
Grapevine Virology in the Third-Generation Sequencing Era: From Virus Detection to Viral Epitranscriptomics

 ,
,  and
and 
Abstract
:1. Introduction
2. Third-Generation Sequencing Platforms
3. The Application of Nanopore Sequencing to Plant Virus Detection
4. Detection of Grapevine RNA Viruses by Nanopore Direct cDNA and RNA Sequencing
5. Detection of Grapevine DNA Viruses by Nanopore Ligation and Rapid Sequencing
6. Adaptive Nanopore Sequencing for Real-Time Virus Detection
7. Viroids and Virusoids
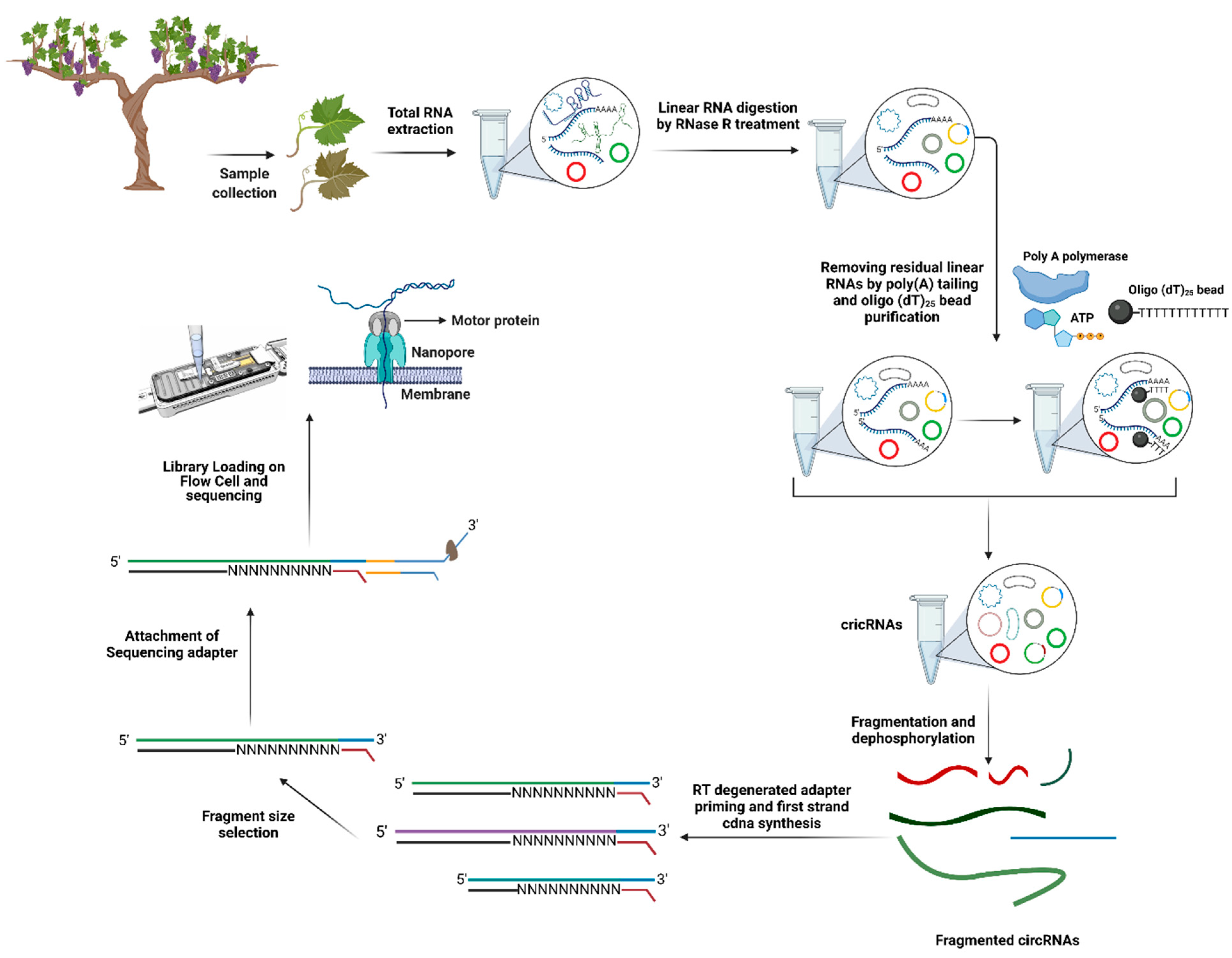
8. Identification of Long Non-Coding RNAs (lncRNAs) and Circular RNAs (circRNAs)
9. Detection of Viral RNA Modifications by Nanopore Sequencing
10. Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Statistical Report on World Vitiviniculture; International Organization of Vine and Wine: Paris, France, 2019.
- Alston, J.M.; Sambucci, O. Grapes in the world economy. In The Grape Genome; Cantu, D., Walker, M.A., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 1–24. [Google Scholar]
- Robinson, J.; Harding, J.; Vouillamoz, J. Wine Grapes: A Complete Guide to 1368 Vine Varieties, Including Their Origins and Flavours; Penguin UK: London, UK, 2013. [Google Scholar]
- Sastry, K.S.; Zitter, T.A. Management of virus and viroid diseases of crops in the tropics. In Plant Virus and Viroid Diseases in the Tropics: Volume 2: Epidemiology and Management; Sastry, K.S., Zitter, T.A., Eds.; Springer: Dordrecht, The Netherlands, 2014; pp. 149–480. [Google Scholar]
- Ricketts, K.D.; Gomez, M.I.; Atallah, S.S.; Fuchs, M.F.; Martinson, T.E.; Battany, M.C.; Bettiga, L.J.; Cooper, M.L.; Verdegaal, P.S.; Smith, R.J. Reducing the economic impact of grapevine leafroll disease in california: Identifying optimal disease management strategies. Am. J. Enol. Vitic. 2015, 66, 138–147. [Google Scholar] [CrossRef]
- Martelli, G.P. Directory of virus and virus-like diseases of the grapevine and their agents. J. Plant Pathol. 2014, 96, 1–136. [Google Scholar]
- Al Rwahnih, M.; Rowhani, A.; Golino, D. First report of grapevine red blotch-associated virus in archival grapevine material from sonoma county, california. Plant Dis. 2015, 99, 895. [Google Scholar] [CrossRef]
- Basso, M.F.; Fajardo, T.V.; Saldarelli, P. Grapevine virus diseases: Economic impact and current advances in viral prospection and management. Rev. Bras. De Frutic. 2017, 39, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, M. Grapevine viruses: A multitude of diverse species with simple but overall poorly adopted management solutions in the vineyard. J. Plant Pathol. 2020, 102, 643–653. [Google Scholar] [CrossRef]
- Giampetruzzi, A.; Roumi, V.; Roberto, R.; Malossini, U.; Yoshikawa, N.; La Notte, P.; Terlizzi, F.; Credi, R.; Saldarelli, P. A new grapevine virus discovered by deep sequencing of virus- and viroid-derived small rnas in cv pinot gris. Virus Res. 2012, 163, 262–268. [Google Scholar] [CrossRef]
- Wallingford, A.K.; Fuchs, M.F.; Martinson, T.; Hesler, S.; Loeb, G.M. Slowing the spread of grapevine leafroll-associated viruses in commercial vineyards with insecticide control of the vector, pseudococcus maritimus (hemiptera: Pseudococcidae). J. Insect Sci. 2015, 15, 112. [Google Scholar] [CrossRef] [PubMed]
- Yadav, N.; Khurana, S.M.P. Plant virus detection and diagnosis: Progress and challenges. In Frontier Discoveries and Innovations in Interdisciplinary Microbiology; Shukla, P., Ed.; Springer: New Delhi, India, 2016; pp. 97–132. [Google Scholar]
- Borges, D.F.; Preising, S.; Ambrósio, M.M.d.Q.; da Silva, W.L. Detection of multiple grapevine viruses in new england vineyards. Crop Prot. 2020, 132. [Google Scholar] [CrossRef]
- Walsh, H.A.; Pietersen, G. Rapid detection of grapevine leafroll-associated virus type 3 using a reverse transcription loop-mediated amplification method. J. Virol. Methods 2013, 194, 308–316. [Google Scholar] [CrossRef] [Green Version]
