
Transcriptomic Analysis Reveals Candidate Genes Responding Maize Gray Leaf Spot Caused by Cercospora zeina

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
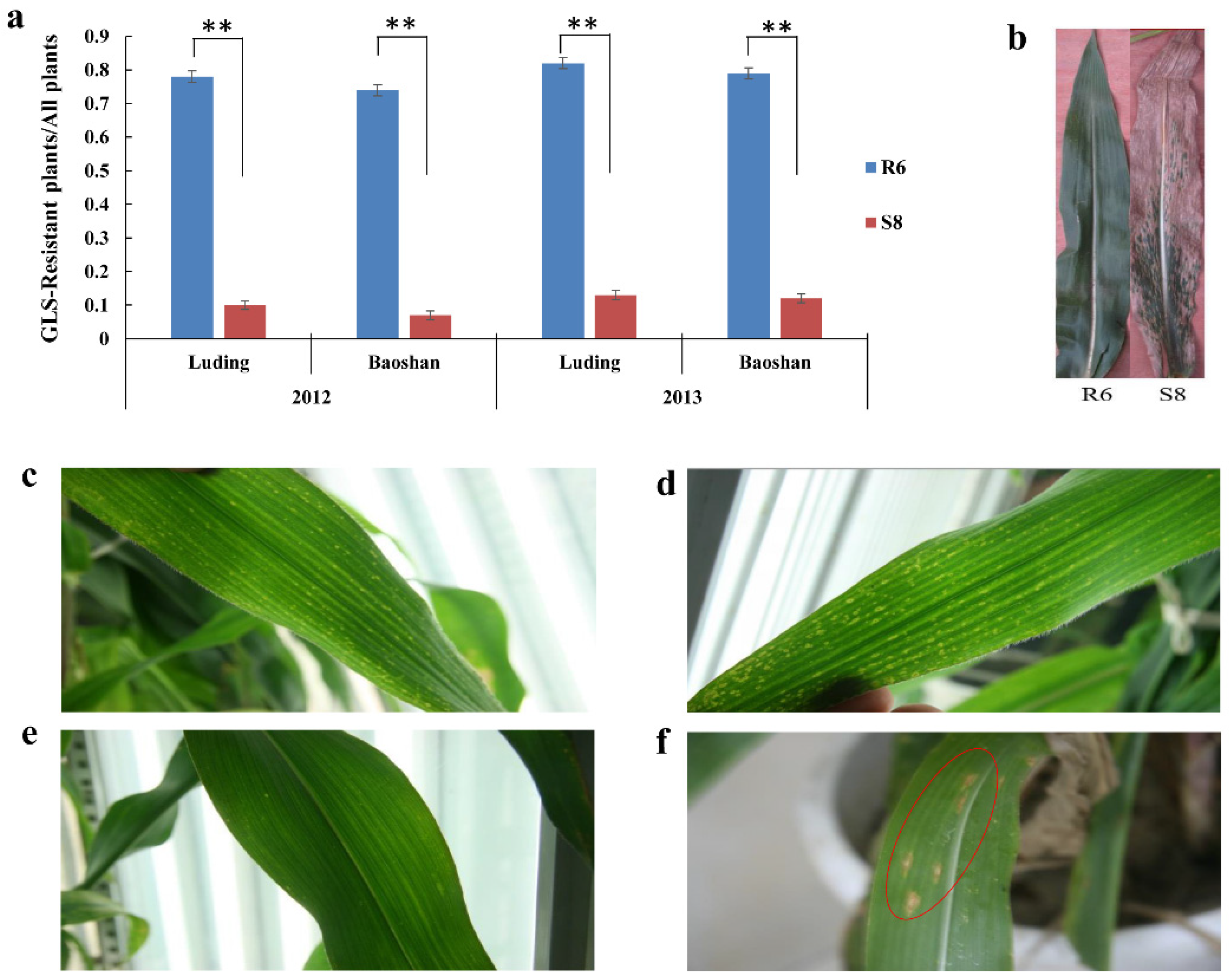
2.1. Phenotypic Evaluation in Response to C. zeina Infection
2.2. Dynamic Changes of Physiological Characteristics of Maize Leaves in Response to Inoculation C. zeina
2.3. Profiling the Maize Leaf Transcriptome Responses to C. zeina Infection
2.4. Real-Time Quantitative Reverse Transcription-PCR (qRT-PCR) Validation
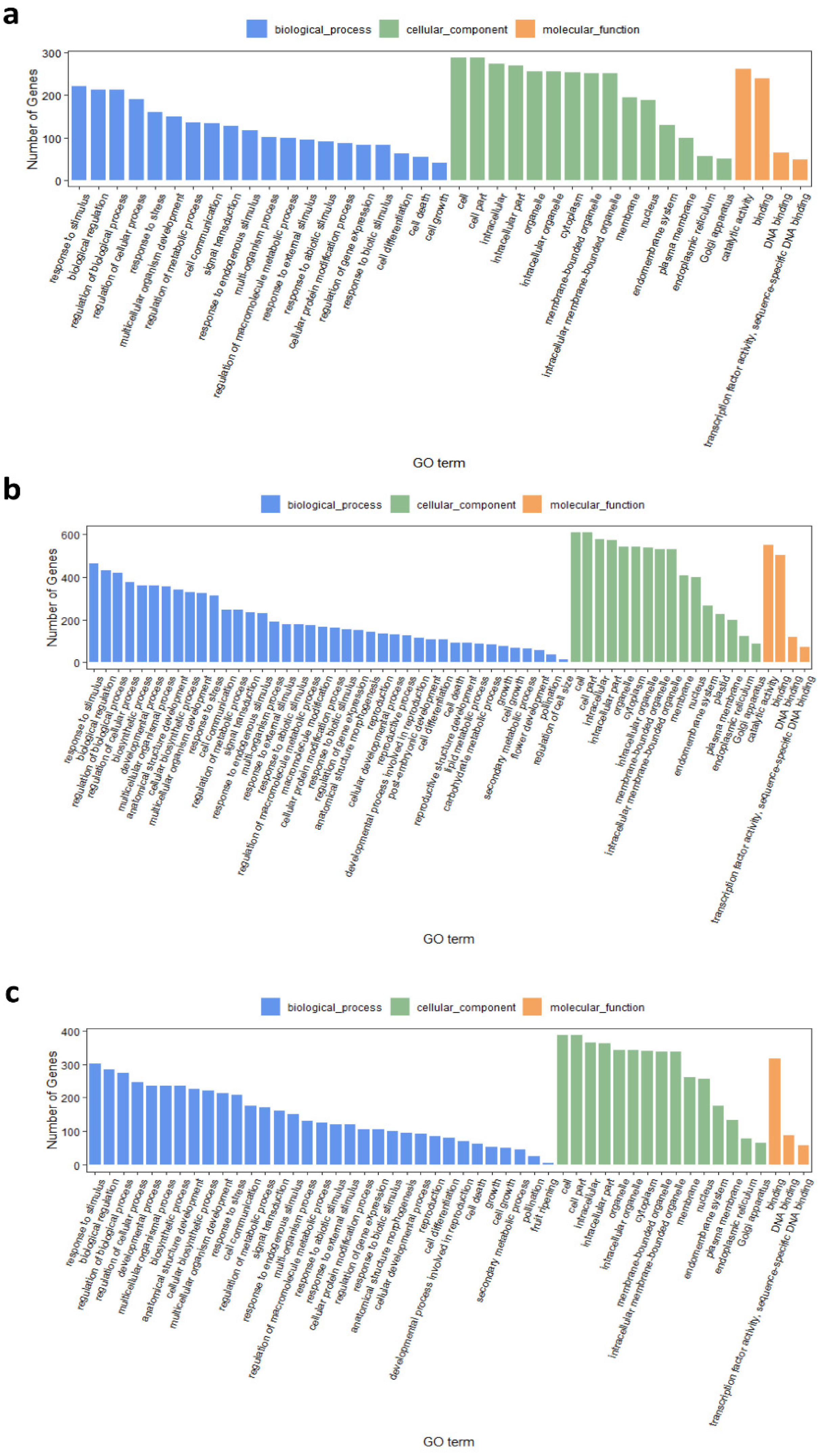
2.5. Gene Ontology and Pathway Enrichment Analyses of DEGs between R6 and S8
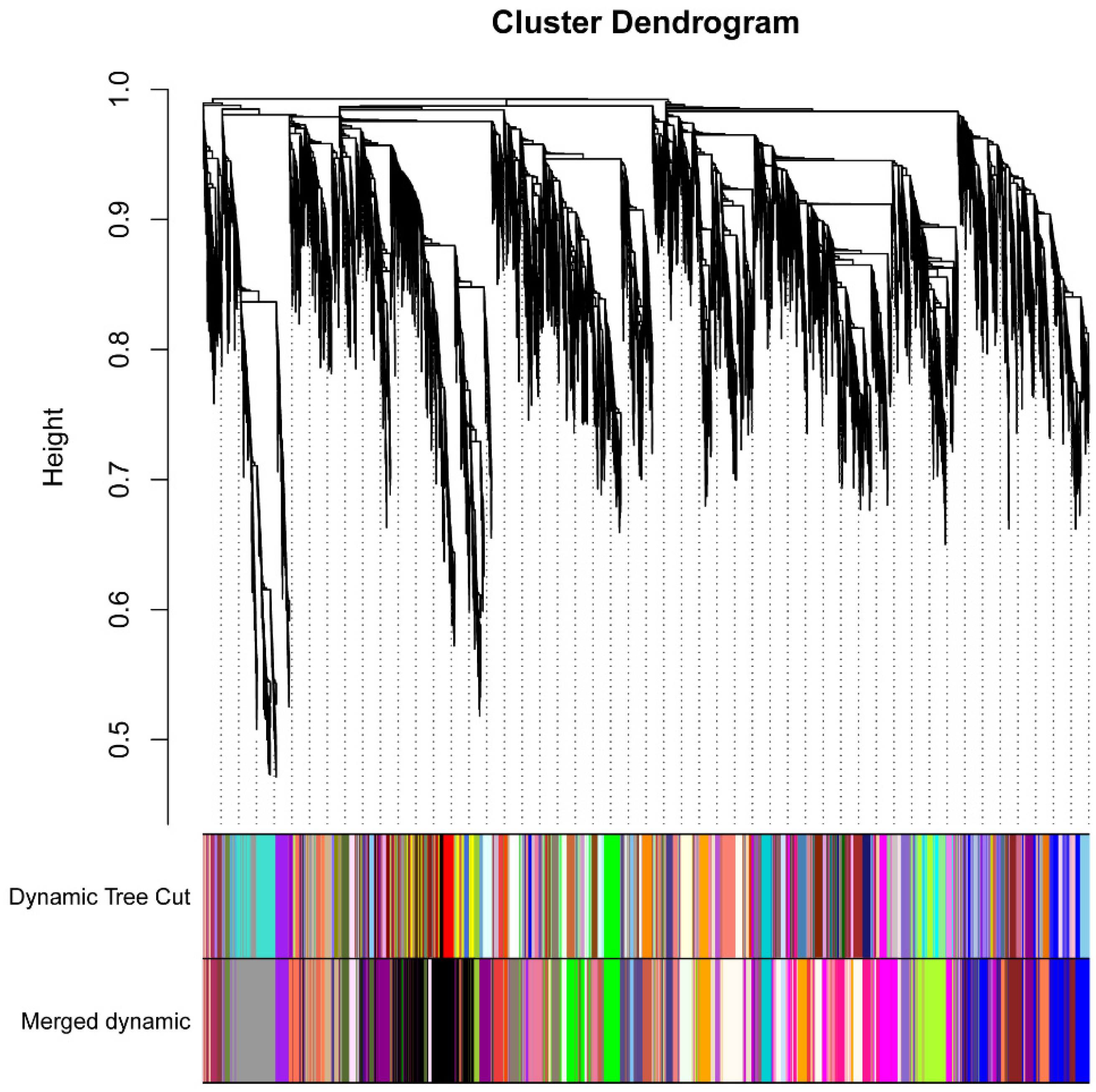
2.6. Co-Expression Network of Genes Related to C. zeina Resistance
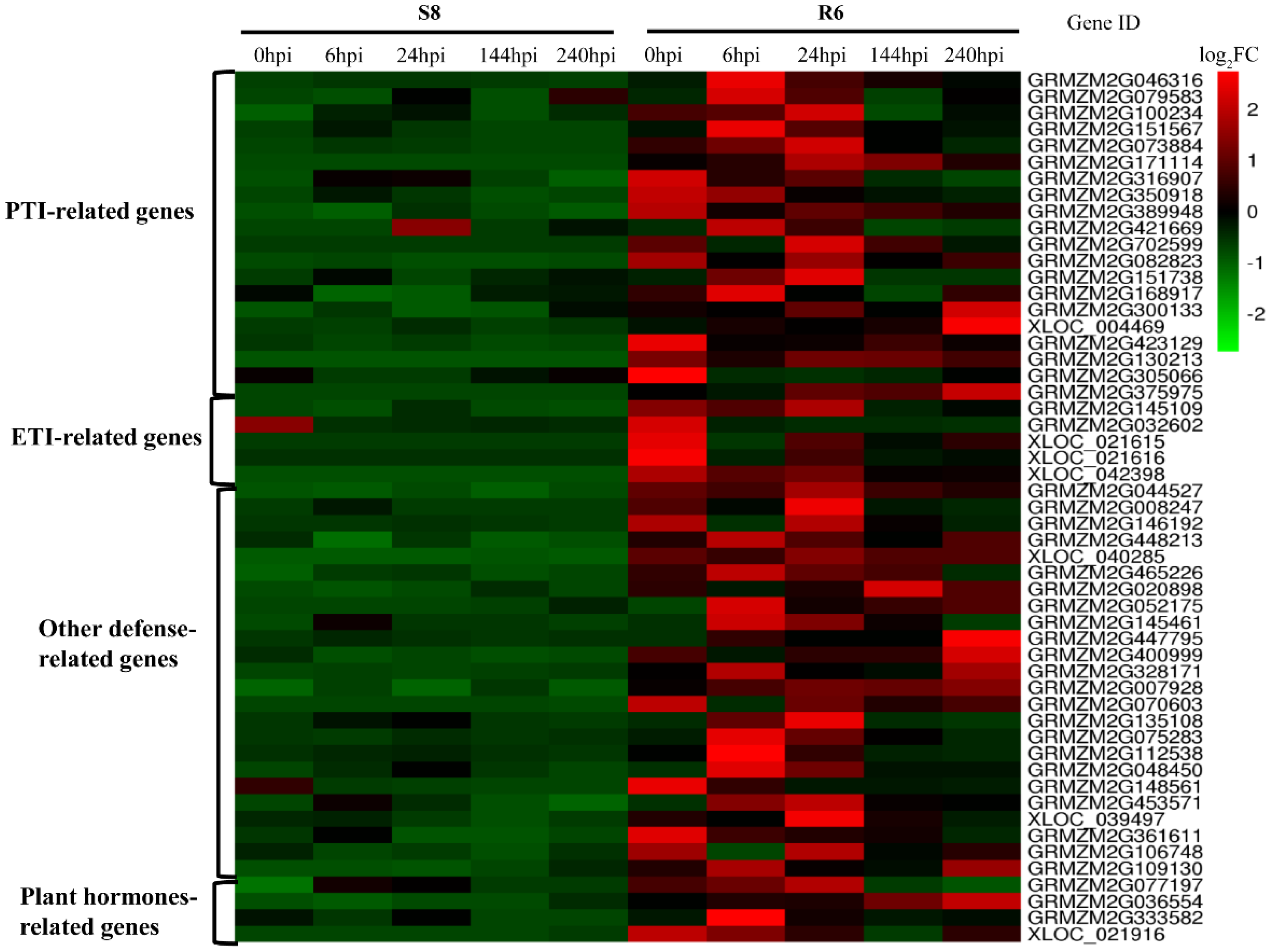
2.7. Identified Resistance-Related Genes Response to C. zeina from DEGs
2.8. Integration of QTL Mapping and DEGs to Find the Candidate Genes’ Response to C. zeina
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Disease Resistance Scoring
4.2. The C. zeina Strain Preparation and Inoculation
4.3. Antioxidant Enzymes Activity and Proline Assay
4.4. Samples Collection and RNA Isolation
4.5. RNA Sequencing and Data Analysis
4.6. Validation of RNA-Seq Results by qRT-PCR
4.7. WGCNA Analysis of RNA-Seq Data from Multiple Timepoints
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tehon, L.R.; Daniels, E. Notes on the Parasitic Fungi of Illinois—II. Mycologia 1925, 17, 240–249. [Google Scholar]
- Payne, G.; Duncan, H.; Adkins, C. Influence of tillage on development of gray leaf spot and number of airborne conidia of Cercospora zeae-maydis. Plant Dis. 1987, 71, 329–332. [Google Scholar] [CrossRef]
- De Nazareno, N.; Lipps, P.; Madden, L. Effect of levels of corn residue on the epidemiology of gray leaf spot of corn in Ohio. Plant Dis. 1993, 77, 67–70. [Google Scholar] [CrossRef]
- Elwinger, G.F.; Johnson, M.W.; Hill, R.R., Jr.; Ayers, J.E. Inheritance of Resistance to Gray Leaf Spot of Corn. Crop Sci. 1990, 30, 350–358. [Google Scholar] [CrossRef]
- Donahue, P.J.; Stromberg, E.L.; Myers, S.L. Inheritance of Reaction to Gray Leaf Spot in a Diallel Cross of 14 Maize Inbreds. Crop Sci. 1991, 31, 926–931. [Google Scholar] [CrossRef]
