
Transcriptome and Proteome Conjoint Analysis Revealed That Exogenous Sulfur Regulates Glucosinolate Synthesis in Cabbage

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Content Analysis of GLS under +S and −S Treatment in Cabbage Seedlings
2.2. Transcriptome Sequencing and Correlation Analysis
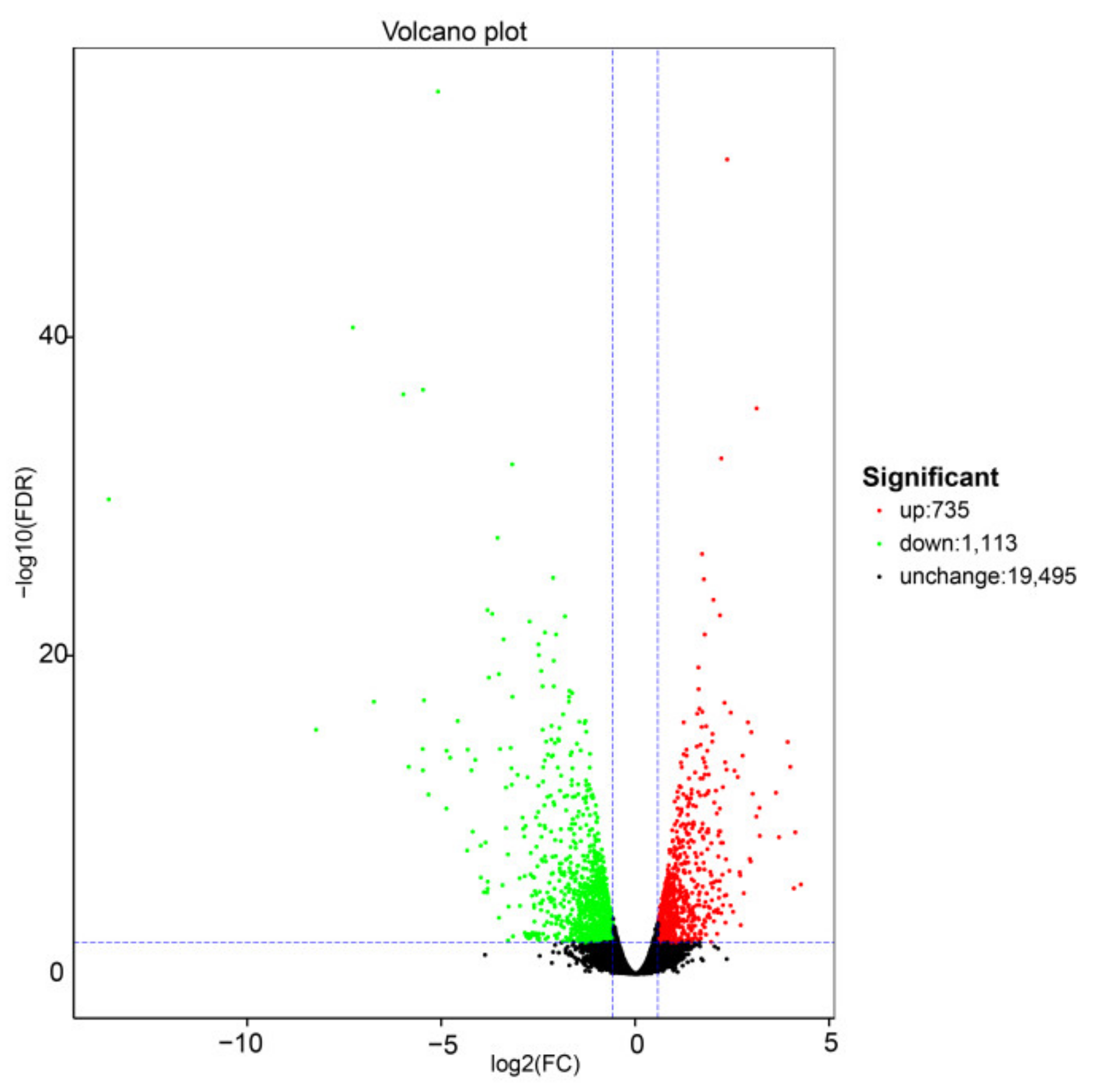
2.2.1. Transcriptome Analysis of Exogenous S Treatments in Cabbage
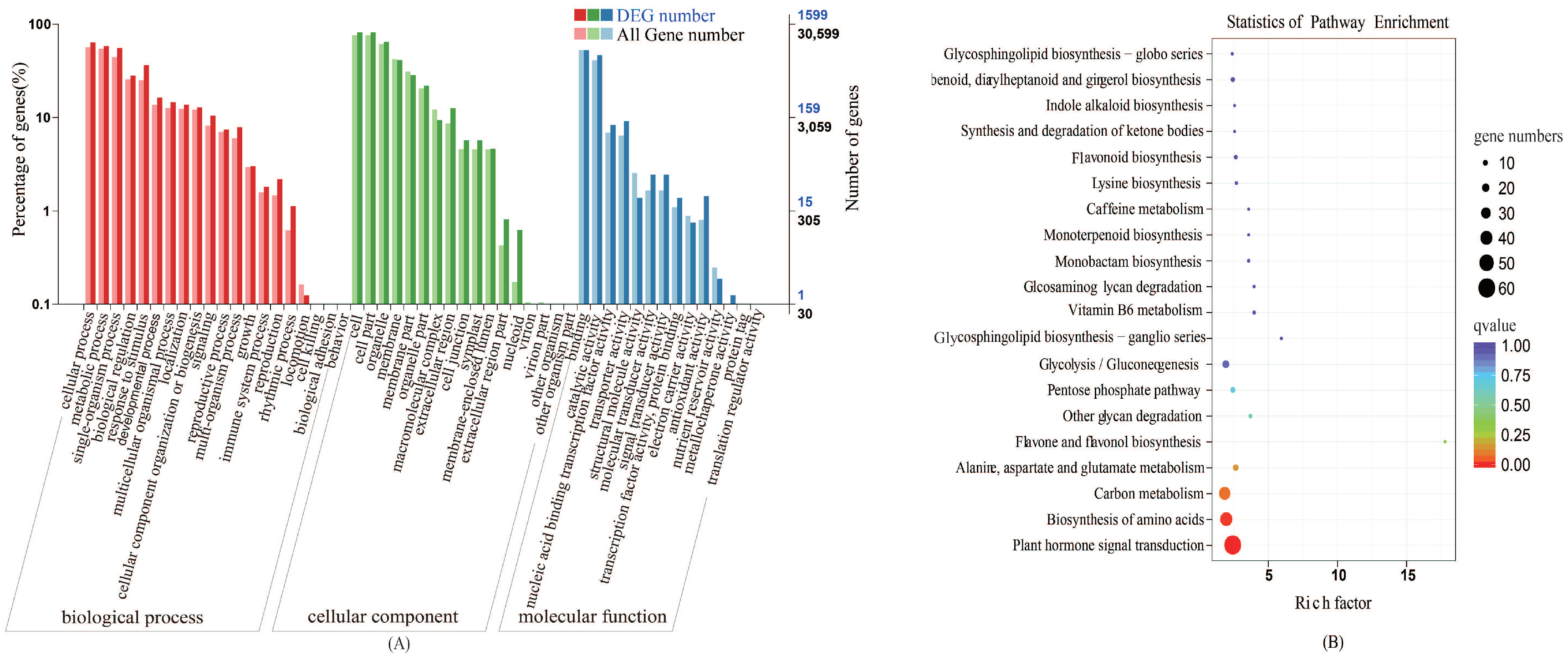
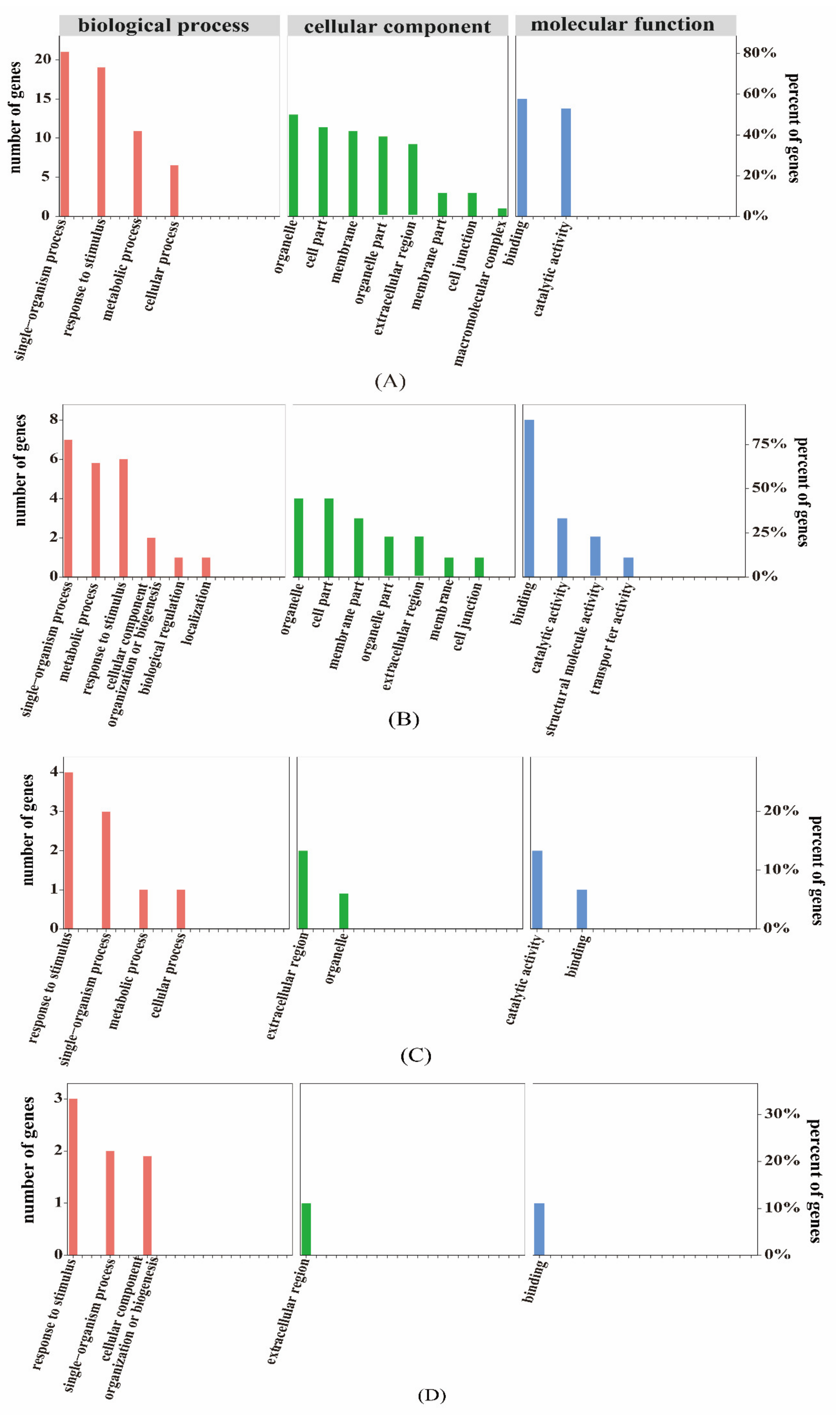
2.2.2. Gene Ontology (GO) Classification Analyses
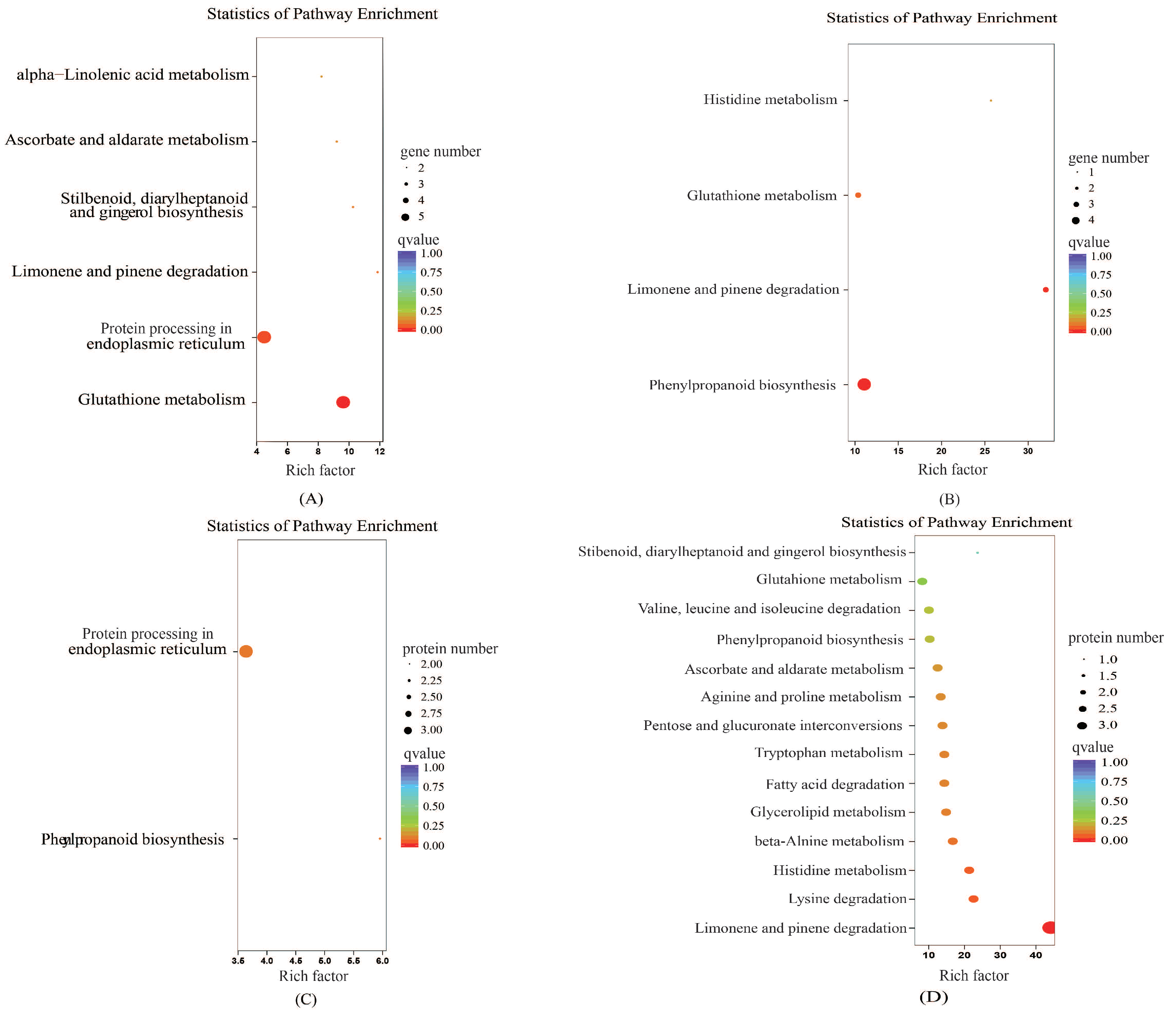
2.2.3. Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Analyses
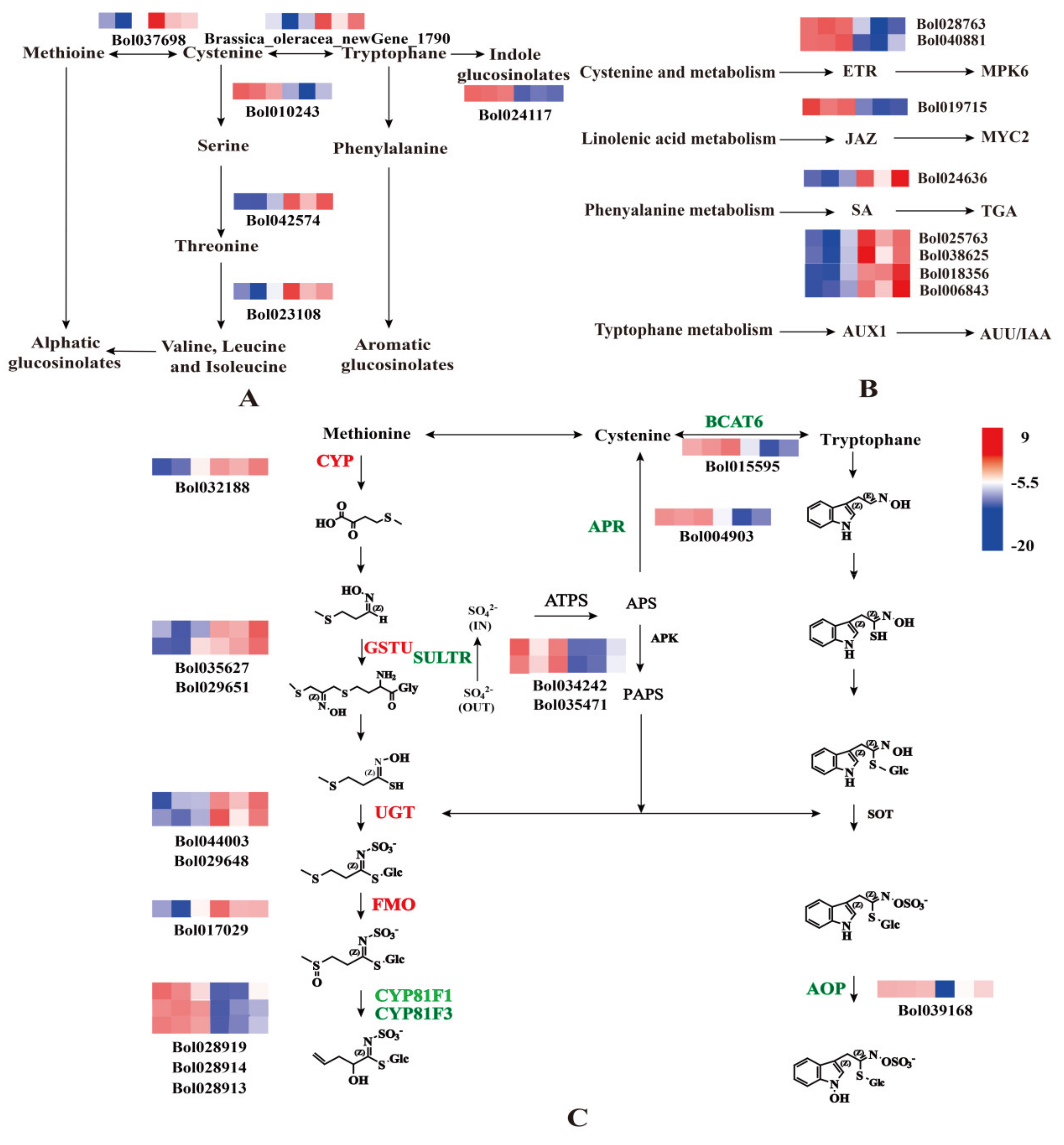
2.3. Sulfur Regulates the Synthesis of Cabbage GLS
2.4. DEG Profiling Validation by qRT-PCR Analysis
2.5. Conjoint Analysis of the Proteome after Exogenous S Treatment in Cabbage
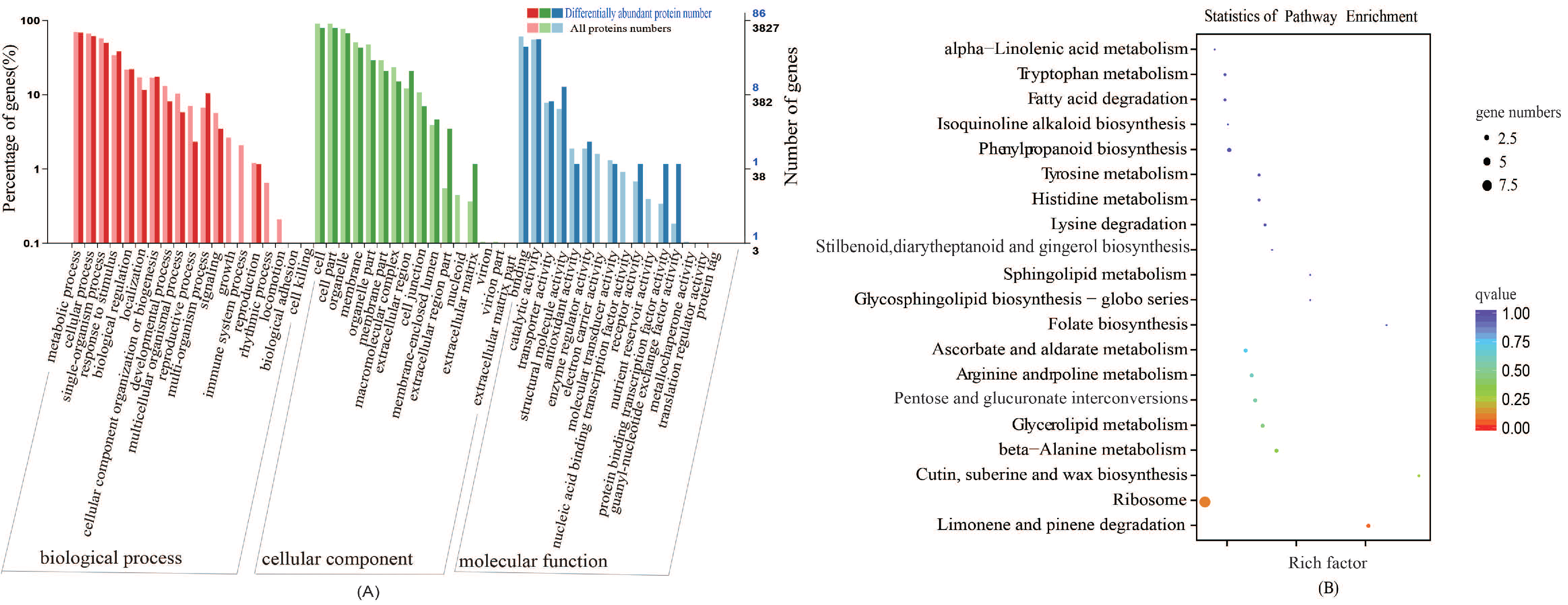
GO Classification and KEGG Pathway Analyses
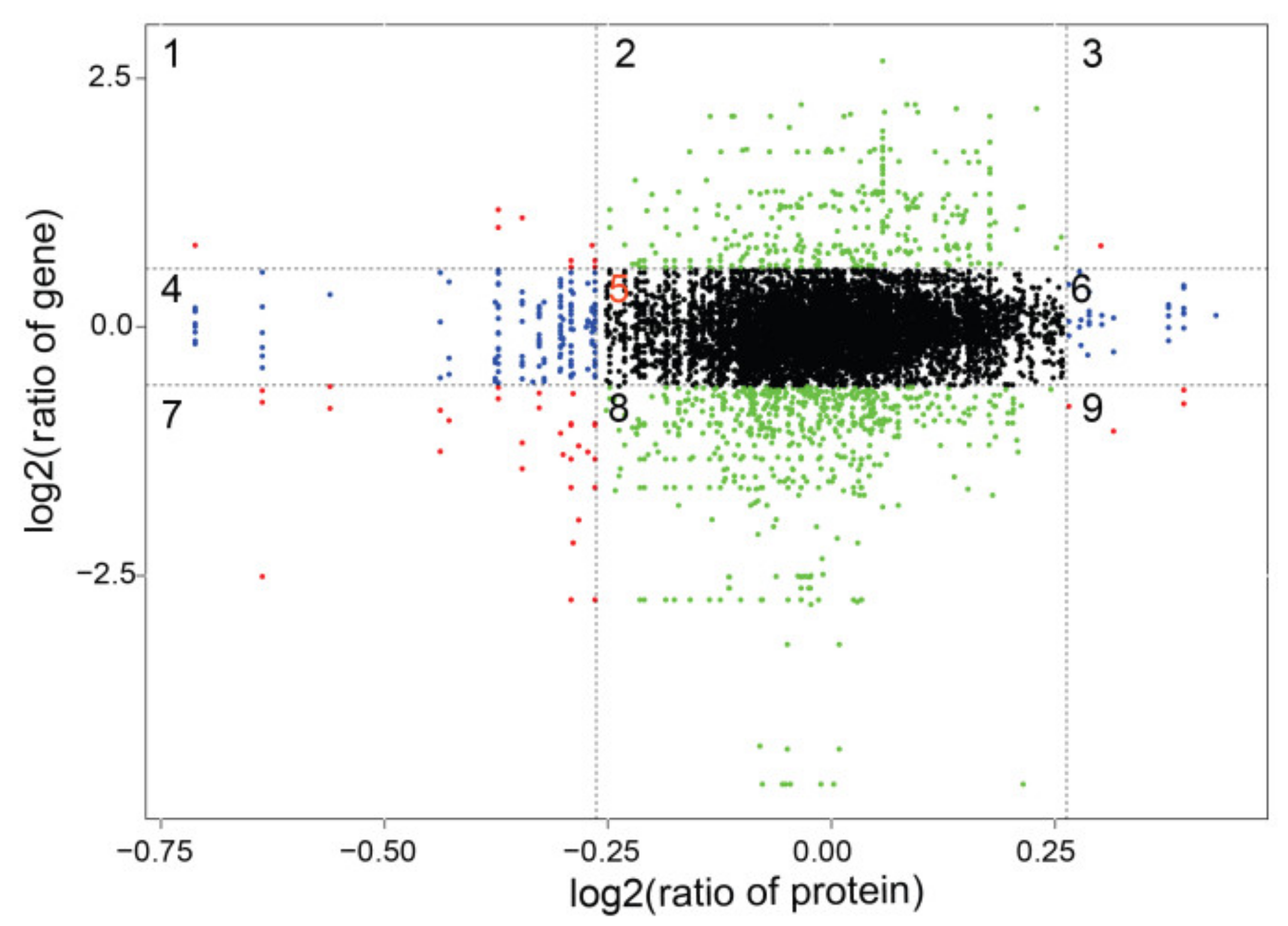
2.6. Association Analysis of the Transcriptome and Proteome
2.6.1. Correlation Analysis of the Transcriptome and Proteome
2.6.2. GO Association Analysis of Proteome and Transcriptome
2.6.3. KEGG Enrichment Analysis of the Proteome and Transcriptome
2.6.4. Associated Differentially Expressed Genes
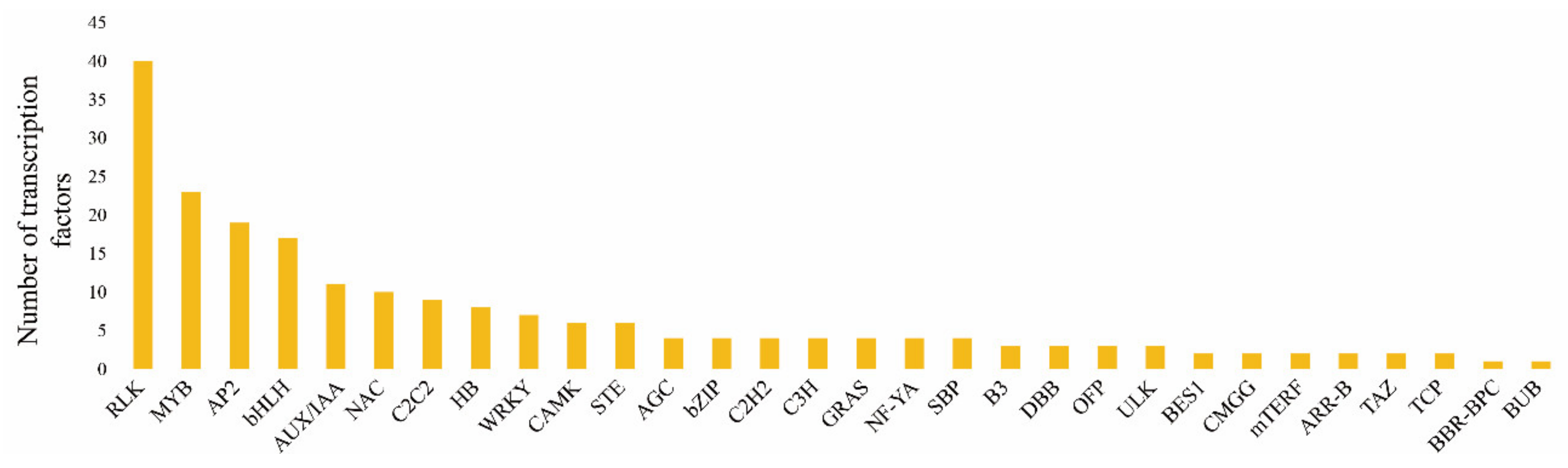
2.7. Transcription Factor Analysis of Cabbage Seedlings Treated with and without S
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Extraction and Identification of Glucosinolates
4.3. RNA Extraction and cDNA Library Construction
4.4. DEG Screening and Functional Analysis
4.5. Proteome Extraction and Profiling
4.6. Validation of DEGs Using qRT-PCR
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lewandowska, M.; Sirko, A. Recent advances in understanding plant response to sulfur-deficiency stress. Acta Biochim. Pol. 2008, 55, 457. [Google Scholar] [CrossRef] [Green Version]
- Kopycki, J.; Wieduwild, E.; Kohlschmidt, J.; Brandt, W.; Stepanova, A.N.; Alonso, J.M.; Pedras, M.S.C.; Abel, S.; Grubb, C.D. Kinetic analysis of Arabidopsis glucosyltransferase UGT74B1 illustrates a general mechanism by which enzymes can escape product inhibition. Biochem. J. 2013, 450, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Saito, K. Sulfur assimilatory metabolism. The long and smelling road. Plant Physiol. 2004, 136, 2443–2450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittstock, U.; Kliebenstein, D.J.; Lambrix, V.; Reichelt, M.; Gershenzon, J. Chapter five glucosinolate hydrolysis and its impact on generalist and specialist insect herbivores. Recent Adv. Phytochem. 2003, 37, 101–125. [Google Scholar]