- Rowhani, A.; Osman, F.; Daubert, S.D.; Al Rwahnih, M.; Saldarelli, P. Polymerase chain reaction methods for the detection of grapevine viruses and viroids. In Grapevine Viruses: Molecular Biology, Diagnostics and Management; Meng, B., Martelli, G.P., Golino, D.A., Fuchs, M., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 431–450. [Google Scholar]
- Hadidi, A.; Czosnek, H.; Barba, M. DNA microarrays and their potential applications for the detection of plant viruses, viroids, and phytoplasmas. J. Plant Pathol. 2004, 86, 97–104. [Google Scholar]
- Engel, E.A.; Escobar, P.F.; Rojas, L.A.; Rivera, P.A.; Fiore, N.; Valenzuela, P.D.T. A diagnostic oligonucleotide microarray for simultaneous detection of grapevine viruses. J. Virol. Methods 2010, 163, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Mahlein, A.-K. Plant disease detection by imaging sensors—Parallels and specific demands for precision agriculture and plant phenotyping. Plant Dis. 2015, 100, 241–251. [Google Scholar] [CrossRef] [Green Version]
- Bendel, N.; Kicherer, A.; Backhaus, A.; Köckerling, J.; Maixner, M.; Bleser, E.; Klück, H.-C.; Seiffert, U.; Voegele, R.T.; Töpfer, R.J.R.S. Detection of grapevine leafroll-associated virus 1 and 3 in white and red grapevine cultivars using hyperspectral imaging. Remote Sens. 2020, 12, 1693. [Google Scholar] [CrossRef]
- Nguyen, C.; Sagan, V.; Maimaitiyiming, M.; Maimaitijiang, M.; Bhadra, S.; Kwasniewski, M.T. Early detection of plant viral disease using hyperspectral imaging and deep learning. Sensors 2021, 21, 742. [Google Scholar] [CrossRef]
- Al Rwahnih, M.; Daubert, S.; Golino, D.; Rowhani, A. Deep sequencing analysis of rnas from a grapevine showing syrah decline symptoms reveals a multiple virus infection that includes a novel virus. Virology 2009, 387, 395–401. [Google Scholar] [CrossRef] [Green Version]
- Fall, M.L.; Xu, D.; Lemoyne, P.; Moussa, I.E.B.; Beaulieu, C.; Carisse, O. A diverse virome of leafroll-infected grapevine unveiled by dsrna sequencing. Viruses 2020, 12, 1142. [Google Scholar] [CrossRef] [PubMed]
- Maliogka, V.I.; Olmos, A.; Pappi, P.G.; Lotos, L.; Efthimiou, K.; Grammatikaki, G.; Candresse, T.; Katis, N.I.; Avgelis, A.D. A novel grapevine badnavirus is associated with the roditis leaf discoloration disease. Virus Res. 2015, 203, 47–55. [Google Scholar] [CrossRef]
- Zhang, Y.; Singh, K.; Kaur, R.; Qiu, W. Association of a novel DNA virus with the grapevine vein-clearing and vine decline syndrome. Phytopathology® 2011, 101, 1081–1090. [Google Scholar] [CrossRef] [Green Version]
- Zherdev, A.V.; Vinogradova, S.V.; Byzova, N.A.; Porotikova, E.V.; Kamionskaya, A.M.; Dzantiev, B.B. Methods for the diagnosis of grapevine viral infections: A review. Agriculture 2018, 8, 195. [Google Scholar] [CrossRef] [Green Version]
- Pop, M.; Salzberg, S.L. Bioinformatics challenges of new sequencing technology. Trends Genet. 2008, 24, 142–149. [Google Scholar] [CrossRef]
- Roossinck, M.J.; Martin, D.P.; Roumagnac, P. Plant virus metagenomics: Advances in virus discovery. Phytopathology® 2015, 105, 716–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Y.; Xu, F.; Wu, J.; Schubert, J.; Li, M.M. Application of next generation sequencing in laboratory medicine. Ann. Lab. Med. 2021, 41, 25–43. [Google Scholar] [CrossRef]
- Hu, T.; Chitnis, N.; Monos, D.; Dinh, A. Next-generation sequencing technologies: An overview. Hum. Immunol. 2021, 82, 801–811. [Google Scholar] [CrossRef]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133. [Google Scholar] [CrossRef]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.-C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Kasianowicz, J.J.; Brandin, E.; Branton, D.; Deamer, D.W. Characterization of individual polynucleotide molecules using a membrane channel. Proc Natl Acad Sci USA 1996, 93, 13770–13773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikheyev, A.S.; Tin, M.M.Y. A first look at the oxford nanopore minion sequencer. Mol. Ecol. Resour. 2014, 14, 1097–1102. [Google Scholar] [CrossRef]