- Shi, L.; Lv, X.; Weng, J.; Zhu, H.; Liu, C.; Hao, Z.; Zhou, Y.; Zhang, D.; Li, M.; Ci, X.; et al. Genetic characterization and linkage disequilibrium mapping of resistance to gray leaf spot in maize (Zea mays L.). Crop J. 2014, 2, 132–143. [Google Scholar] [CrossRef] [Green Version]
- Latterell, F.M.; Rossi, A.E. Gray leaf spot of corn: A disease on the move. Plant Dis. 1983, 67, 842–847. [Google Scholar] [CrossRef]
- Lehmensiek, A.; Esterhuizen, A.M.; van Staden, D.; Nelson, S.W.; Retief, A.E. Genetic mapping of gray leaf spot (GLS) resistance genes in maize. Theor. Appl. Genet. 2001, 103, 797–803. [Google Scholar] [CrossRef]
- Ward, J.M.J.; Stromberg, E.L.; Nowell, D.C.; Nutter, F.W. Gray leaf Spot: A Disease of Global Importance in Maize Production. Plant Dis. 1999, 83, 884–895. [Google Scholar] [CrossRef] [Green Version]
- Berger, D.K.; Carstens, M.; Korsman, J.N.; Middleton, F.; Kloppers, F.J.; Tongoona, P.; Myburg, A.A. Mapping QTL conferring resistance in maize to gray leaf spot disease caused by Cercospora zeina. BMC Genet. 2014, 15, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Benson, J.M.; Poland, J.A.; Benson, B.M.; Stromberg, E.L.; Nelson, R.J. Resistance to Gray Leaf Spot of Maize: Genetic Architecture and Mechanisms Elucidated through Nested Association Mapping and Near-Isogenic Line Analysis. PLoS Genet. 2015, 11, e1005045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mammadov, J.; Sun, X.; Gao, Y.; Ochsenfeld, C.; Bakker, E.; Ren, R.; Flora, J.; Wang, X.; Kumpatla, S.; Meyer, D.; et al. Combining powers of linkage and association mapping for precise dissection of QTL controlling resistance to gray leaf spot disease in maize (Zea mays L.). BMC Genom. 2015, 16, 916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuki, M.C.; Scapim, C.A.; Rossi, E.S.; Mangolin, C.A.; Amaral Júnior, A.T.d.; Pinto, R.J.B. Genome wide association study for gray leaf spot resistance in tropical maize core. PLoS ONE 2018, 13, e0199539. [Google Scholar] [CrossRef] [PubMed]