- Rouzaud, G.; Rabot, S.; Ratcliffe, B.; Duncan, A.J. Influence of plant and bacterial myrosinase activity on the metabolic fate of glucosinolates in gnotobiotic rats. Br. J. Nutr. 2003, 90, 395–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, T.A.; Fahey, J.W.; Wade, K.L.; Stephenson, K.K.; Talalay, P. Chemoprotective glucosinolates and isothiocyanates of Broccoli sprouts. Cancer Epidemiol. Prev. Biomark. 2001, 10, 501–508. [Google Scholar]
- Hanschen, F.S.; Rohn, S.; Mewis, I.; Schreiner, M.; Kroh, L.W. Influence of the chemical structure on the thermal degradation of the glucosinolates in broccoli sprouts. Food Chem. 2012, 130, 1–8. [Google Scholar] [CrossRef]
- Fahey, J.W.; Zalcmann, A.T.; Talalay, P. The chemical diversity and distribution of glucosinolates and isothiocyanates among plants. Phytochemistry 2001, 56, 5–51. [Google Scholar] [CrossRef]
- Hesse, H. Molecular aspects of methionine biosynthesis. Trends Plant Sci. 2003, 8, 259–262. [Google Scholar] [CrossRef]
- Jiang, X.; He, H. Application of methionine and methyl jasmonate to affect glucosinolates of chinese kale. Acta Agric. Boreali-Sin. 2009, 24, 31–35. [Google Scholar]
- Hu, K.; Zhu, Z. Effects of IAA with different concentrations on glucosinolate contents in pakchoi. Agric. Res. Arid. Areas 2015, 33, 37–41. [Google Scholar]
- Knill, T.; Schuster, J.; Reichelt, M.; Gershenzon, J.; Binder, S. Arabidopsis branched-chain aminotransferase 3 functions in both amino acid and glucosinolate biosynthesis. Plant Physiol. 2008, 146, 1028–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostafa, I.; Zhu, N.; Yoo, M.-J.; Balmant, K.M.; Misra, B.B.; Dufresne, C.; Abou-Hashem, M.; Chen, S.; El-Domiaty, M. New nodes and edges in the glucosinolate molecular network revealed by proteomics and metabolomics of Arabidopsis myb28/29 and cyp79B2/B3 glucosinolate mutants. J. Proteom. 2016, 138, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Glawischnig, E.; Jørgensen, K.; Naur, P.; Jørgensen, B.; Olsen, C.E.; Hansen, C.H.; Rasmussen, H.; Pickett, J.A.; Halkier, B.A. CYP79F1 and CYP79F2 have distinct functions in the biosynthesis of aliphatic glucosinolates in Arabidopsis. Plant J. 2003, 33, 923–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Kaminaga, Y.; Cooper, B.; Pichersky, E.; Dudareva, N.; Chapple, C. Benzoylation and sinapoylation of glucosinolate R-groups in Arabidopsis. Plant J. 2012, 72, 411–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruyama-Nakashita, A. Metabolic changes sustain the plant life in low-sulfur environments. Curr. Opin. Plant Biol. 2017, 39, 144–151. [Google Scholar] [CrossRef]
- Schwanhäusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Correction: Corrigendum: Global quantification of mammalian gene expression control. Nat. Cell Biol. 2013, 495, 126–127. [Google Scholar] [CrossRef] [Green Version]
- Bones, A.; Rossiter, J. The enzymic and chemically induced decomposition of glucosinolates. Phytochemistry 2006, 67, 1053–1067. [Google Scholar] [CrossRef] [PubMed]
- Kopriva, S. Regulation of sulfate assimilation in Arabidopsis and beyond. Ann. Bot. 2006, 97, 479–495. [Google Scholar] [CrossRef] [Green Version]
- Mugford, S.G.; Lee, B.-R.; Koprivova, A.; Matthewman, C.; Kopriva, S. Control of sulfur partitioning between primary and secondary metabolism. Plant J. 2011, 65, 96–105. [Google Scholar] [CrossRef]
- Falk, K.L.; Tokuhisa, J.; Gershenzon, J. The effect of sulfur nutrition on plant glucosinolate content: Physiology and molecular mechanisms. Plant Biol. 2007, 9, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-J.; Matsuo, T.; Watanabe, M.; Watanabe, Y. Effect of nitrogen and sulphur application on the glucosinolate content in vegetable turnip rape (Brassica rapa L.). Soil Sci. Plant Nutr. 2002, 48, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Blake-Kalff, M.M.; Harrison, K.R.; Hawkesford, M.; Zhao, F.J.; McGrath, S.P. Distribution of sulfur within oilseed rape leaves in response to sulfur deficiency during vegetative growth. Plant Physiol. 1998, 118, 1337–1344. [Google Scholar] [CrossRef] [Green Version]
- Kushad, M.M.; Cloyd, R.; Babadoost, M. Distribution of glucosinolates in ornamental cabbage and kale cultivars. Sci. Hortic. 2004, 101, 215–221. [Google Scholar] [CrossRef]
- Zhai, Z.; Guo, S. The effects of amino acids on glucosinolates of cabbage. J. Plant Nutr. Fertil. 2009, 15, 447–452. [Google Scholar]
- Doughty, K.J.; Kiddle, G.A.; Pye, B.J. Selective induction of glucosinolates oilseed rape leaves by methyl jasmonate. Phytochemistry 1995, 38, 347–350. [Google Scholar] [CrossRef]
