival. Blood 2003, 101, 2393–2400. [Google Scholar] [CrossRef]
- Murphy, B.M.; O’Neill, A.J.; Adrain, C.; Watson, R.W.G.; Martin, S.J. The Apoptosome Pathway to Caspase Activation in Primary Human Neutrophils Exhibits Dramatically Reduced Requirements for Cytochrome c. J. Exp. Med. 2003, 197, 625–632. [Google Scholar] [CrossRef]
- Altznauer, F.; Martinelli, S.; Yousefi, S.; Thürig, C.; Schmid, I.; Conway, E.M.; Schöni, M.H.; Vogt, P.; Mueller, C.; Fey, M.F.; et al. Inflammation-associated Cell Cycle–independent Block of Apoptosis by Survivin in Terminally Differentiated Neutrophils. J. Exp. Med. 2004, 199, 1343–1354. [Google Scholar] [CrossRef] [PubMed]
- Weinmann, P.; Gaehtgens, P.; Walzog, B. Bcl-Xl- and Bax-alpha-mediated regulation of apoptosis of human neutrophils via caspase-3. Blood 1999, 93, 3106–3115. [Google Scholar] [PubMed]
- Santos-Beneit, A.M.; Mollinedo, F. Expression of genes involved in initiation, regulation, and execution of apoptosis in human neutrophils and during neutrophil differentiation of HL-60 cells. J. Leukoc. Biol. 2000, 67, 712–724. [Google Scholar] [CrossRef] [PubMed]
- Moulding, D.A.; Akgul, C.; Derouet, M.; White, M.R.; Edwards, S.W. BCL-2 family expression in human neutrophils during delayed and accelerated apoptosis. J. Leukoc. Biol. 2001, 70, 783–792. [Google Scholar] [PubMed]
- Kasahara, Y.; Iwai, K.; Yachie, A.; Ohta, K.; Konno, A.; Seki, H.; Miyawaki, T.; Taniguchi, N. Involvement of reactive oxygen intermediates in spontaneous and CD95 (Fas/APO-1)-mediated apoptosis of neutrophils. Blood 1997, 89, 1748–1753. [Google Scholar]
- Aoshiba, K.; Yasui, S.; Hayashi, M.; Tamaoki, J.; Nagai, A. Role of p38-mitogen-activated protein kinase in spontaneous apoptosis of human neutrophils. J. Immunol. 1999, 162, 1692–1700. [Google Scholar] [PubMed]
- Zhang, B.; Hirahashi, J.; Cullere, X.; Mayadas, T.N. Elucidation of molecular events leading to neutrophil apoptosis following phagocytosis. Cross-talk between caspase 8, reactive oxygen species, and MAPK/ERK activation. J. Biol. Chem. 2003, 278, 28443–28454. [Google Scholar] [CrossRef] [PubMed]
- Takei, H.; Araki, A.; Watanabe, H.; Ichinose, A.; Sendo, F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. J. Leukoc. Biol. 1996, 59, 229–240. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Von Köckritz-Blickwede, M.; Goldmann, O.; Thulin, P.; Heinemann, K.; Norrby-Teglund, A.; Rohde, M.; Medina, E. Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood 2008, 111, 3070–3080. [Google Scholar] [CrossRef]
- Von Köckritz-Blickwede, M.; Chow, O.A.; Nizet, V. Fetal calf serum contains heat-stable nucleases that degrade neutrophil extracellular traps. Blood 2009, 114, 5245–5246. [Google Scholar] [CrossRef] [PubMed]
- Yipp, B.G.; Kubes, P. NETosis: How vital is it? Blood 2013, 122, 2784–2794. [Google Scholar] [CrossRef] [PubMed]
- Berghe, T.V.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Iba, T.; Hashiguchi, N.; Nagaoka, I.; Tabe, Y.; Murai, M. Neutrophil cell death in response to infection and its relation to coagulation. J. Intensive Care 2013, 1, 13. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Remijsen, Q.; Berghe, T.V.; Wirawan, E.; Asselbergh, B.; Parthoens, E.; De Rycke, R.; Noppen, S.; Delforge, M.; Willems, J.; Vandenabeele, P. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 2011, 21, 290. [Google Scholar] [CrossRef]
- Azzouz, D.; Palaniyar, N. ApoNETosis: Discovery of a novel form of neutrophil death with concomitant apoptosis and NETosis. Cell Death Dis. 2018, 9, 839. [Google Scholar] [CrossRef]
- Burg, N.D.; Pillinger, M.H. The neutrophil: Function and regulation in innate and humoral immunity. Clin. Immunol. 2001, 99, 7–17. [Google Scholar] [CrossRef]
- Rada, B.; Leto, T. Oxidative innate immune defenses by Nox/Duox Family NADPH oxidases. Contrib. Microbiol. 2008, 15, 164–187. [Google Scholar]
- Brown, D.I.; Griendling, K.K. Nox proteins in signal transduction. Free Radic. Biol. Med. 2009, 47, 1239–1253. [Google Scholar] [CrossRef]