- Juliatti, F.C.; Pedrosa, M.G.; Silva, H.D.; da Silva, J.V.C. Genetic mapping for resistance to gray leaf spot in maize. Euphytica 2009, 169, 227–238. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, L.; Fan, X.; Tan, J.; Chen, W.; Xu, M. QTL mapping of resistance to gray leaf spot in maize. Theor. Appl. Genet. 2012, 125, 1797–1808. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, Y.D.; Li, H.Y.; Bi, Y.Q.; Yu, L.J.; Fan, X.M.; Tan, J.; Jeffers, D.P.; Kang, M.S. QTL Mapping for Gray Leaf Spot Resistance in a Tropical Maize Population. Plant Dis. 2015, 100, 304–312. [Google Scholar] [CrossRef] [Green Version]
- Welgemoed, T.; Pierneef, R.; Sterck, L.; Van de Peer, Y.; Swart, V.; Scheepers, K.D.; Berger, D.K. De novo Assembly of Transcriptomes From a B73 Maize Line Introgressed With a QTL for Resistance to Gray Leaf Spot Disease Reveals a Candidate Allele of a Lectin Receptor-Like Kinase. Front. Plant Sci. 2020, 11, 191. [Google Scholar] [CrossRef] [PubMed]
- Kibe, M.; Nair, S.K.; Das, B.; Bright, J.M.; Makumbi, D.; Kinyua, J.; Suresh, L.M.; Beyene, Y.; Olsen, M.S.; Prasanna, B.M.; et al. Genetic Dissection of Resistance to Gray Leaf Spot by Combining Genome-Wide Association, Linkage Mapping, and Genomic Prediction in Tropical Maize Germplasm. Front. Plant Sci. 2020, 11, 1602. [Google Scholar] [CrossRef]
- Du, L.; Yu, F.; Zhang, H.; Wang, B.; Ma, K.; Yu, C.; Xin, W.; Huang, X.; Liu, Y.; Liu, K. Genetic mapping of quantitative trait loci and a major locus for resistance to grey leaf spot in maize. Theor. Appl. Genet. 2020, 133, 2521–2533. [Google Scholar] [CrossRef]
- Chen, L.; Liu, L.; Li, Z.; Zhang, Y.; Kang, M.S.; Wang, Y.; Fan, X. High-density mapping for gray leaf spot resistance using two related tropical maize recombinant inbred line populations. Mol. Biol. Rep. 2021, 48, 3379–3392. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, Y.; Shao, S.; Chen, W.; Tan, J.; Zhu, M.; Zhong, T.; Fan, X.; Xu, M. High-resolution mapping and characterization of qRgls2, a major quantitative trait locus involved in maize resistance to gray leaf spot. BMC Plant Biol. 2014, 14, 230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Yang, Q.; Rucker, E.; Thomason, W.; Balint-Kurti, P. Fine mapping of a quantitative resistance gene for gray leaf spot of maize (Zea mays L.) derived from teosinte (Z. mays ssp. parviglumis). Theor. Appl. Genet. 2017, 130, 1285–1295. [Google Scholar] [CrossRef]