- Mewis, I.; Appel, H.M.; Hom, A.; Raina, R.; Schultz, J. Major signaling pathways modulate arabidopsis glucosinolate accumulation and response to both phloem-feeding and chewing insects. Plant Physiol. 2005, 138, 1149–1162. [Google Scholar] [CrossRef] [Green Version]
- Mikkelsen, M.D.; Petersen, B.L.; Olsen, C.E.; Halkier, B.A. Biosynthesis and metabolic engineering of glucosinolates. Amino Acids 2002, 22, 279–295. [Google Scholar] [CrossRef] [PubMed]
- Bano, A.; Khan, N. Nutritive values of Brassica campestris L. oil as affected by growth regulator treatments. J. Chem. Soc. Pak. 2009, 31, 819–822. [Google Scholar]
- Bender, J.; Celenza, J.L. Indolic glucosinolates at the crossroads of tryptophan metabolism. Phytochem. Rev. 2009, 8, 25–37. [Google Scholar] [CrossRef]
- Sønderby, I.E.; Geu-Flores, F.; Halkier, B. Biosynthesis of glucosinolates—Gene discovery and beyond. Trends Plant Sci. 2010, 15, 283–290. [Google Scholar] [CrossRef]
- Hansen, C.H.; Du, L.; Naur, P.; Olsen, C.E.; Axelsen, K.; Hick, A.J.; Pickett, J.A.; Halkier, B. CYP83B1 is the oxime-metabolizing enzyme in the glucosinolate pathway in arabidopsis. J. Biol. Chem. 2001, 276, 24790–24796. [Google Scholar] [CrossRef] [Green Version]
- Schlaeppi, K.; Bodenhausen, N.; Buchala, A.; Mauch, F.; Reymond, P. The glutathione-deficient mutant pad2-1 accumulates lower amounts of glucosinolates and is more susceptible to the insect herbivore Spodoptera littoralis. Plant J. 2008, 55, 774–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piotrowski, M.; Schemenewitz, A.; Lopukhina, A.; Müller, A.; Janowitz, T.; Weiler, E.W.; Oecking, C. Desulfoglucosinolate sulfotransferases from arabidopsis thaliana catalyze the final step in the biosynthesis of the glucosinolate core structure. J. Biol. Chem. 2004, 279, 50717–50725. [Google Scholar] [CrossRef] [Green Version]
- Grubb, C.D.; Zipp, B.J.; Ludwig-Müller, J.; Masuno, M.N.; Molinski, T.F.; Abel, S. Arabidopsis glucosyltransferase UGT74B1 functions in glucosinolate biosynthesis and auxin homeostasis. Plant J. 2004, 40, 893–908. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Liu, G. Glucosinolate profile and content analysis of different cabbage (Brassica oleracea L. var. capitata L.) varieties. China Veg. 2015, 6, 41–47. [Google Scholar]
- Hu, L.; Ye, M.; Kuai, P.; Ye, M.; Erb, M.; Lou, Y. OsLRR-RLK1, an early responsive leucine-rich repeat receptor-like kinase, initiates rice defense responses against a chewing herbivore. New Phytol. 2018, 219, 1097–1111. [Google Scholar] [CrossRef] [Green Version]