- Phannareth, T.; Nunziata, S.O.; Stulberg, M.J.; Galvez, M.E.; Rivera, Y. Comparison of nanopore sequencing protocols and real-time analysis for phytopathogen diagnostics. Plant Health Prog. 2021, 22, 31–36. [Google Scholar] [CrossRef]
- Silvestre-Ryan, J.; Holmes, I. Pair consensus decoding improves accuracy of neural network basecallers for nanopore sequencing. Genome Biol. 2021, 22, 38. [Google Scholar] [CrossRef]
- Boykin, L.M.; Sseruwagi, P.; Alicai, T.; Ateka, E.; Mohammed, I.U.; Stanton, J.-A.L.; Kayuki, C.; Mark, D.; Fute, T.; Erasto, J.; et al. Tree lab: Portable genomics for early detection of plant viruses and pests in sub-saharan africa. Genes 2019, 10, 632. [Google Scholar] [CrossRef] [Green Version]
- Stenger, D.C.; Burbank, L.P.; Wang, R.; Stewart, A.A.; Mathias, C.; Goodin, M.M. Lost and found: Rediscovery and genomic characterization of sowthistle yellow vein virus after a 30+ year hiatus. Virus Res. 2020, 284, 197987. [Google Scholar] [CrossRef] [PubMed]
- Naito, F.Y.B.; Melo, F.L.; Fonseca, M.E.N.; Santos, C.A.F.; Chanes, C.R.; Ribeiro, B.M.; Gilbertson, R.L.; Boiteux, L.S.; de Cássia Pereira-Carvalho, R. Nanopore sequencing of a novel bipartite new world begomovirus infecting cowpea. Arch. Virol. 2019, 164, 1907–1910. [Google Scholar] [CrossRef]
- Bronzato Badial, A.; Sherman, D.; Stone, A.; Gopakumar, A.; Wilson, V.; Schneider, W.; King, J. Nanopore sequencing as a surveillance tool for plant pathogens in plant and insect tissues. Plant Dis. 2018, 102, 1648–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fellers, J.P.; Webb, C.; Fellers, M.C.; Shoup Rupp, J.; De Wolf, E. Wheat virus identification within infected tissue using nanopore sequencing technology. Plant Dis. 2019, 103, 2199–2203. [Google Scholar] [CrossRef]
- Chalupowicz, L.; Dombrovsky, A.; Gaba, V.; Luria, N.; Reuven, M.; Beerman, A.; Lachman, O.; Dror, O.; Nissan, G.; Manulis-Sasson, S. Diagnosis of plant diseases using the nanopore sequencing platform. Plant Pathol. 2019, 68, 229–238. [Google Scholar] [CrossRef]
- Filloux, D.; Fernandez, E.; Loire, E.; Claude, L.; Galzi, S.; Candresse, T.; Winter, S.; Jeeva, M.L.; Makeshkumar, T.; Martin, D.P.; et al. Nanopore-based detection and characterization of yam viruses. Sci. Rep. 2018, 8, 17879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Della Bartola, M.; Byrne, S.; Mullins, E. Characterization of potato virus y isolates and assessment of nanopore sequencing to detect and genotype potato viruses. Viruses 2020, 12, 478. [Google Scholar] [CrossRef] [Green Version]
- Kesanakurti, P.; Belton, M.; Saeed, H.; Rast, H.; Boyes, I.; Rott, M. Screening for plant viruses by next generation sequencing using a modified double strand rna extraction protocol with an internal amplification control. J. Virol. Methods 2016, 236, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Dodds, J.A.; Morris, T.J.; Jordan, R.L. Plant viral double-stranded rna. Annu. Rev. Phytopathol. 1984, 22, 151–168. [Google Scholar] [CrossRef]
- Weber, F.; Wagner, V.; Rasmussen Simon, B.; Hartmann, R.; Paludan Søren, R. Double-stranded rna is produced by positive-strand rna viruses and DNA viruses but not in detectable amounts by negative-strand rna viruses. J. Virol. 2006, 80, 5059–5064. [Google Scholar] [CrossRef] [Green Version]
- Balázs, Z.; Tombácz, D.; Csabai, Z.; Moldován, N.; Snyder, M.; Boldogkői, Z. Template-switching artifacts resemble alternative polyadenylation. BMC Genom. 2019, 20, 824. [Google Scholar] [CrossRef] [Green Version]
- Moldován, N.; Tombácz, D.; Szűcs, A.; Csabai, Z.; Balázs, Z.; Kis, E.; Molnár, J.; Boldogkői, Z. Third-generation sequencing reveals extensive polycistronism and transcriptional overlapping in a baculovirus. Sci. Rep. 2018, 8, 8604. [Google Scholar] [CrossRef]
- Young, K.T.; Lahmers, K.K.; Sellers, H.S.; Stallknecht, D.E.; Poulson, R.L.; Saliki, J.T.; Tompkins, S.M.; Padykula, I.; Siepker, C.; Howerth, E.W.; et al. Randomly primed, strand-switching, minion-based sequencing for the detection and characterization of cultured rna viruses. J. Vet. Diagn. Investig. 2021, 33, 202–215. [Google Scholar] [CrossRef] [PubMed]