- Karlsson, A.; Nixon, J.B.; McPhail, L.C. Phorbol myristate acetate induces neutrophil NADPH-oxidase activity by two separate signal transduction pathways: Dependent or independent of phosphatidylinositol 3-kinase. J. Leukoc. Biol. 2000, 67, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Farahvash, A.; Douda, D.N.; Licht, J.-C.C.; Grasemann, H.; Sweezey, N.; Palaniyar, N. JNK Activation Turns on LPS-And Gram-Negative Bacteria-Induced NADPH Oxidase-Dependent Suicidal NETosis. Sci. Rep. 2017, 7, 3409. [Google Scholar] [CrossRef] [PubMed]
- Metzler, K.D.; Goosmann, C.; Lubojemska, A.; Zychlinsky, A.; Papayannopoulos, V. Myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep. 2014, 8, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Nauseef, W.M. How human neutrophils kill and degrade microbes: An integrated view. Immunol. Rev. 2007, 219, 88–102. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Philip, L.M.; Cheung, G.; Vadakepeedika, S.; Grasemann, H.; Sweezey, N.; Palaniyar, N. Regulating NETosis: Increasing pH Promotes NADPH Oxidase-Dependent NETosis. Front. Med. 2018, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- De Souza, C.N.; Breda, L.C.D.; Khan, M.A.; de Almeida, S.R.; Câmara, N.O.S.; Sweezey, N.; Palaniyar, N. Alkaline pH promotes NADPH oxidase-independent neutrophil extracellular trap formation: A matter of mitochondrial reactive oxygen species generation and citrullination and cleavage of histone. Front. Immunol. 2018, 8, 1849. [Google Scholar] [CrossRef] [PubMed]
- Vorobjeva, N.V.; Pinegin, B.V. Neutrophil Extracellular Traps: Mechanisms of formation and role in health and disease. Biochemistry 2014, 79, 1286–1296. [Google Scholar] [CrossRef]
- Pilsczek, F.H.; Salina, D.; Poon, K.K.H.; Fahey, C.; Yipp, B.G.; Sibley, C.D.; Robbins, S.M.; Green, F.H.Y.; Surette, M.G.; Sugai, M.; et al. A Novel Mechanism of Rapid Nuclear Neutrophil Extracellular Trap Formation in Response to Staphylococcus aureus. J. Immunol. 2010, 185, 7413–7425. [Google Scholar] [CrossRef]
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.V.; Zbytnuik, L.D.; Pittman, K.; Asaduzzaman, M.; Wu, K.; Meijndert, H.C.; et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 2012, 18, 1386–1393. [Google Scholar] [CrossRef]
- Roos, D.; Voetman, A.A.; Meerhof, L.J. Functional activity of enucleated human polymorphonuclear leukocytes. J. Cell Biol. 1983, 97, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Giaglis, S.; Hasler, P.; Hahn, S. Efficient neutrophil extracellular trap induction requires mobilization of both intracellular and extracellular calcium pools and is modulated by cyclosporine A. PLoS ONE 2014, 9, e97088. [Google Scholar] [CrossRef] [PubMed]
- Slaba, I.; Wang, J.; Kolaczkowska, E.; Mcdonald, B.; Lee, W.Y.; Kubes, P. Imaging the dynamic platelet-neutrophil response in sterile liver injury and repair in mice. Hepatology 2015, 62, 1593–1605. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.; Kubes, P. Innate Immune Cell Trafficking and Function During Sterile Inflammation of the Liver. Gastroenterology 2016, 151, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Christoffersson, G.; Henriksnäs, J.; Johansson, L.; Rolny, C.; Ahlström, H.; Caballero-Corbalan, J.; Segersvärd, R.; Permert, J.; Korsgren, O.; Carlsson, P.O.; et al. Clinical and experimental pancreatic islet transplantation to striated muscle: Establishment of a vascular system similar to that in native islets. Diabetes 2010, 59, 2569–2578. [Google Scholar] [CrossRef]
- Azzouz, L.; Cherry, A.; Riedl, M.; Khan, M.; Pluthero, F.G.; Kahr, W.H.A.; Palaniyar, N.; Licht, C. Relative antibacterial functions of complement and NETs: NETs trap and complement effectively kills bacteria. Mol. Immunol. 2018, 97, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Zychlinsky, A. Neutrophil extracellular traps: Is immunity the second function of chromatin? J. Cell Biol. 2012, 198, 773–783. [Google Scholar] [CrossRef]
- Bianchi, M.; Hakkim, A.; Brinkmann, V.; Siler, U.; Seger, R.A.; Zychlinsky, A.; Reichenbach, J. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood 2009, 114, 2619–2622. [Google Scholar] [CrossRef]
- Jones, J.E.; Causey, C.P.; Knuckley, B.; Slack-Noyes, J.L.; Thompson, P.R. Protein arginine deiminase 4 (PADI4 ): Current understanding and future therapeutic potential. Curr. Opin. Drug Discov. Dev. 2009, 12, 616. [Google Scholar]
- Neeli, I.; Khan, S.N.; Radic, M. Histone Deimination As a Response to Inflammatory Stimuli in Neutrophils. J. Immunol. 2014, 180, 1895–1902. [Google Scholar] [CrossRef]