- Lv, X.; Song, M.; Cheng, Z.; Yang, X.; Zhang, X.; Zhou, Z.; Zhang, C.; Zheng, L.; Li, Y.; Lei, K.; et al. qGLS1.02, a novel major locus for resistance to gray leaf spot in maize. Mol. Breed. 2020, 40, 59. [Google Scholar] [CrossRef]
- Sun, H.; Zhai, L.; Teng, F.; Li, Z.; Zhang, Z. qRgls1.06, a major QTL conferring resistance to gray leaf spot disease in maize. Crop J. 2021, 9, 342–350. [Google Scholar] [CrossRef]
- Dodds, P.N.; Rathjen, J.P. Plant immunity: Towards an integrated view of plant–pathogen interactions. Nat. Rev. Genet. 2010, 11, 539–548. [Google Scholar] [CrossRef]
- Mauch-Mani, B.; Slusarenko, A.J. Production of Salicylic Acid Precursors Is a Major Function of Phenylalanine Ammonia-Lyase in the Resistance of Arabidopsis to Peronospora parasitica. Plant Cell 1996, 8, 203–212. [Google Scholar] [CrossRef]
- Vogt, T. Phenylpropanoid Biosynthesis. Mol. Plant 2010, 3, 2–20. [Google Scholar] [CrossRef] [Green Version]
- Zhai, K.; Deng, Y.; Liang, D.; Tang, J.; Liu, J.; Yan, B.; Yin, X.; Lin, H.; Chen, F.; Yang, D.; et al. RRM Transcription Factors Interact with NLRs and Regulate Broad-Spectrum Blast Resistance in Rice. Mol. Cell 2019, 74, 996–1009.e7. [Google Scholar] [CrossRef]
- Zhou, X.; Liao, H.; Chern, M.; Yin, J.; Chen, Y.; Wang, J.; Zhu, X.; Chen, Z.; Yuan, C.; Zhao, W.; et al. Loss of function of a rice TPR-domain RNA-binding protein confers broad-spectrum disease resistance. Proc. Natl. Acad. Sci. USA 2018, 115, 3174. [Google Scholar] [CrossRef] [Green Version]
- Apel, K.; Hirt, H. REACTIVE OXYGEN SPECIES: Metabolism, Oxidative Stress, and Signal Transduction. Annu. Rev. Plant Biol. 2004, 55, 373–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittler, R.; Vanderauwera, S.; Suzuki, N.; Miller, G.; Tognetti, V.B.; Vandepoele, K.; Gollery, M.; Shulaev, V.; Van Breusegem, F. ROS signaling: The new wave? Trends Plant Sci. 2011, 16, 300–309. [Google Scholar] [CrossRef]
- Zhang, J.; Sun, X. Recent advances in polyphenol oxidase-mediated plant stress responses. Phytochemistry 2021, 181, 112588. [Google Scholar] [CrossRef]
- Kaur, G.; Asthir, B. Proline: A key player in plant abiotic stress tolerance. Biol. Plant. 2015, 59, 609–619. [Google Scholar] [CrossRef]