- Rang, F.J.; Kloosterman, W.P.; de Ridder, J. From squiggle to basepair: Computational approaches for improving nanopore sequencing read accuracy. Genome Biol. 2018, 19, 90. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.A.; Ersavas, T.; Ferguson, J.M.; Liu, H.; Lucas, M.C.; Begik, O.; Bojarski, L.; Barton, K.; Novoa, E.M. Molecular barcoding of native rnas using nanopore sequencing and deep learning. Genome Res. 2020, 30, 1345–1353. [Google Scholar] [CrossRef]
- Boonham, N.; Walsh, K.; Smith, P.; Madagan, K.; Graham, I.; Barker, I. Detection of potato viruses using microarray technology: Towards a generic method for plant viral disease diagnosis. J. Virol. Methods 2003, 108, 181–187. [Google Scholar] [CrossRef]
- Li, R.; Ren, X.; Ding, Q.; Bi, Y.; Xie, D.; Zhao, Z. Direct full-length rna sequencing reveals unexpected transcriptome complexity during caenorhabditis elegans development. Genome Res. 2020, 30, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Harel, N.; Meir, M.; Gophna, U.; Stern, A. Direct sequencing of rna with minion nanopore: Detecting mutations based on associations. Nucleic Acids Res. 2019, 47, e148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Li, J.; Chu, X.; Liu, H.; Liu, W.; Liu, D. Nanopore sequencing: A rapid solution for infectious disease epidemics. Sci. China Life Sci. 2019, 62, 1101–1103. [Google Scholar] [CrossRef] [Green Version]
- Leigh, D.M.; Schefer, C.; Cornejo, C. Determining the suitability of minion’s direct rna and DNA amplicon sequencing for viral subtype identification. Viruses 2020, 12, 801. [Google Scholar] [CrossRef]
- Fritz, A.; Bremges, A.; Deng, Z.-L.; Lesker, T.R.; Götting, J.; Ganzenmueller, T.; Sczyrba, A.; Dilthey, A.; Klawonn, F.; McHardy, A.C. Haploflow: Strain-resolved de novo assembly of viral genomes. Genome Biol. 2021, 22, 212. [Google Scholar] [CrossRef]
- Deng, Z.-L.; Dhingra, A.; Fritz, A.; Götting, J.; Münch, P.C.; Steinbrück, L.; Schulz, T.F.; Ganzenmüller, T.; McHardy, A.C. Evaluating assembly and variant calling software for strain-resolved analysis of large DNA viruses. Brief Bioinform. 2021, 22. [Google Scholar] [CrossRef]
- Tan, S.; Dvorak, C.M.T.; Murtaugh, M.P. Rapid, unbiased prrsv strain detection using minion direct rna sequencing and bioinformatics tools. Viruses 2019, 11, 1132. [Google Scholar] [CrossRef] [Green Version]
- Alabi, O.J.; Appel, D.N.; McBride, S.; Al Rwahnih, M.; Pontasch, F.M. Complete genome sequence analysis of a genetic variant of grapevine virus l from the grapevine cultivar blanc du bois. Arch. Virol. 2020, 165, 1905–1909. [Google Scholar] [CrossRef] [PubMed]
- Loose, M.; Malla, S.; Stout, M. Real-time selective sequencing using nanopore technology. Nat. Methods 2016, 13, 751–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Li, H.; Jain, C.; Cheng, H.; Au, K.F.; Li, H.; Aluru, S. Real-time mapping of nanopore raw signals. Bioinformatics 2021, 37, i477–i483. [Google Scholar] [CrossRef]
- Payne, A.; Holmes, N.; Clarke, T.; Munro, R.; Debebe, B.J.; Loose, M. Readfish enables targeted nanopore sequencing of gigabase-sized genomes. Nat. Biotechnol. 2021, 39, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, N.; Bujarski, J.J. Long noncoding rnas in plant viroids and viruses: A review. Pathogens 2020, 9, 765. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Adkar-Purushothama, C.R.; Lemoyne, P.; Perreault, J.P.; Fall, M. First report of grapevine yellow speckle viroid 1 infecting grapevine (Vitis vinifera L.) in canada. Plant Dis. 2021. [Google Scholar] [CrossRef]
- Flores, R.; Di Serio, F.; Navarro, B.; Duran-Vila, N.; Owens, R.A. Viroids and viroid diseases of plants. Stud. Viral Ecol. 2021, 231–273. [Google Scholar]
- Ding, B.; Kwon, M.-O.; Hammond, R.; Owens, R. Cell-to-cell movement of potato spindle tuber viroid. Plant J. 1997, 12, 931–936. [Google Scholar] [CrossRef] [PubMed]