- Luo, Y.; Arita, K.; Bhatia, M.; Knuckley, B.; Lee, Y.H.; Stallcup, M.R.; Sato, M.; Thompson, P.R. Inhibitors and inactivators of protein arginine deiminase 4: Functional and structural characterization. Biochemistry 2006, 45, 11727–11736. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, M.; Stadler, S.; Correll, S.; Li, P.; Wang, D.; Hayama, R.; Leonelli, L.; Han, H.; Grigoryev, S.A.; et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 2009, 184, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PADI4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Li, R.H.L.; Ng, G.; Tablin, F. Lipopolysaccharide-induced neutrophil extracellular trap formation in canine neutrophils is dependent on histone H3 citrullination by peptidylarginine deiminase. Vet. Immunol. Immunopathol. 2017, 193, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Neeli, I.; Radic, M. Opposition between PKC isoforms regulates histone deimination and neutrophil extracellular chromatin release. Front. Immunol. 2013, 4, 38. [Google Scholar] [CrossRef] [PubMed]
- Kenny, E.F.; Herzig, A.; Krüger, R.; Muth, A.; Mondal, S.; Thompson, P.R.; Brinkmann, V.; von Bernuth, H.; Zychlinsky, A. Diverse stimuli engage different neutrophil extracellular trap pathways. eLife 2017, 6, e24437. [Google Scholar] [CrossRef] [PubMed]
- Fischle, W.; Franz, H.; Jacobs, S.A.; Allis, C.D.; Khorasanizadeh, S. Specificity of the chromodomain Y chromosome family of chromodomains for lysine-methylated ARK(S/T) motifs. J. Biol. Chem. 2008, 283, 19626–19635. [Google Scholar] [CrossRef] [PubMed]
- Byvoet, P.; Shepherd, G.R.; Hardin, J.M.; Noland, B.J. The distribution and turnover of labeled methyl groups in histone fractions of cultured mammalian cells. Arch. Biochem. Biophys. 1972, 148, 558–567. [Google Scholar] [CrossRef]
- Murray, K. The Occurrence of epsilon-N-Methyl Lysine in Histones. Biochemistry 1963, 3, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef]
- Rea, S.; Eisenhaber, F.; O’Carroll, D.; Strahl, B.D.; Sun, Z.W.; Schmid, M.; Opravil, S.; Mechtier, K.; Ponting, C.P.; Allis, C.D.; et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 2000, 406, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Wang, H.; Ng, H.H.; Erdjument-Bromage, H.; Tempst, P.; Struhl, K.; Zhang, Y. Methylation of H3-lysine 79 is mediated by a new family of HMTases without a SET domain. Curr. Biol. 2002, 12, 1052–1058. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Cloos, P.A.C.; Christensen, J.; Agger, K.; Maiolica, A.; Rappsilber, J.; Antal, T.; Hansen, K.H.; Helin, K. The putative oncogene GASC1 demethylates tri- and dimethylated lysine 9 on histone H3. Nature 2006, 442, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Whetstine, J.R.; Nottke, A.; Lan, F.; Huarte, M.; Smolikov, S.; Chen, Z.; Spooner, E.; Li, E.; Zhang, G.; Colaiacovo, M.; et al. Reversal of Histone Lysine Trimethylation by the JMJD2 Family of Histone Demethylases. Cell 2006, 125, 467–481. [Google Scholar] [CrossRef]
- Chang, B.; Chen, Y.; Zhao, Y.; Bruick, R.K. JMJD6 is a histone arginine demethylase. Science 2007, 318, 444–447. [Google Scholar] [CrossRef] [PubMed]
- Webby, C.J.; Wolf, A.; Gromak, N.; Dreger, M.; Kramer, H.; Kessler, B.; Nielsen, M.L.; Schmitz, C.; Butler, D.S.; Yates, J.R.; et al. Jmjd6 catalyses lysyl-hydroxylation of U2AF65, a protein associated with RNA splicing. Science 2009, 325, 90–93. [Google Scholar] [CrossRef]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef]
- Pieterse, E.; Hofstra, J.; Berden, J.; Herrmann, M.; Dieker, J.; van der Vlag, J. Acetylated histones contribute to the immunostimulatory potential of neutrophil extracellular traps in systemic lupus erythematosus. Clin. Exp. Immunol. 2015, 179, 68–74. [Google Scholar] [CrossRef]
- Liu, C.L.; Tangsombatvisit, S.; Rosenberg, J.M.; Mandelbaum, G.; Gillespie, E.C.; Gozani, O.P.; Alizadeh, A.A.; Utz, P.J. Specific post-translational histone modifications of neutrophil extracellular traps as immunogens and potential targets of lupus autoantibodies. Arthritis Res. Ther. 2012, 14, R25. [Google Scholar] [CrossRef]