- Shiu, S.-H.; Bleecke, A.B. Plant receptor-like kinase gene family: Diversity, function, and signaling. STKE 2001, 113, re22. [Google Scholar] [CrossRef] [PubMed]
- Chu, S.; Wang, J.; Zhu, Y.; Liu, S.; Zhou, X.; Zhang, H.; Wang, C.-E.; Yang, W.; Tian, Z.; Cheng, H.; et al. An R2R3-type MYB transcription factor, GmMYB29, regulates isoflavone biosynthesis in soybean. PLoS Genet. 2017, 13, e1006770. [Google Scholar] [CrossRef]
- Zhou, M.; Memelink, J. Jasmonate-responsive transcription factors regulating plant secondary metabolism. Biotechnol. Adv. 2016, 34, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Patra, B.; Pattanaik, S.; Schluttenhofer, C.; Yuan, L. A network of jasmonate-responsive bHLH factors modulate monoterpenoid indole alkaloid biosynthesis in Catharanthus roseus. New Phytol. 2018, 217, 1566–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavy, M.; Estelle, M. Mechanisms of auxin signaling. Development 2016, 143, 3226–3229. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Yan, T.; Shen, Q.; Lu, X.; Pan, Q.; Huang, Y.; Tang, Y.; Fu, X.; Liu, M.; Jiang, W.; et al. Glandular trichome—Specific WRKY 1 promotes artemisinin biosynthesis in Artemisia annua. New Phytol. 2017, 214, 304–316. [Google Scholar] [CrossRef]
- Frerigmann, H.; Gigolashvili, T. MYB34, MYB51 and MYB122 distinctly regulate indolic glucosinolate biosynthesis in Arabidopsis thaliana. Mol. Plant 2014, 7, 814–828. [Google Scholar] [CrossRef] [Green Version]
- Gigolashvili, T.; Berger, B.; Flügge, U.-I. Specific and coordinated control of indolic and aliphatic glucosinolate biosynthesis by R2R3-MYB transcription factors in Arabidopsis thaliana. Phytochem. Rev. 2009, 8, 3–13. [Google Scholar] [CrossRef]
- Yin, L.; Chen, H.; Cao, B.; Lei, J.; Chen, G. Molecular characterization of MYB28 involved in aliphatic glucosinolate biosynthesis in Chinese kale (Brassica oleracea var. alboglabra Bailey). Front. Plant Sci. 2017, 8, 1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feller, A.; Hernandez, J.M.; Grotewold, E. An ACT-like domain participates in the dimerization of several plant basic-helix-loop-helix transcription factors. J. Biol. Chem. 2006, 281, 28964–28974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittstock, U.; Halkier, B.A. Glucosinolate research in the Arabidopsis era. Trends Plant Sci. 2002, 7, 263–270. [Google Scholar] [CrossRef]