- Kim, K.H.; Kang, Y.J.; Kim, D.H.; Yoon, M.Y.; Moon, J.-K.; Kim, M.Y.; Van, K.; Lee, S.-H. RNA-Seq Analysis of a Soybean Near-Isogenic Line Carrying Bacterial Leaf Pustule-Resistant and -Susceptible Alleles. DNA Res. 2011, 18, 483–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagnaresi, P.; Biselli, C.; Orrù, L.; Urso, S.; Crispino, L.; Abbruscato, P.; Piffanelli, P.; Lupotto, E.; Cattivelli, L.; Valè, G. Comparative Transcriptome Profiling of the Early Response to Magnaporthe oryzae in Durable Resistant vs Susceptible Rice (Oryza sativa L.) Genotypes. PLoS ONE 2012, 7, e51609. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Guo, Y.; Ma, C.; Zhang, D.; Wang, C.; Yang, Q. Transcriptome analysis of maize resistance to Fusarium graminearum. BMC Genom. 2016, 17, 477. [Google Scholar]
- Wu, X.-j.; Xu, L.; Zhao, P.-f.; Li, N.; Wu, L.; He, Y.; Wang, S.-c. Comparative transcriptome profiling of two maize near-isogenic lines differing in the allelic state for bacterial brown spot disease resistance. J. Integr. Agric. 2015, 14, 610–621. [Google Scholar] [CrossRef]
- Meyer, J.; Berger, D.K.; Christensen, S.A.; Murray, S.L. RNA-Seq analysis of resistant and susceptible sub-tropical maize lines reveals a role for kauralexins in resistance to grey leaf spot disease, caused by Cercospora zeina. BMC Plant Biol. 2017, 17, 197. [Google Scholar] [CrossRef] [Green Version]
- Kebede, A.Z.; Johnston, A.; Schneiderman, D.; Bosnich, W.; Harris, L.J. Transcriptome profiling of two maize inbreds with distinct responses to Gibberella ear rot disease to identify candidate resistance genes. BMC Genom. 2018, 19, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Shi, J.; Li, X.; Liu, J.; Geng, Q.; Shi, H.; Ke, Y.; Sun, Q. Transcriptome analysis reveals the molecular mechanisms of the defense response to gray leaf spot disease in maize. BMC Genom. 2018, 19, 742. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Liu, S.; Zhao, Y.; Wang, W.; Kong, Z.; Tang, D. ENHANCED DISEASE RESISTANCE4 Associates with CLATHRIN HEAVY CHAIN2 and Modulates Plant Immunity by Regulating Relocation of EDR1 in Arabidopsis. Plant Cell 2015, 27, 857–873. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Grubb, L.E.; Wang, J.; Liang, X.; Li, L.; Gao, C.; Ma, M.; Feng, F.; Li, M.; Li, L.; et al. A Regulatory Module Controlling Homeostasis of a Plant Immune Kinase. Mol. Cell 2018, 69, 493–504.e6. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.-J.D.; Liebrand, T.W.H.; Yadeta, K.A.; Coaker, G. PBL13 Is a Serine/Threonine Protein Kinase That Negatively Regulates Arabidopsis Immune Responses. Plant Physiol. 2015, 169, 2950–2962. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wen, J.; Lease, K.A.; Doke, J.T.; Tax, F.E.; Walker, J.C. BAK1, an Arabidopsis LRR Receptor-like Protein Kinase, Interacts with BRI1 and Modulates Brassinosteroid Signaling. Cell 2002, 110, 213–222. [Google Scholar] [CrossRef] [Green Version]
- Roux, M.; Schwessinger, B.; Albrecht, C.; Chinchilla, D.; Jones, A.; Holton, N.; Malinovsky, F.G.; Tör, M.; de Vries, S.; Zipfel, C. The Arabidopsis Leucine-Rich Repeat Receptor–Like Kinases BAK1/SERK3 and BKK1/SERK4 Are Required for Innate Immunity to Hemibiotrophic and Biotrophic Pathogens. Plant Cell 2011, 23, 2440–2455. [Google Scholar] [CrossRef] [Green Version]
- Heitz, T.; Widemann, E.; Lugan, R.; Miesch, L.; Ullmann, P.; Désaubry, L.; Holder, E.; Grausem, B.; Kandel, S.; Miesch, M.; et al. Cytochromes P450 CYP94C1 and CYP94B3 Catalyze Two Successive Oxidation Steps of Plant Hormone Jasmonoyl-isoleucine for Catabolic Turnover. J. Biol. Chem. 2012, 287, 6296–6306. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Yu, Y.; Liu, Z.; Li, S.; Wang, Z.; Xiang, F. Membrane-bound NAC transcription factors in maize and their contribution to the oxidative stress response. Plant Sci. 2016, 250, 30–39. [Google Scholar] [CrossRef]
- Li, G.; Zhou, J.; Jia, H.; Gao, Z.; Fan, M.; Luo, Y.; Zhao, P.; Xue, S.; Li, N.; Yuan, Y.; et al. Mutation of a histidine-rich calcium-binding-protein gene in wheat confers resistance to Fusarium head blight. Nat. Genet. 2019, 51, 1106–1112. [Google Scholar] [CrossRef]
- He, W.; Yang, L.; Leng, Y.; Zhang, B.; Yang, J.; Li, L.; Chen, Y.; Kang, J.; Tang, H.; Deng, L.; et al. QTL mapping for resistance of maize to grey leaf spot. J. Phytopathol. 2018, 166, 167–176. [Google Scholar] [CrossRef]
- Meng, X.; Zhang, S. MAPK Cascades in Plant Disease Resistance Signaling. Annu. Rev. Phytopathol. 2013, 51, 245–266. [Google Scholar] [CrossRef]