- Cedar, H.; Bergman, Y. Linking DNA methylation and histone modification: Patterns and paradigms. Nat. Rev. Genet. 2009, 10, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Rajakumara, E.; Law, J.A.; Simanshu, D.K.; Voigt, P.; Johnson, L.M.; Reinberg, D.; Patel, D.J.; Jacobsen, S.E. A dual flip-out mechanism for 5mC recognition by the Arabidopsis SUVH5 SRA domain and its impact on DNA methylation and H3K9 dimethylation in vivo. Genes Dev. 2011, 25, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Hashimshony, T.; Zhang, J.; Keshet, I.; Bustin, M.; Cedar, H. The role of DNA methylation in setting up chromatin structure during development. Nat. Genet. 2003, 34, 187. [Google Scholar] [CrossRef] [PubMed]
- Lande-Diner, L.; Zhang, J.; Ben-Porath, I.; Amariglio, N.; Keshet, I.; Hecht, M.; Azuara, V.; Fisher, A.G.; Rechavi, G.; Cedar, H. Role of DNA methylation in stable gene repression. J. Biol. Chem. 2007, 282, 12194–12200. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wysocka, J.; Sayegh, J.; Lee, Y.H.; Pertin, J.R.; Leonelli, L.; Sonbuchner, L.S.; McDonald, C.H.; Cook, R.G.; Dou, Y.; et al. Human PADI4 regulates histone arginine methylation levels via demethylimination. Science 2004, 306, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Raijmakers, R.; Zendman, A.J.W.; Egberts, W.V.; Vossenaar, E.R.; Raats, J.; Soede-Huijbregts, C.; Rutjes, F.P.J.T.; van Veelen, P.A.; Drijfhout, J.W.; Pruijn, G.J.M. Methylation of Arginine Residues Interferes with Citrullination by Peptidylarginine Deiminases in vitro. J. Mol. Biol. 2007, 367, 1118–1129. [Google Scholar] [CrossRef] [PubMed]
- Cuthbert, G.L.; Daujat, S.; Snowden, A.W.; Erdjument-Bromage, H.; Hagiwara, T.; Yamada, M.; Schneider, R.; Gregory, P.D.; Tempst, P.; Bannister, A.J.; et al. Histone deimination antagonizes arginine methylation. Cell 2004, 118, 545–553. [Google Scholar] [CrossRef]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone acetyltransferases. Histone Acetyltransferases 2001, 70, 81–120. [Google Scholar] [CrossRef]
- Hollands, A.; Corriden, R.; Gysler, G.; Dahesh, S.; Olson, J.; Ali, S.R.; Kunkel, M.T.; Lin, A.E.; Forli, S.; Newton, A.C.; et al. Natural product anacardic acid from cashew nut shells stimulates neutrophil extracellular trap production and bactericidal activity. J. Biol. Chem. 2016, 291, 13964–13973. [Google Scholar] [CrossRef]
- Pandey, D.; Chen, F.; Patel, A.; Wang, C.Y.; Dimitropoulou, C.; Patel, V.S.; Rudic, R.D.; Stepp, D.W.; Fulton, D.J. SUMO1 negatively regulates reactive oxygen species production from NADPH oxidases. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1634–1642. [Google Scholar] [CrossRef]
- Johnstone, R.W.; Licht, J.D. Histone deacetylase inhibitors in cancer therapy: Is transcription the primary target? Cancer Cell 2003, 4, 13–18. [Google Scholar] [CrossRef]
- Yang, X.-J.; Grégoire, S. Class II histone deacetylases: From sequence to function, regulation, and clinical implication. Mol. Cell. Biol. 2005, 25, 2873–2884. [Google Scholar] [CrossRef] [PubMed]
- Chen, L. Medicinal Chemistry of Sirtuin Inhibitors. Curr. Med. Chem. 2011, 18, 1936–1946. [Google Scholar] [CrossRef] [PubMed]
- Kankaanranta, H.; Janka-Junttila, M.; Ilmarinen-Salo, P.; Ito, K.; Jalonen, U.; Ito, M.; Adcock, I.M.; Moilanen, E.; Zhang, X. Histone deacetylase inhibitors induce apoptosis in human eosinophils and neutrophils. J. Inflamm. 2010, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yu, C.-W.; Duan, J.; Luo, M.; Wang, K.; Tian, G.; Cui, Y.; Wu, K. HDA6 Directly Interacts with DNA Methyltransferase MET1 and Maintains Transposable Element Silencing in Arabidopsis. Plant Physiol. 2012, 158, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Vaissière, T.; Sawan, C.; Herceg, Z. Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat. Res. Rev. Mutat. Res. 2008, 659, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-H.; Coonrod, S.A.; Kraus, W.L.; Jelinek, M.A.; Stallcup, M.R. Regulation of coactivator complex assembly and function by protein arginine methylation and demethylimination. Proc. Natl. Acad. Sci. USA 2005, 102, 3611–3616. [Google Scholar] [CrossRef]
- Fuhrmann, J.; Thompson, P.R. Protein Arginine Methylation and Citrullination in Epigenetic Regulation. ACS Chem. Biol. 2016, 11, 654–668. [Google Scholar] [CrossRef]
- Denis, H.; Deplus, R.; Putmans, P.; Yamada, M.; Metivier, R.; Fuks, F. Functional Connection between Deimination and Deacetylation of Histones. Mol. Cell. Biol. 2009, 29, 4982–4993. [Google Scholar] [CrossRef]