- Butelli, E.; Titta, L.; Giorgio, M.; Mock, H.-P.; Matros, A.; Peterek, S.; Schijlen, E.G.W.M.; Hall, R.D.; Bovy, A.G.; Luo, J.; et al. Enrichment of tomato fruit with health-promoting anthocyanins by expression of select transcription factors. Nat. Biotechnol. 2008, 26, 1301–1308. [Google Scholar] [CrossRef]
- Schweizer, F.; Calvo, P.F.; Zander, M.; Diez-Diaz, M.; Fonseca, S.; Glauser, G.; Lewsey, M.; Ecker, J.; Solano, R.; Reymond, P. Arabidopsis basic helix-loop-helix transcription factors MYC2, MYC3, and MYC4 regulate glucosinolate biosynthesis, insect performance, and feeding behavior. Plant Cell 2013, 25, 3117–3132. [Google Scholar] [CrossRef] [Green Version]
- Schön, M.; Töller, A.; Diezel, C.; Roth, C.; Westphal, L.; Wiermer, M.; Somssich, I.E. Analyses of wrky18 wrky40 plants reveal critical roles of SA/EDS1 signaling and indole-glucosinolate biosynthesis for Golovinomyces orontii resistance and a loss-of resistance towards Pseudomonas syringae pv. tomato AvrRPS4. Mol. Plant-Microbe Interact. 2013, 26, 758–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Zhu, Z.; Gerendás, J. Interactive effects of phosphorus supply and light intensity on glucosinolates in pakchoi (Brassica campestris L. ssp. chinensis var. communis). Plant Soil 2009, 323, 323–333. [Google Scholar] [CrossRef]
- Zhao, C.; Zhou, X.; Qiu, J.; Xin, D.; Li, T.; Chu, X.; Yuan, H.; Wang, H.; Wang, Z.; Wang, D. Exosomes derived from bone marrow mesenchymal stem cells inhibit complement activation in rats with spinal cord injury. Drug Des. Dev. Ther. 2019, 13, 3693–3704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Shi, H.; Li, S.; Cao, Z.; Yang, H.; Wang, Y. Integrative hepatic metabolomics and proteomics reveal insights into the mechanism of different feed efficiency with high or low dietary forage levels in Holstein heifers. J. Proteom. 2018, 194, 1–13. [Google Scholar] [CrossRef]
- Zhang, B.; Chamba, Y.; Shang, P.; Wang, Z.; Ma, J.; Wang, L.; Zhang, H. Comparative transcriptomic and proteomic analyses provide insights into the key genes involved in high-altitude adaptation in the Tibetan pig. Sci. Rep. 2017, 7, 3654. [Google Scholar] [CrossRef] [Green Version]
- Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 383–385. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Alphatic Glucosinolates/μmol·g-1 (DW) | Indole Glucosinolates/μmol·g-1 (DW) | |||||
|---|---|---|---|---|---|---|---|
| Progoitrin | sinigrin | gluconapoleiferin | 4-Hydroxyglucobrassicin | Glucobrassicin | 4-Methoxyglucobrassicin | 1-Methoxyglucobrassicin | |