- Adkar-Purushothama, C.R.; Perreault, J.-P. Current overview on viroid–host interactions. WIREs RNA 2020, 11, e1570. [Google Scholar] [CrossRef]
- Rotunno, S.; Vaira, A.M.; Marian, D.; Schneider, A.; Raimondi, S.; Di Serio, F.; Navarro, B.; Miozzi, L. First report of grapevine latent viroid infecting grapevine (Vitis vinifera) in italy. Plant Dis. 2018, 102, 1672. [Google Scholar] [CrossRef]
- Di Serio, F.; Izadpanah, K.; Hajizadeh, M.; Navarro, B. Viroids infecting the grapevine. In Grapevine Viruses: Molecular Biology, Diagnostics and Management; Meng, B., Martelli, G.P., Golino, D.A., Fuchs, M., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 373–392. [Google Scholar]
- Navarro, B.; Rubino, L.; Di Serio, F. Chapter 61—Small circular satellite rnas. In Viroids and Satellites; Hadidi, A., Flores, R., Randles, J.W., Palukaitis, P., Eds.; Academic Press: Boston, MA, USA, 2017; pp. 659–669. [Google Scholar]
- Chay, C.A.; Guan, X.; Bruening, G. Formation of circular satellite tobacco ringspot virus rna in protoplasts transiently expressing the linear rna. Virology 1997, 239, 413–425. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, H.; Fan, M.; He, Y.; Guo, P. Genome-wide identification of long non-coding rnas and circular rnas reveal their cerna networks in response to cucumber green mottle mosaic virus infection in watermelon. Arch. Virol. 2020, 165, 1177–1190. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Eichten, S.R.; Shimizu, R.; Petsch, K.; Yeh, C.-T.; Wu, W.; Chettoor, A.M.; Givan, S.A.; Cole, R.A.; Fowler, J.E.; et al. Genome-wide discovery and characterization of maize long non-coding rnas. Genome Biol. 2014, 15, R40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wierzbicki, A.T.; Haag, J.R.; Pikaard, C.S. Noncoding transcription by rna polymerase pol ivb/pol v mediates transcriptional silencing of overlapping and adjacent genes. Cell 2008, 135, 635–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Okada, T.; Fukushima, T.; Tsudzuki, T.; Sugiura, M.; Yukawa, Y. A novel hypoxic stress-responsive long non-coding rna transcribed by rna polymerase iii in arabidopsis. RNA Biol. 2012, 9, 302–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Chung, P.J.; Liu, J.; Jang, I.-C.; Kean, M.J.; Xu, J.; Chua, N.-H. Genome-wide identification of long noncoding natural antisense transcripts and their responses to light in arabidopsis. Genome Res. 2014, 24, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding rnas: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Shao, C.; Jin, Y.; Wang, H.; Meng, Y. Long non-coding rnas: A novel endogenous source for the generation of dicer-like 1-dependent small rnas in arabidopsis thaliana. RNA Biol 2014, 11, 373–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, R.; Liu, P.; Irwanto, N.; Loh, R.; Wong, S.M. Upregulation of linc-ap2 is negatively correlated with ap2 gene expression with turnip crinkle virus infection in arabidopsis thaliana. Plant Cell Rep. 2016, 35, 2257–2267. [Google Scholar] [CrossRef]
- Wang, J.; Yang, Y.; Jin, L.; Ling, X.; Liu, T.; Chen, T.; Ji, Y.; Yu, W.; Zhang, B. Re-analysis of long non-coding rnas and prediction of circrnas reveal their novel roles in susceptible tomato following tylcv infection. BMC Plant Biol. 2018, 18, 104. [Google Scholar] [CrossRef]
- Wang, J.; Yu, W.; Yang, Y.; Li, X.; Chen, T.; Liu, T.; Ma, N.; Yang, X.; Liu, R.; Zhang, B. Genome-wide analysis of tomato long non-coding rnas and identification as endogenous target mimic for microrna in response to tylcv infection. Sci. Rep. 2015, 5, 16946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Z.; Li, J.; Luo, M.; Li, H.; Chen, Q.; Wang, L.; Song, S.; Zhao, L.; Xu, W.; Zhang, C.; et al. Characterization and cloning of grape circular rnas identified the cold resistance-related vv-circats1. Plant Physiol. 2019, 180, 966–985. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Guo, S.; Wang, Y.; Wang, L.; Shu, S.; Sun, J. Systematic identification and analysis of heat-stress-responsive lncrnas, circrnas and mirnas with associated co-expression and cerna networks in cucumber (Cucumis sativus L.). Physiol. Plant. 