- Dokmanovic, M.; Marks, P.A. Prospects: Histone deacetylase inhibitors. J. Cell. Biochem. 2005, 96, 293–304. [Google Scholar] [CrossRef]
- Marks, P.A.; Dokmanovic, M. Histone deacetylase inhibitors: Discovery and development as anticancer agents. Expert Opin. Investig. Drugs 2005, 14, 1497–1511. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, P.M.; Cole, K.E.; Dowling, D.P.; Christianson, D.W. Structure, mechanism, and inhibition of histone deacetylases and related metalloenzymes. Curr. Opin. Struct. Biol. 2011, 21, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Eom, G.H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam Med. J. 2016, 52, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA Approval Summary: Vorinostat for Treatment of Advanced Primary Cutaneous T-Cell Lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A.; Xu, W.S. Histone deacetylase inhibitors: Potential in cancer therapy. J. Cell. Biochem. 2009, 107, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Poole, R.M. Belinostat: First global approval. Drugs 2014, 74, 1445–1453. [Google Scholar] [CrossRef]
- Molife, L.R.; de Bono, J.S. Belinostat: Clinical applications in solid tumors and lymphoma. Expert Opin. Investig. Drugs 2011, 20, 1723–1732. [Google Scholar] [CrossRef]
- Plumb, J.A.; Finn, P.W.; Williams, R.J.; Bandara, M.J.; Romero, M.R.; Watkins, C.J.; La Thangue, N.B.; Brown, R. Pharmacodynamic response and inhibition of growth of human tumor xenografts by the novel histone deacetylase inhibitor PXD101. Mol. Cancer Ther. 2003, 2, 721–728. [Google Scholar]
- Steele, N.L.; Plumb, J.A.; Vidal, L.; Tjørnelund, J.; Knoblauch, P.; Rasmussen, A.; Ooi, C.E.; Buhl-Jensen, P.; Brown, R.; Evans, T.R.J.; et al. A phase 1 pharmacokinetic and pharmacodynamic study of the histone deacetylase inhibitor belinostat in patients with advanced solid tumors. Clin. Cancer Res. 2008, 14, 804–810. [Google Scholar] [CrossRef]
- Tumber, A.; Collins, L.S.; Petersen, K.D.; Thougaard, A.; Christiansen, S.J.; Dejligbjerg, M.; Jensen, P.B.; Sehested, M.; Ritchie, J.W.A. The histone deacetylase inhibitor PXD101 synergises with 5-fluorouracil to inhibit colon cancer cell growth in vitro and in vivo. Cancer Chemother. Pharmacol. 2007, 60, 275–283. [Google Scholar] [CrossRef]
- Kong, L.R.; Tan, T.Z.; Ong, W.R.; Bi, C.; Huynh, H.; Lee, S.C.; Chng, W.J.; Eichhorn, P.J.A.; Goh, B.C. Belinostat exerts antitumor cytotoxicity through the ubiquitin-proteasome pathway in lung squamous cell carcinoma. Mol. Oncol. 2017, 11, 965–980. [Google Scholar] [CrossRef] [PubMed]
- Valiuliene, G.; Stirblyte, I.; Cicenaite, D.; Kaupinis, A.; Valius, M.; Navakauskiene, R. Belinostat, a potent HDACi, exerts antileukaemic effect in human acute promyelocytic leukaemia cells via chromatin remodeling. J. Cell. Mol. Med. 2015, 19, 1742–1755. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Ara, G.; Mills, E.; LaRochelle, W.J.; Lichenstein, H.S.; Jeffers, M. Activity of the histone deacetylase inhibitor belinostat (PXD101) in preclinical models of prostate cancer. Int. J. Cancer 2008, 122, 1400–1410. [Google Scholar] [CrossRef] [PubMed]
- Qian, X. Activity of PXD101, a histone deacetylase inhibitor, in preclinical ovarian cancer studies. Mol. Cancer Ther. 2006, 5, 2086–2095. [Google Scholar] [CrossRef] [PubMed]
- Buckley, M.T.; Yoon, J.; Yee, H.; Chiriboga, L.; Liebes, L.; Ara, G.; Qian, X.; Bajorin, D.F.; Sun, T.-T.; Wu, X.-R.; et al. The histone deacetylase inhibitor belinostat (PXD101) suppresses bladder cancer cell growth in vitro and in vivo. J. Transl. Med. 2007, 5, 49. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Z.; Kwitkowski, V.E.; Del Valle, P.L.; Ricci, M.S.; Saber, H.; Habtemariam, B.A.; Bullock, J.; Bloomquist, E.; Shen, Y.L.; Chen, X.H.; et al. FDA approval: Belinostat for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma. Clin. Cancer Res. 2015, 21, 2666–2670. [Google Scholar] [CrossRef]
- (FDA), U.S.F. & D. administration Press Announcements - FDA approves Farydak for treatment of multiple myeloma. Available online: https://www.cancer.org/latest-news/fda-approves-farydak-panobinostat-for-multiple-myeloma.html (accessed on 25 June 2019).