- Shigenaga, A.M.; Berens, M.L.; Tsuda, K.; Argueso, C.T. Towards engineering of hormonal crosstalk in plant immunity. Curr. Opin. Plant Biol. 2017, 38, 164–172. [Google Scholar] [CrossRef]
- Ahmad, S.; Veyrat, N.; Gordon-Weeks, R.; Zhang, Y.; Martin, J.; Smart, L.; Glauser, G.; Erb, M.; Flors, V.; Frey, M.; et al. Benzoxazinoid Metabolites Regulate Innate Immunity against Aphids and Fungi in Maize. Plant Physiol. 2011, 157, 317–327. [Google Scholar] [CrossRef] [Green Version]
- Dong, X. SA, JA, ethylene, and disease resistance in plants. Curr. Opin. Plant Biol. 1998, 1, 316–323. [Google Scholar] [CrossRef]
- Jones, J.D.G.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlot, A.C.; Dempsey, D.M.A.; Klessig, D.F. Salicylic Acid, a Multifaceted Hormone to Combat Disease. Annu. Rev. Phytopathol. 2009, 47, 177–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanseverino, W.; Roma, G.; De Simone, M.; Faino, L.; Melito, S.; Stupka, E.; Frusciante, L.; Ercolano, M.R. PRGdb: A bioinformatics platform for plant resistance gene analysis. Nucleic Acids Res. 2009, 38 (Suppl. 1), D814–D821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehmany, A.P.; Gordon, A.; Rose, L.E.; Allen, R.L.; Armstrong, M.R.; Whisson, S.C.; Kamoun, S.; Tyler, B.M.; Birch, P.R.J.; Beynon, J.L. Differential Recognition of Highly Divergent Downy Mildew Avirulence Gene Alleles by RPP1 Resistance Genes from Two Arabidopsis Lines. Plant Cell 2005, 17, 1839–1850. [Google Scholar] [CrossRef] [Green Version]
- Mackey, D.; Holt, B.F.; Wiig, A.; Dangl, J.L. RIN4 Interacts with Pseudomonas syringae Type III Effector Molecules and Is Required for RPM1-Mediated Resistance in Arabidopsis. Cell 2002, 108, 743–754. [Google Scholar] [CrossRef] [Green Version]
- Serrano, M.; Hubert, D.A.; Dangl, J.L.; Schulze-Lefert, P.; Kombrink, E. A chemical screen for suppressors of the avrRpm1-RPM1-dependent hypersensitive cell death response in Arabidopsis thaliana. Planta 2010, 231, 1013–1023. [Google Scholar] [CrossRef] [Green Version]
- Eulgem, T.; Somssich, I.E. Networks of WRKY transcription factors in defense signaling. Curr. Opin. Plant Biol. 2007, 10, 366–371. [Google Scholar] [CrossRef] [Green Version]