2020, 168, 736–754. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Hu, J.; Gao, C.; Chen, G.; Wang, B.; Lin, C.; Song, L.; Ding, Y.; Zhou, G. Genome-wide analysis of long non-coding rnas unveils the regulatory roles in the heat tolerance of chinese cabbage (Brassica rapa ssp.Chinensis). Sci. Rep. 2019, 9, 5002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Liu, T.; Shen, D.; Wang, J.; Ling, X.; Hu, Z.; Chen, T.; Hu, J.; Huang, J.; Yu, W.; et al. Tomato yellow leaf curl virus intergenic sirnas target a host long noncoding rna to modulate disease symptoms. PLOS Pathog. 2019, 15, e1007534. [Google Scholar] [CrossRef] [PubMed]
- Ebbesen, K.K.; Hansen, T.B.; Kjems, J. Insights into circular rna biology. RNA Biol. 2017, 14, 1035–1045. [Google Scholar] [CrossRef]
- Ye, C.-Y.; Chen, L.; Liu, C.; Zhu, Q.-H.; Fan, L. Widespread noncoding circular rnas in plants. New Phytol. 2015, 208, 88–95. [Google Scholar] [CrossRef]
- Jeck, W.R.; Sharpless, N.E. Detecting and characterizing circular rnas. Nat. Biotechnol. 2014, 32, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Lasda, E.; Parker, R. Circular rnas: Diversity of form and function. RNA 2014, 20, 1829–1842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.-T.; Niu, Z.-M.; Zheng, Z.-Y.; Lv, J.-J.; Chen, Q.-Y.; Liu, J.-Q.; Wan, D.-S. Contrasting origins, expression patterns and functions of circrnas between salt-sensitive and salt-tolerant poplars. Environ. Exp. Bot. 2021, 185, 104403. [Google Scholar] [CrossRef]
- Pan, T.; Sun, X.; Liu, Y.; Li, H.; Deng, G.; Lin, H.; Wang, S. Heat stress alters genome-wide profiles of circular rnas in arabidopsis. Plant Mol. Biol. 2018, 96, 217–229. [Google Scholar] [CrossRef]
- Tong, W.; Yu, J.; Hou, Y.; Li, F.; Zhou, Q.; Wei, C.; Bennetzen, J.L. Circular rna architecture and differentiation during leaf bud to young leaf development in tea (camellia sinensis). Planta 2018, 248, 1417–1429. [Google Scholar] [CrossRef]
- Chu, Q.; Bai, P.; Zhu, X.; Zhang, X.; Mao, L.; Zhu, Q.-H.; Fan, L.; Ye, C.-Y. Characteristics of plant circular rnas. Brief Bioinform. 2020, 21, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Conn, V.M.; Hugouvieux, V.; Nayak, A.; Conos, S.A.; Capovilla, G.; Cildir, G.; Jourdain, A.; Tergaonkar, V.; Schmid, M.; Zubieta, C.; et al. A circrna from sepallata3 regulates splicing of its cognate mrna through r-loop formation. Nat. Plants 2017, 3, 17053. [Google Scholar] [CrossRef]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural rna circles function as efficient microrna sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, G.; Sharma, S.; Upadhyay, S.K.; Singh, K. Long non-coding rnas coordinate developmental transitions and other key biological processes in grapevine. Sci. Rep. 2019, 9, 3552. [Google Scholar] [CrossRef] [Green Version]
- Ghorbani, A.; Izadpanah, K.; Peters, J.R.; Dietzgen, R.G.; Mitter, N. Detection and profiling of circular rnas in uninfected and maize iranian mosaic virus-infected maize. Plant Sci. 2018, 274, 402–409. [Google Scholar] [CrossRef] [Green Version]
- Saleembhasha, A.; Mishra, S. Novel molecules lncrnas, trfs and circrnas deciphered from next-generation sequencing/rna sequencing: Computational databases and tools. Brief Funct. Genom. 2018, 17, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.-Y.; Chen, M.-X.; Ye, N.-H.; Shi, L.; Ma, K.-L.; Yang, J.-F.; Cao, Y.-Y.; Zhang, Y.; Yoshida, T.; Fernie, A.R.; et al. Proteogenomic analysis reveals alternative splicing and translation as part of the abscisic acid response in arabidopsis seedlings. Plant J. 2017, 91, 518–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirov, I.; Dudnikov, M.; Merkulov, P.; Shingaliev, A.; Omarov, M.; Kolganova, E.; Sigaeva, A.; Karlov, G.; Soloviev, A. Nanopore rna sequencing revealed long non-coding and ltr retrotransposon-related rnas expressed at early stages of triticale seed development. Plants 2020, 9, 1794. [Google Scholar] [CrossRef]