- Garnock-Jones, K.P. Panobinostat: First global approval. Drugs 2015, 75, 695–704. [Google Scholar] [CrossRef]
- Atadja, P. Development of the pan-DAC inhibitor panobinostat (LBH589): Successes and challenges. Cancer Lett. 2009, 280, 233–241. [Google Scholar] [CrossRef]
- Cheng, T.; Grasse, L.; Shah, J.; Chandra, J. Panobinostat, a pan-histone deacetylase inhibitor: Rationale for and application to treatment of multiple myeloma. Drugs Today 2015, 51, 491–504. [Google Scholar] [CrossRef]
- Moore, D. Panobinostat (Farydak): A Novel Option for the Treatment of Relapsed or Relapsed and Refractory Multiple Myeloma. Pharm. Ther. 2016, 41, 296. [Google Scholar]
- Cashen, A.; Juckett, M.; Jumonville, A.; Litzow, M.; Flynn, P.J.; Eckardt, J.; LaPlant, B.; Laumann, K.; Erlichman, C.; DiPersio, J. Phase II study of the histone deacetylase inhibitor belinostat (PXD101) for the treatment of myelodysplastic syndrome (MDS). Ann. Hematol. 2012, 91, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Dincman, T.A.; Beare, J.E.; Ohri, S.S.; Gallo, V.; Hetman, M.; Whittemore, S.R. Histone deacetylase inhibition is cytotoxic to oligodendrocyte precursor cells in vitro and in vivo. Int. J. Dev. Neurosci. 2016, 54, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Savickiene, J.; Treigyte, G.; Valiuliene, G.; Stirblyte, I.; Navakauskiene, R. Epigenetic and molecular mechanisms underlying the antileukemic activity of the histone deacetylase inhibitor belinostat in human acute promyelocytic leukemia cells. Anticancer Drugs 2014, 25, 938–949. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.J.; Huang, K.K.; Yang, M.; Qiao, L.; Wang, Q.; Ye, J.Y.; Zhou, H.S.; Yi, Z.S.; Wu, F.Q.; Wang, Z.X.; et al. Synergistic effect of panobinostat and bortezomib on chemoresistant acute myelogenous leukemia cells via AKT and NF-κB pathways. Cancer Lett. 2012, 326, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Matthay, M.A.; Ware, L.B.; Zimmerman, G.A. The acute respiratory distress syndrome. J. Clin. Investig. 2012, 122, 2731–2740. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Su, X.; Pan, P.; Zhang, L.; Hu, Y.; Tan, H.; Wu, D.; Liu, B.; Li, H.; Li, H.; et al. Neutrophil extracellular traps are indirectly triggered by lipopolysaccharide and contribute to acute lung injury. Sci. Rep. 2016, 6, 37252. [Google Scholar] [CrossRef]
- Ota, C.; Yamada, M.; Fujino, N.; Motohashi, H.; Tando, Y.; Takei, Y.; Suzuki, T.; Takahashi, T.; Kamata, S.; Makiguchi, T.; et al. Histone deacetylase inhibitor restores surfactant protein-C expression in alveolar-epithelial type II cells and attenuates bleomycin-induced pulmonary fibrosis in vivo. Exp. Lung Res. 2015, 41, 422–434. [Google Scholar] [CrossRef]
- Sperling, C.; Fischer, M.; Maitz, M.F.; Werner, C. Neutrophil extracellular trap formation upon exposure of hydrophobic materials to human whole blood causes thrombogenic reactions. Biomater. Sci. 2017, 5, 1998–2008. [Google Scholar] [CrossRef]
- Martinod, K.; Demers, M.; Fuchs, T.A.; Wong, S.L.; Brill, A.; Gallant, M.; Hu, J.; Wang, Y.; Wagner, D.D. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 8674–8679. [Google Scholar] [CrossRef]
- Lipinska-Gediga, M. Neutrophils, NETs, NETosis—Old or new factors in sepsis and septic shock? Anestezjol. Intens. Ter. 2017, 49, 235–240. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, X.; Pelayo, R.; Monestier, M.; Ammollo, C.T.; Semeraro, F.; Taylor, F.B.; Esmon, N.L.; Lupu, F.; Esmon, C.T. Extracellular histones are major mediators of death in sepsis. Nat. Med. 2009, 15, 1318. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463. [Google Scholar] [CrossRef] [PubMed]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: A predominant role of histones. PLoS ONE 2012, 7, e32366. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, B.M.; Fisher, B.J.; Kraskauskas, D.; Farkas, D.; Brophy, D.F.; Fowler, A.A.; Natarajan, R. Vitamin C: A novel regulator of neutrophil extracellular trap formation. Nutrients 2013, 5, 3131–3150. [Google Scholar] [CrossRef] [PubMed]
- Claushuis, T.A.M.; van der Donk, L.E.H.; Luitse, A.L.; van Veen, H.A.; van der Wel, N.N.; van Vught, L.A.; Roelofs, J.J.T.H.; de Boer, O.J.; Lankelma, J.M.; Boon, L.; et al. Role of Peptidylarginine Deiminase 4 in Neutrophil Extracellular Trap Formation and Host Defense during Klebsiella pneumoniae—Induced Pneumonia-Derived Sepsis. J. Immunol. 2018, 201, 1241–1252. [Google Scholar] [CrossRef]
- Nomura, K.; Miyashita, T.; Yamamoto, Y.; Munesue, S.; Harashima, A.; Takayama, H.; Fushida, S.; Ohta, T. Citrullinated Histone H3: Early Biomarker of Neutrophil Extracellular Traps in Septic Liver Damage. J. Surg. Res. 2019, 234, 132–138. [Google Scholar] [CrossRef]