| +S | 1.25± 0.10 a | 6.17 ± 0.98 a | 0.28 ± 0.1 a | 3.79 ± 0.51 a | 9.01 ± 1.29 a | 1.37 ± 0.2 a | 1.82 ± 0.24 a |
| −S | 0.32 ± 0.03 b | 1.82 ± 0.67 b | 0 b | 1.35 ± 0.48 b | 2.5 2± 0.86 b | 0.22 ± 0.05 b | 0.33 ± 0.1 b |
| Treatment | Total Reads | Clean Reads | Q30 | Map Read | GC Content |
|---|---|---|---|---|---|
| +S | 42,384,400 | 2,119,200 | 94.26% | 32,970,027 (77.79%) | 47.67% |
| 58,336,300 | 29,168,150 | 94.37% | 45,400,252 (77.83%) | 47.96% | |
| 47,468,320 | 23,734,160 | 94.12% | 36,767,459 (77.46%) | 47.65% | |
| −S | 44,430,344 | 22,215,172 | 93.96% | 34,403,152 (77.43%) | 47.60% |
| 58,451,572 | 29,225,786 | 94.70% | 58,451,519 (77.86%) | 47.59% | |
| 40,308,466 | 20,154,233 | 94.83% | 31,383,916 (77.86%) | 47.62% |
| Protein Name | Regulation | Gene Name | Regulation | GO Annotation | KEGG Annotation |
|---|---|---|---|---|---|
| gene_Bol013541 | down | gene_Bol013541 | down | Molecular Function | K14709 |
| gene_Bol009168 | down | gene_Bol009168 | down | Molecular Function | K03152 |
| gene_Bol027549 | down | gene_Bol027549 | down | Molecular Function | K09580 |
| Bo_newGene_2279 | down | Bo_newGene_2279 | down | Molecular Function | - |
| gene_Bol028304 | down | gene_Bol028304 | down | Cellular Component | - |
| gene_Bol014966 | down | gene_Bol014966 | down | - | - |
| gene_Bol016517 | down | gene_Bol016517 | down | Molecular Function | K00799 |
| gene_Bol040974 | down | gene_Bol040974 | down | Molecular Function | K14709 |
| gene_Bol031335 | down | gene_Bol031335 | down | Molecular Function | K08245 |
| gene_Bol008941 | down | gene_Bol008941 | down | Biological Process | K01366 |
| gene_Bol016209 | down | gene_Bol016209 | down | Molecular Function | K12657 |
| gene_Bol039726 | down | gene_Bol039726 | down | Cellular Component | K13496 |
| Bo_newGene_299 | down | Bo_newGene_299 | down | - | - |
| gene_Bol028307 | down | gene_Bol028307 | down | Cellular Component | - |
| gene_Bol029650 | down | gene_Bol029650 | down | Biological Process | K01657 |
| gene_Bol029161 | down | gene_Bol029161 | down | Cellular Component | K10525 |
| gene_Bol041097 | down | gene_Bol041097 | down | Cellular Component | - |
| gene_Bol021589 | down | gene_Bol021589 | down | Molecular Function | - |
| gene_Bol025300 | down | gene_Bol025300 | down | Molecular Function | - |
| gene_Bol043996 | down | gene_Bol043996 | down | Molecular Function | - |
| gene_Bol022426 | down | gene_Bol022426 | down | Biological Process | K01057 |