- Palazzo, A.F.; Koonin, E.V. Functional long non-coding rnas evolve from junk transcripts. Cell 2020, 183, 1151–1161. [Google Scholar] [CrossRef]
- Lanciano, S.; Cristofari, G. Measuring and interpreting transposable element expression. Nat. Rev. Genet. 2020, 21, 721–736. [Google Scholar] [CrossRef]
- Saville, L.; Cheng, Y.; Gollen, B.; Mitchell, L.; Stuart-Edwards, M.; Haight, T.; Mohajerani, M.; Zovoilis, A. Nerd-seq: A novel approach of nanopore direct rna sequencing that expands representation of non-coding rnas. bioRxiv 2021. [Google Scholar] [CrossRef]
- Garalde, D.R.; Snell, E.A.; Jachimowicz, D.; Sipos, B.; Lloyd, J.H.; Bruce, M.; Pantic, N.; Admassu, T.; James, P.; Warland, A.; et al. Highly parallel direct rna sequencing on an array of nanopores. Nat. Methods 2018, 15, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Boivin, V.; Deschamps-Francoeur, G.; Couture, S.; Nottingham, R.M.; Bouchard-Bourelle, P.; Lambowitz, A.M.; Scott, M.S.; Abou-Elela, S. Simultaneous sequencing of coding and noncoding rna reveals a human transcriptome dominated by a small number of highly expressed noncoding genes. RNA 2018, 24, 950–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Wang, J.; Zhao, F. Ciri: An efficient and unbiased algorithm for de novo circular rna identification. Genome Biol. 2015, 16, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, L.; Morey, R.; Palpant, N.J.; Wang, P.L.; Afari, N.; Jiang, C.; Parast, M.M.; Murry, C.E.; Laurent, L.C.; Salzman, J. Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular rna during human fetal development. Genome Biol. 2015, 16, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.-O.; Wang, H.-B.; Zhang, Y.; Lu, X.; Chen, L.-L.; Yang, L. Complementary sequence-mediated exon circularization. Cell 2014, 159, 134–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Wang, J.; Zheng, Y.; Zhang, J.; Chen, S.; Zhao, F. Comprehensive identification of internal structure and alternative splicing events in circular rnas. Nat. Commun. 2016, 7, 12060. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Hou, L.; Zuo, Z.; Ji, P.; Zhang, X.; Xue, Y.; Zhao, F. Comprehensive profiling of circular rnas with nanopore sequencing and ciri-long. Nat. Biotechnol. 2021, 39, 836–845. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, H.; Xi, F.; Wang, H.; Han, X.; Wei, W.; Zhang, H.; Zhang, Q.; Zheng, Y.; Zhu, Q.; et al. Profiling of circular rna n6-methyladenosine in moso bamboo (phyllostachys edulis) using nanopore-based direct rna sequencing. J. Integr. Plant Biol. 2020, 62, 1823–1838. [Google Scholar] [CrossRef] [PubMed]
- Courtney, D.G. Post-transcriptional regulation of viral rna through epitranscriptional modification. Cells 2021, 10, 1129. [Google Scholar] [CrossRef]
- Ito, S.; Horikawa, S.; Suzuki, T.; Kawauchi, H.; Tanaka, Y.; Suzuki, T.; Suzuki, T. Human nat10 is an atp-dependent rna acetyltransferase responsible for n4-acetylcytidine formation in 18 s ribosomal rna (rrna) *. J. Biol. Chem. 2014, 289, 35724–35730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Netzband, R.; Pager, C.T. Epitranscriptomic marks: Emerging modulators of rna virus gene expression. WIREs RNA 2020, 11, e1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courtney, D.G.; Kennedy, E.M.; Dumm, R.E.; Bogerd, H.P.; Tsai, K.; Heaton, N.S.; Cullen, B.R. Epitranscriptomic enhancement of influenza a virus gene expression and replication. Cell Host Microbe 2017, 22, 377–386.e375. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Pérez, M.; Aparicio, F.; López-Gresa, M.P.; Bellés, J.M.; Sánchez-Navarro, J.A.; Pallás, V. Arabidopsis m6a demethylase activity modulates viral infection of a plant virus and the m6a abundance in its genomic rnas. Proc. Natl. Acad. Sci. USA 2017, 114, 10755. [Google Scholar] [CrossRef] [Green Version]