- Warford, J.; Lamport, A.C.; Kennedy, B.; Easton, A.S. Human Brain Chemokine and Cytokine Expression in Sepsis: A Report of Three Cases. Can. J. Neurol. Sci. 2017, 44, 96–104. [Google Scholar] [CrossRef]
- Alamdari, N.; Smith, I.J.; Aversa, Z.; Hasselgren, P.-O. Sepsis and glucocorticoids upregulate p300 and downregulate HDAC6 expression and activity in skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 299, R509–R520. [Google Scholar] [CrossRef]
- Cheng, F.; Lienlaf, M.; Perez-Villarroel, P.; Wang, H.W.; Lee, C.; Woan, K.; Woods, D.; Knox, T.; Bergman, J.; Pinilla-Ibarz, J.; et al. Divergent roles of histone deacetylase 6 (HDAC6) and histone deacetylase 11 (HDAC11) on the transcriptional regulation of IL10 in antigen presenting cells. Mol. Immunol. 2014, 60, 44–53. [Google Scholar] [CrossRef]
- von Knethen, A.; Brüne, B. Histone Deacetylation Inhibitors as Therapy Concept in Sepsis. Int. J. Mol. Sci. 2019, 20, 346. [Google Scholar] [CrossRef]
- Wang, H.; Cheng, F.; Woan, K.; Sahakian, E.; Merino, O.; Rock-Klotz, J.; Vicente-Suarez, I.; Pinilla-Ibarz, J.; Wright, K.L.; Seto, E.; et al. Histone Deacetylase Inhibitor LAQ824 Augments Inflammatory Responses in Macrophages through Transcriptional Regulation of IL-10. J. Immunol. 2011, 186, 3986–3996. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jin, S.; Wang, C.; Jiang, R.; Wan, J. Histone deacetylase inhibitors attenuate acute lung injury during cecal ligation and puncture-induced polymicrobial sepsis. World J. Surg. 2010, 34, 1676–1683. [Google Scholar] [CrossRef] [PubMed]
- Ji, M.H.; Li, G.M.; Jia, M.; Zhu, S.H.; Gao, D.P.; Fan, Y.X.; Wu, J.; Yang, J.J. Valproic acid attenuates lipopolysaccharide-induced acute lung injury in mice. Inflammation 2013, 36, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Zhang, Y. Regulation of histone methylation by demethylimination and demethylation. Nat. Rev. Mol. Cell Biol. 2007, 8, 307. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zang, C.; Cui, K.; Schones, D.E.; Barski, A.; Peng, W.; Zhao, K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 2009, 138, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Gibot, S.; Alauzet, C.; Massin, F.; Sennoune, N.; Faure, G.C.; Béné, M.; Lozniewski, A.; Bollaert, P.; Lévy, B. Modulation of the Triggering Receptor Expressed on Myeloid Cells—1 Pathway during Pneumonia in Rats. J. Infect. Dis. 2006, 194, 975–983. [Google Scholar] [CrossRef]
- Yuan, Z.; Syed, M.A.; Panchal, D.; Rogers, D.; Joo, M.; Sadikot, R.T. Curcumin mediated epigenetic modulation inhibits TREM-1 expression in response to lipopolysaccharide. Int. J. Biochem. Cell Biol. 2012, 44, 2032–2043. [Google Scholar] [CrossRef]
- Rahman, A.; Isenberg, D.A. Systemic Lupus Erythematosus. N. Engl. J. Med. 2008, 358, 929–939. [Google Scholar] [CrossRef]
- Hakkim, A.; Furnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA 2010, 107, 9813–9818. [Google Scholar] [CrossRef]
- Leffler, J.; Martin, M.; Gullstrand, B.; Tyden, H.; Lood, C.; Truedsson, L.; Bengtsson, A.A.; Blom, A.M. Neutrophil Extracellular Traps That Are Not Degraded in Systemic Lupus Erythematosus Activate Complement Exacerbating the Disease. J. Immunol. 2012, 188, 3522–3531. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, N.; Upadhyay, J.; Neeli, I.; Khan, S.; Pattanaik, D.; Myers, L.; Kirou, K.A.; Hellmich, B.; Knuckley, B.; Thompson, P.R.; et al. Felty’s syndrome autoantibodies bind to deiminated histones and neutrophil extracellular chromatin traps. Arthritis Rheum. 2012, 64, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, N.; Neeli, I.; Schall, N.; Wan, H.; Desiderio, D.M.; Csernok, E.; Thompson, P.R.; Dali, H.; Briand, J.P.; Muller, S.; et al. Deimination of linker histones links neutrophil extracellular trap release with autoantibodies in systemic autoimmunity. FASEB J. 2014, 28, 2840–2851. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.S.; Zhao, W.; Luo, W.; Subramanian, V.; O’Dell, A.A.; Yalavarthi, S.; Hodgin, J.B.; Eitzman, D.T.; Thompson, P.R.; Kaplan, M.J. Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. J. Clin. Investig. 2013, 123, 2981–2993. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.S.; Subramanian, V.; O’Dell, A.A.; Yalavarthi, S.; Zhao, W.; Smith, C.K.; Hodgin, J.B.; Thompson, P.R.; Kaplan, M.J. Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice. Ann. Rheum. Dis. 2015, 74, 2199–2206. [Google Scholar] [CrossRef] [PubMed]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef] [PubMed]