- Casey, R.; Hughes, R.K. Recombinant Lipoxygenases and Oxylipin Metabolism in Relation to Food Quality. Food Biotechnol. 2004, 18, 135–170. [Google Scholar] [CrossRef]
- Solano, R.; Gimenez-Ibanez, S. Nuclear jasmonate and salicylate signaling and crosstalk in defense against pathogens. Front. Plant Sci. 2013, 4, 72. [Google Scholar]
- Maroof, M.S.; Van Scoyoc, S.; Yu, Y.; Stromberg, E. Gray leaf spot disease of maize: Rating methodology and inbred line evaluation. Plant Dis. 1993, 77, 583–587. [Google Scholar] [CrossRef]
- Wang, Z.; Li, J.Y.; Jia, C.H.; Li, J.P.; Xu, B.Y.; Jin, Z.Q. Molecular cloning and expression of four phenylalanine ammonia lyase genes from banana interacting with Fusarium oxysporum. Biol. Plant. 2016, 60, 459–468. [Google Scholar] [CrossRef]
- Beers, R.F.; Sizer, I.W. A spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. J. Biol. Chem. 1952, 195, 133–140. [Google Scholar] [CrossRef]
- Upadhyaya, A.; Sankhla, D.; Davis, T.D.; Sankhla, N.; Smith, B.N. Effect of Paclobutrazol on the Activities of some Enzymes of Activated Oxygen Metabolism and Lipid Peroxidation in Senescing Soybean Leaves. J. Plant Physiol. 1985, 121, 453–461. [Google Scholar] [CrossRef]
- Bates, L.S.; Waldren, R.P.; Teare, I.D. Rapid determination of free proline for water-stress studies. Plant Soil 1973, 39, 205–207. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, W.; Zhu, Y.; Leng, Y.; Yang, L.; Zhang, B.; Yang, J.; Zhang, X.; Lan, H.; Tang, H.; Chen, J.; et al. Transcriptomic Analysis Reveals Candidate Genes Responding Maize Gray Leaf Spot Caused by Cercospora zeina. Plants 2021, 10, 2257. https://doi.org/10.3390/plants10112257
He W, Zhu Y, Leng Y, Yang L, Zhang B, Yang J, Zhang X, Lan H, Tang H, Chen J, et al. Transcriptomic Analysis Reveals Candidate Genes Responding Maize Gray Leaf Spot Caused by Cercospora zeina. Plants. 2021; 10(11):2257. https://doi.org/10.3390/plants10112257
Chicago/Turabian StyleHe, Wenzhu, Yonghui Zhu, Yifeng Leng, Lin Yang, Biao Zhang, Junpin Yang, Xiao Zhang, Hai Lan, Haitao Tang, Jie Chen, and et al. 2021. "Transcriptomic Analysis Reveals Candidate Genes Responding Maize Gray Leaf Spot Caused by Cercospora zeina" Plants 10, no. 11: 2257. https://doi.org/10.3390/plants10112257