| gene_Bol022099 | down | gene_Bol022099 | down | Cellular Component | - |
| gene_Bol028238 | down | gene_Bol028238 | down | Cellular Component | - |
| gene_Bol004625 | down | gene_Bol004625 | down | Molecular Function | K00799 |
| gene_Bol024581 | down | gene_Bol024581 | down | Cellular Component | K07407 |
| gene_Bol033864 | down | gene_Bol033864 | down | Molecular Function | - |
| gene_Bol025335 | down | gene_Bol025335 | down | - | - |
| gene_Bol036090 | down | gene_Bol036090 | down | Molecular Function | K00423 |
| gene_Bol038487 | down | gene_Bol038487 | down | Cellular Component | - |
| gene_Bol037300 | down | gene_Bol037300 | down | - | - |
| gene_Bol031446 | down | gene_Bol031446 | down | Molecular Function | - |
| gene_Bol024085 | down | gene_Bol024085 | up | Molecular Function | K00128 |
| gene_Bol042093 | up | gene_Bol042093 | up | Molecular Function | K09872 |
| gene_Bol011158 | up | gene_Bol011158 | up | Cellular Component | - |
| gene_Bol029420 | up | gene_Bol029420 | up | Molecular Function | K00008 |
| gene_Bol033052 | up | gene_Bol033052 | up | Cellular Component | - |
| gene_Bol040405 | up | gene_Bol040405 | up | Molecular Function | - |
| gene_Bol033653 | up | gene_Bol033653 | up | Molecular Function | K08235 |
| gene_Bol031534 | up | gene_Bol031534 | up | Cellular Component | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, L.; Zhang, H.; Chai, X.; Wei, S.; Luo, S.; Wang, H.; Lv, J.; Yu, J.; Liu, Z. Transcriptome and Proteome Conjoint Analysis Revealed That Exogenous Sulfur Regulates Glucosinolate Synthesis in Cabbage. Plants 2021, 10, 2104. https://doi.org/10.3390/plants10102104
Li L, Zhang H, Chai X, Wei S, Luo S, Wang H, Lv J, Yu J, Liu Z. Transcriptome and Proteome Conjoint Analysis Revealed That Exogenous Sulfur Regulates Glucosinolate Synthesis in Cabbage. Plants. 2021; 10(10):2104. https://doi.org/10.3390/plants10102104
Chicago/Turabian StyleLi, Lushan, Hui Zhang, Xiaohong Chai, Shouhui Wei, Shilei Luo, Huiping Wang, Jian Lv, Jihua Yu, and Zeci Liu. 2021. "Transcriptome and Proteome Conjoint Analysis Revealed That Exogenous Sulfur Regulates Glucosinolate Synthesis in Cabbage" Plants 10, no. 10: 2104. https://doi.org/10.3390/plants10102104
APA StyleLi, L., Zhang, H., Chai, X., Wei, S., Luo, S., Wang, H., Lv, J., Yu, J., & Liu, Z. (2021). Transcriptome and Proteome Conjoint Analysis Revealed That Exogenous Sulfur Regulates Glucosinolate Synthesis in Cabbage. Plants, 10(10), 2104. https://doi.org/10.3390/plants10102104