- Viehweger, A.; Krautwurst, S.; Lamkiewicz, K.; Madhugiri, R.; Ziebuhr, J.; Hölzer, M.; Marz, M. Direct rna nanopore sequencing of full-length coronavirus genomes provides novel insights into structural variants and enables modification analysis. Genome Res. 2019, 29, 1545–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arribas-Hernández, L.; Brodersen, P. Occurrence and functions of m6a and other covalent modifications in plant mrna1 [open]. Plant Physiol. 2020, 182, 79–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, A.S.N.; Huang, J.; Syed, N.H.; Ben-Hur, A.; Dong, S.; Gu, L. Decoding co-/post-transcriptional complexities of plant transcriptomes and epitranscriptome using next-generation sequencing technologies. Biochem. Soc. Trans. 2020, 48, 2399–2414. [Google Scholar] [CrossRef] [PubMed]
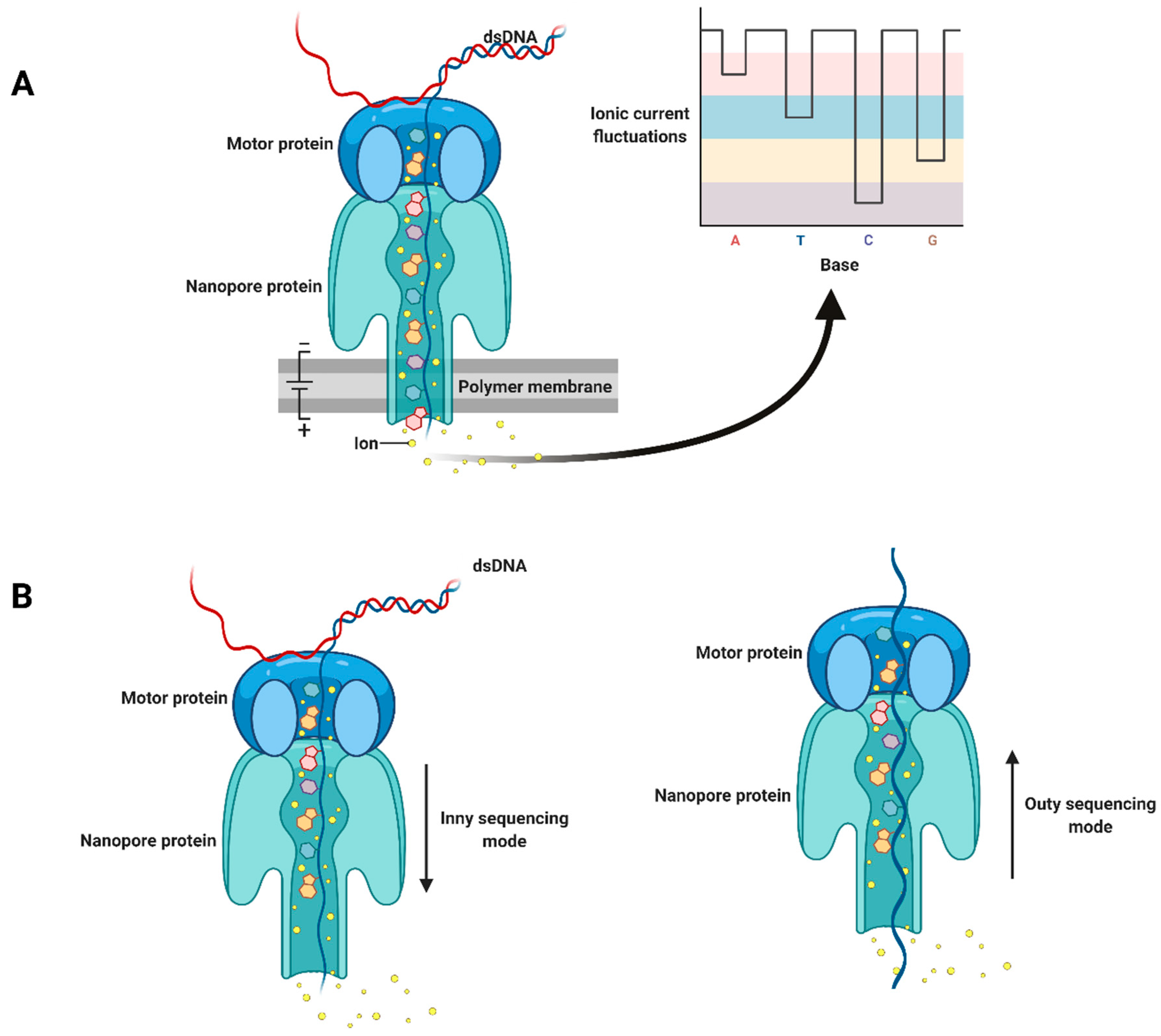




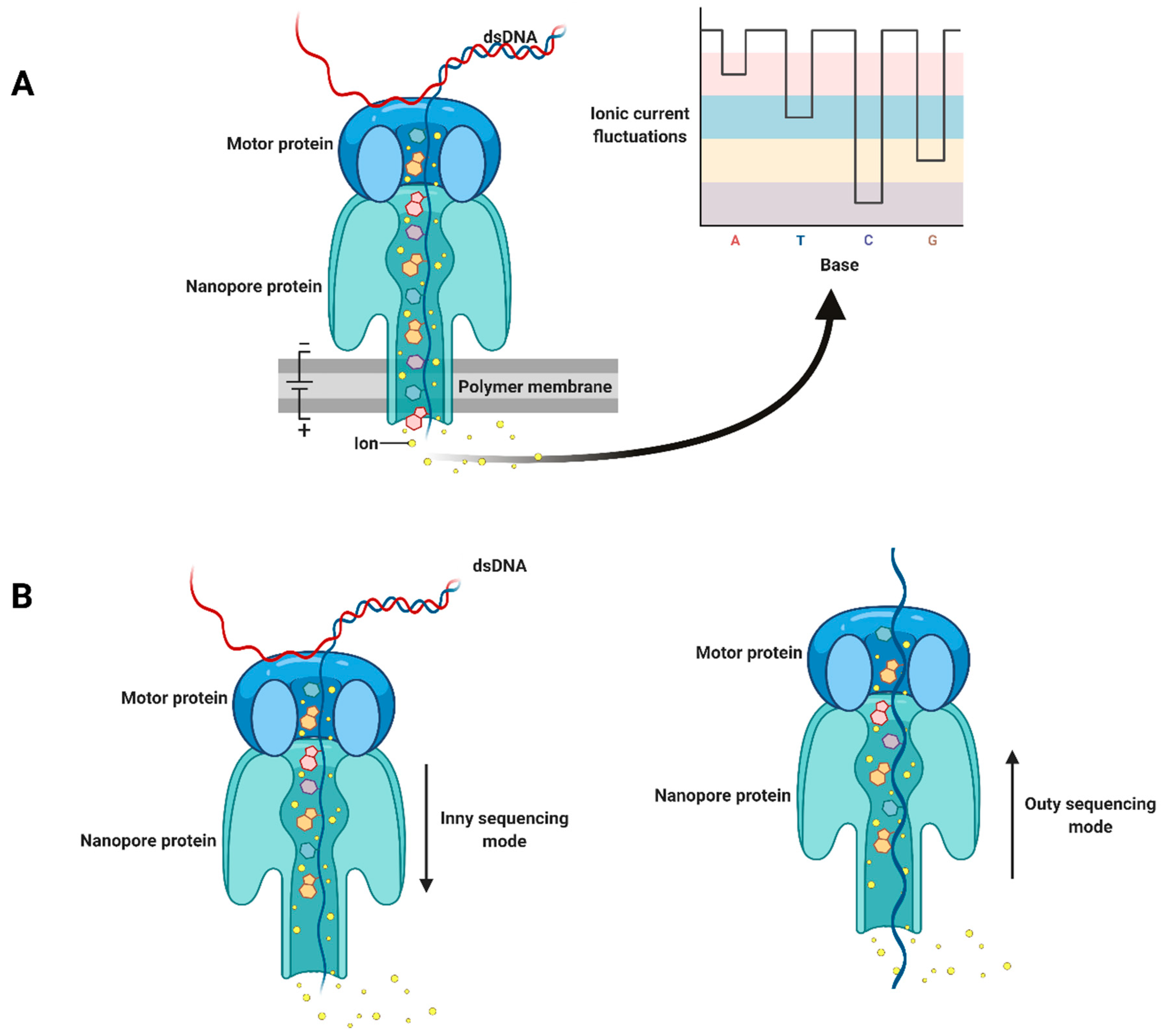{kind=link}
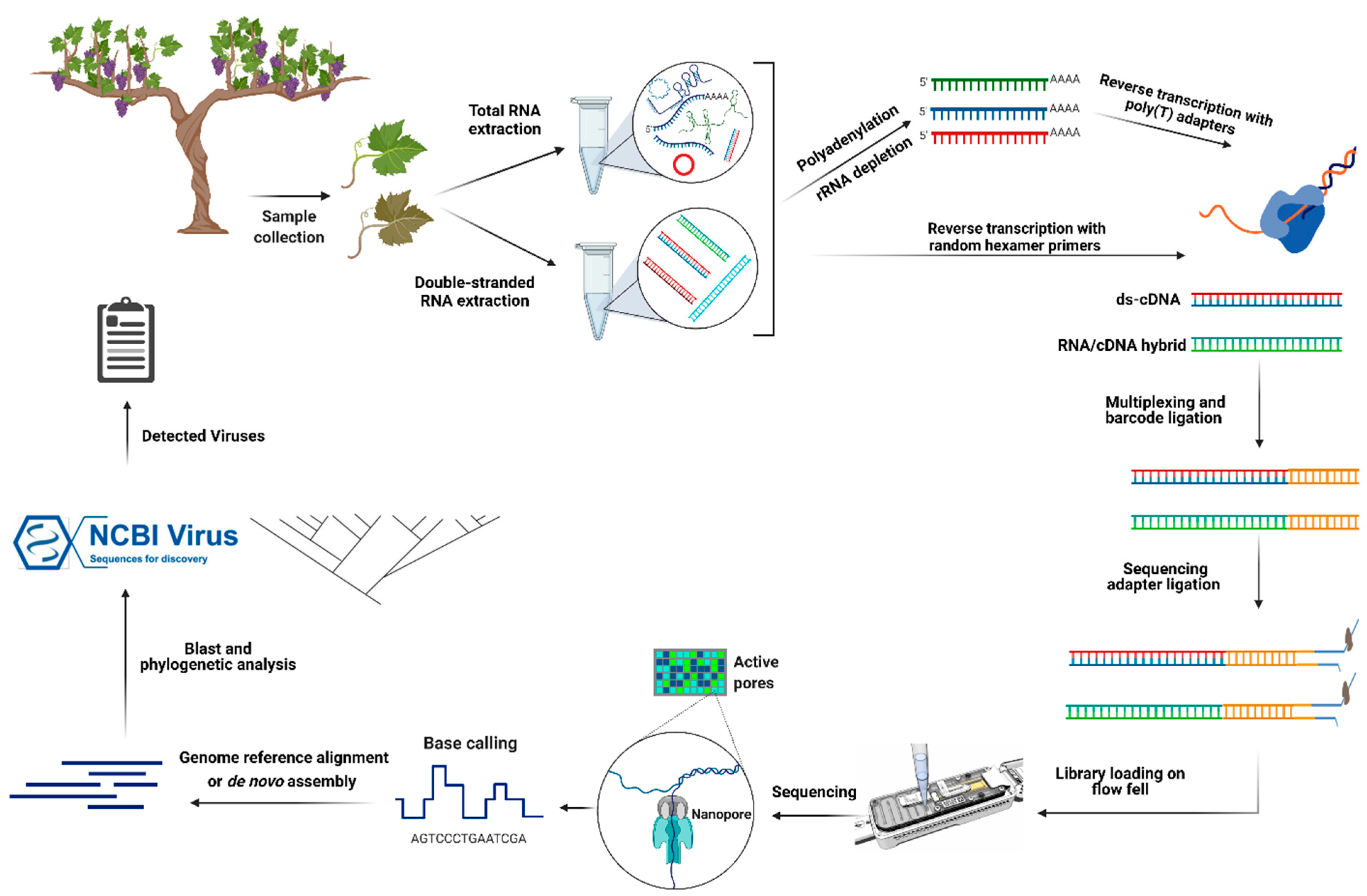{kind=link}
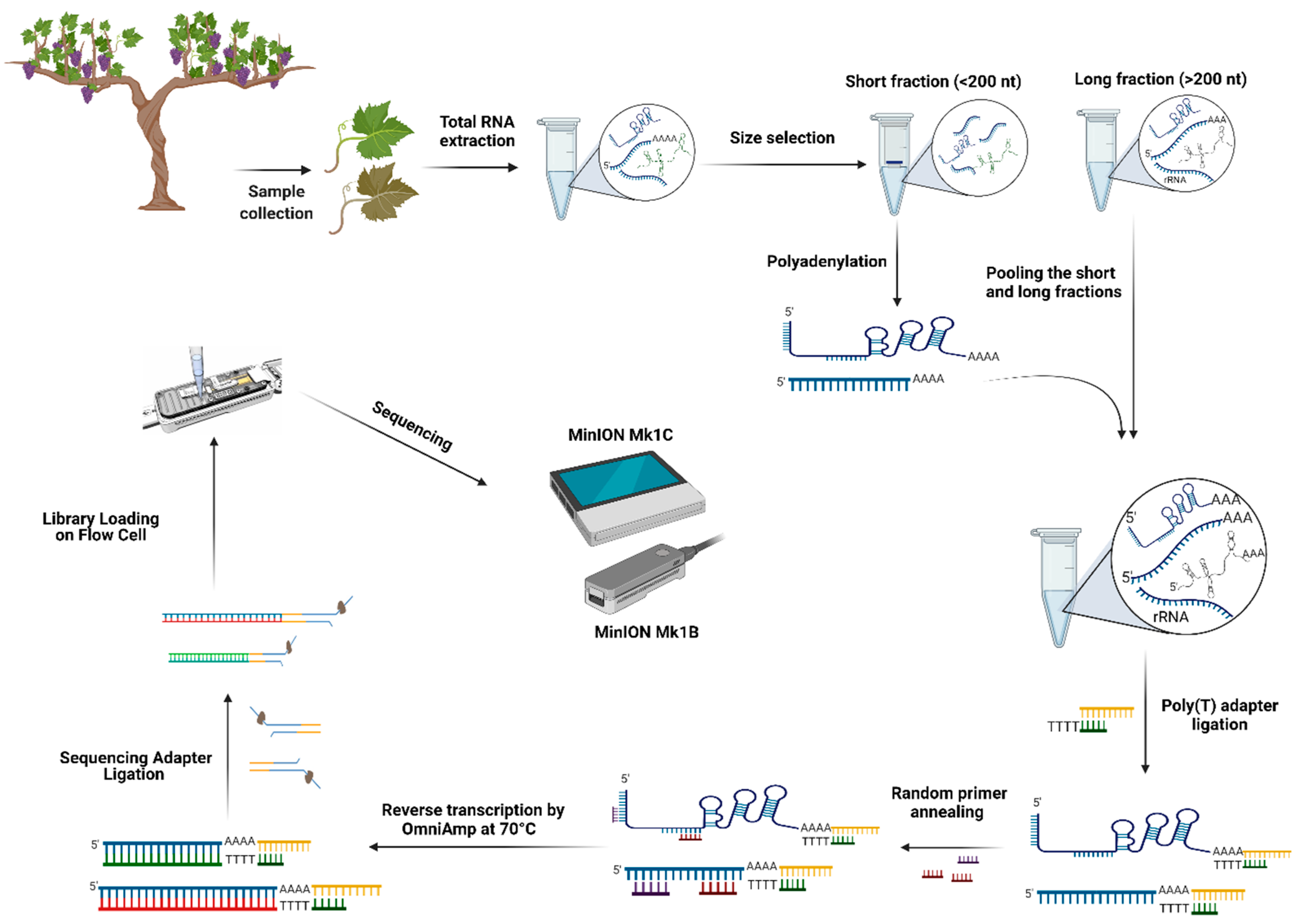{kind=link}
{kind=link}
| Sequencer | Flow Cell | Maximum Yield per Flow Cell (Gb) | Flow Cell Number in Each Running | Maximum Running Time (Hours) | Nanopore Channel Number in Each Flow Cell |
|---|---|---|---|---|---|
| MinION Mk1B/Mk1C | MinION (R9.4.1 or R10.3) /Flongle | 50 for MinION/2.8 for Flongle | 1 | 72 for MinION/16 for Flongle | 512 for MinION/126 for Flongle |
| GridION | MinION (R9.4.1 or R10.3)/Flongle | 50 for MinION/2.8 for Flongle | 5 | 72 for MinION/16 for Flongle | 512 for MinION/126 for Flongle |
| PromethION 24/48 | PromethION | 290 | 24/48 | 72 | 2675 |
| Virus Name | Host Plant | Nucleic Acid Extraction | Library Preparation Kit | References |
|---|---|---|---|---|
| Sowthistle yellow vein virus (SYVV) | Sonchus oleraceus L. | Total RNA | Direct RNA Sequencing | [37] |
| Cowpea bright yellow mosaic virus (CoBYMV) | Vigna unguiculata | Total DNA | Ligation Sequencing | [38] |
| Plum pox virus | Prunus persica | Total RNA | Ligation Sequencing | [39] |
| Wheat streak mosaic virus (WSMV) Triticum mosaic virus (TriMV) Barley yellow dwarf virus (BYDV) | Wheat | Total RNA | Ligation Sequencing | [40] |
| Tomato yellow leaf curl virus (TYLCV) Watermelon chlorotic stunt virus (WmCSV) Tomato brown rugose fruit virus (ToBRFV) Cucumber green mottle mosaic virus (CGMMV) Zucchini yellow mosaic virus (ZYMV) | Solanum lycopersicum Cucumis sativus Citrullus lanatus Cucurbita moschata | Total RNA Total DNA | Direct RNA Sequencing Ligation Sequencing | [41] |
| Dioscorea bacilliform viruses (DBVs) Yam mild mosaic virus (YMMV) Yam chlorotic necrosis virus (YCNV) | Dioscorea alata | Total RNA | PCR-cDNA Sequencing Kit | [42] |
| Potato virus Y (PVY) Potato virus X (PVX) Potato virus S (PVS) Potato leafroll virus (PLRV) | Solanum tuberosum L. | Total RNA | Ligation Sequencing Kit | [43] |
| East African cassava mosaic virus (EACMV) African cassava mosaic virus (ACMV) Tobacco leaf curl virus (TLCV) | Manihot esculenta | Total DNA | Rapid Barcoding Kit | [36] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Javaran, V.J.; Moffett, P.; Lemoyne, P.; Xu, D.; Adkar-Purushothama, C.R.; Fall, M.L. Grapevine Virology in the Third-Generation Sequencing Era: From Virus Detection to Viral Epitranscriptomics. Plants 2021, 10, 2355. https://doi.org/10.3390/plants10112355
Javaran VJ, Moffett P, Lemoyne P, Xu D, Adkar-Purushothama CR, Fall ML. Grapevine Virology in the Third-Generation Sequencing Era: From Virus Detection to Viral Epitranscriptomics. Plants. 2021; 10(11):2355. https://doi.org/10.3390/plants10112355
Chicago/Turabian StyleJavaran, Vahid Jalali, Peter Moffett, Pierre Lemoyne, Dong Xu, Charith Raj Adkar-Purushothama, and Mamadou Lamine Fall. 2021. "Grapevine Virology in the Third-Generation Sequencing Era: From Virus Detection to Viral Epitranscriptomics" Plants 10, no. 11: 2355. https://doi.org/10.3390/plants10112355
APA StyleJavaran, V. J., Moffett, P., Lemoyne, P., Xu, D., Adkar-Purushothama, C. R., & Fall, M. L. (2021). Grapevine Virology in the Third-Generation Sequencing Era: From Virus Detection to Viral Epitranscriptomics. Plants, 10(11), 2355. https://doi.org/10.3390/plants10112355