- Fiskus, W.; Ren, Y.; Mohapatra, A.; Bali, P.; Mandawat, A.; Rao, R.; Herger, B.; Yang, Y.; Atadja, P.; Wu, J.; et al. Hydroxamic acid analogue histone deacetylase inhibitors attenuate estrogen receptor-α levels and transcriptional activity: A result of hyperacetylation and inhibition of chaperone function of heat shock protein 90. Clin. Cancer Res. 2007, 13, 4882–4890. [Google Scholar] [CrossRef] [PubMed]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.W. Histone-deacetylase inhibitors: Novel drugs for the treatment of cancer. Nat. Rev. Drug Discov. 2002, 1, 287–299. [Google Scholar] [CrossRef]
- Park, J.H.; Jung, Y.; Kim, T.Y.; Kim, S.G.; Jong, H.S.; Lee, J.W.; Kim, D.K.; Lee, J.S.; Kim, N.K.; Kim, T.Y.; et al. Class I histone deacetylase-selective novel synthetic inhibitors potently inhibit human tumor proliferation. Clin. Cancer Res. 2004, 10, 5271–5281. [Google Scholar] [CrossRef]
- Huang, B.H.; Laban, M.; Leung, C.H.-W.; Lee, L.; Lee, C.K.; Salto-Tellez, M.; Raju, G.C.; Hooi, S.C. Inhibition of histone deacetylase 2 increases apoptosis and p21Cip1/WAF1 expression, independent of histone deacetylase 1. Cell Death Differ. 2005, 12, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Swierczak, A.; Mouchemore, K.A.; Hamilton, J.A.; Anderson, R.L. Neutrophils: Important contributors to tumor progression and metastasis. Cancer Metastasis Rev. 2015, 34, 735–751. [Google Scholar] [CrossRef] [PubMed]
- Demers, M.; Wong, S.L.; Martinod, K.; Gallant, M.; Cabral, J.E.; Wang, Y.; Wagner, D.D. Priming of neutrophils toward NETosis promotes tumor growth. Oncoimmunology 2016, 5, e1134073. [Google Scholar] [CrossRef] [PubMed]
- Pieterse, E.; Rother, N.; Garsen, M.; Hofstra, J.M.; Satchell, S.C.; Hoffmann, M.; Loeven, M.A.; Knaapen, H.K.; Van Der Heijden, O.W.H.; Berden, J.H.M.; et al. Neutrophil Extracellular Traps Drive Endothelial-to-Mesenchymal Transition. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1371–1379. [Google Scholar] [CrossRef] [PubMed]
- Cools-Lartigue, J.; Spicer, J.; McDonald, B.; Gowing, S.; Chow, S.; Giannias, B.; Bourdeau, F.; Kubes, P.; Ferri, L. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J. Clin. 2013, 123, 3446–3458. [Google Scholar] [CrossRef] [PubMed]
- Thålin, C.; Lundström, S.; Seignez, C.; Daleskog, M.; Lundström, A.; Henriksson, P.; Helleday, T.; Phillipson, M.; Wallén, H.; Demers, M. Citrullinated histone H3 as a novel prognostic blood marker in patients with advanced cancer. PLoS ONE 2018, 13, e0191231. [Google Scholar] [CrossRef] [PubMed]
- Sen, G.S.; Mohanty, S.; Hossain, D.M.S.; Bhattacharyya, S.; Banerjee, S.; Chakraborty, J.; Saha, S.; Ray, P.; Bhattacharjee, P.; Mandal, D.; et al. Curcumin enhances the efficacy of chemotherapy by tailoring p65NFκB-p300 cross-talk in favor of p53-p300 in breast cancer. J. Biol. Chem. 2011, 286, 42232–42247. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Wang, Y.; Zhu, D.; Xue, Z.; Mao, H. Alteration of histone H3 lysine 9 dimethylation in peripheral white blood cells of septic patients with trauma and cancer. Mol. Med. Rep. 2016, 14, 5467–5474. [Google Scholar] [CrossRef]
- Simiele, F.; Recchiuti, A.; Patruno, S.; Plebani, R.; Pierdomenico, A.M.; Codagnone, M.; Romano, M. Epigenetic regulation of the formyl peptide receptor 2 gene. Biochim. Biophys. Acta Gene Regul. Mech. 2016, 1859, 1252–1258. [Google Scholar] [CrossRef]
- Wang, B.; Wang, X.B.; Chen, L.Y.; Huang, L.; Dong, R.Z. Belinostat-induced apoptosis and growth inhibition in pancreatic cancer cells involve activation of TAK1-AMPK signaling axis. Biochem. Biophys. Res. Commun. 2013, 437, 1–6. [Google Scholar] [CrossRef]
- Lin, S.F.; Lin, J.D.; Chou, T.C.; Huang, Y.Y.; Wong, R.J. Utility of a Histone Deacetylase Inhibitor (PXD101) for Thyroid Cancer Treatment. PLoS ONE 2013, 8, e77684. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamam, H.J.; Palaniyar, N. Post-Translational Modifications in NETosis and NETs-Mediated Diseases. Biomolecules 2019, 9, 369. https://doi.org/10.3390/biom9080369
Hamam HJ, Palaniyar N. Post-Translational Modifications in NETosis and NETs-Mediated Diseases. Biomolecules. 2019; 9(8):369. https://doi.org/10.3390/biom9080369
Chicago/Turabian StyleHamam, Hussein J., and Nades Palaniyar. 2019. "Post-Translational Modifications in NETosis and NETs-Mediated Diseases" Biomolecules 9, no. 8: 369. https://doi.org/10.3390/biom9080369
APA StyleHamam, H. J., & Palaniyar, N. (2019). Post-Translational Modifications in NETosis and NETs-Mediated Diseases. Biomolecules, 9(8), 369. https://doi.org/10.3390/biom9080369