
Cold Acclimation in Brachypodium Is Accompanied by Changes in Above-Ground Bacterial and Fungal Communities


Abstract
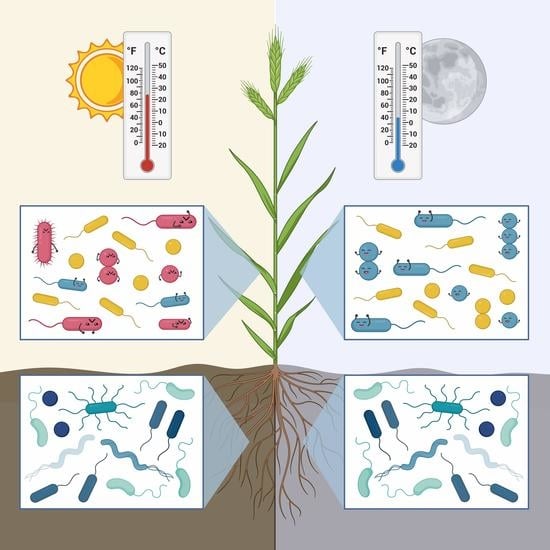
:
1. Introduction
2. Materials and Methods
2.1. Soil Inoculation and Preparation
2.2. Plant Material and Growth Conditions
2.3. Microbiome Extraction and Preparation
2.4. Shotgun Metagenomics Library Preparation and Sequencing
2.5. Preprocessing and Quality Control
2.6. Taxonomic Classification
2.7. Core and Functional Microbiome
2.8. Amplicon Sequencing
2.9. Amplicon Sequence Processing
2.10. Statistical Analysis
3. Results
3.1. Pre-Processing, Shotgun Sequencing, and Kit Controls
3.2. Compatible Results with Shotgun and Amplicon Sequencing
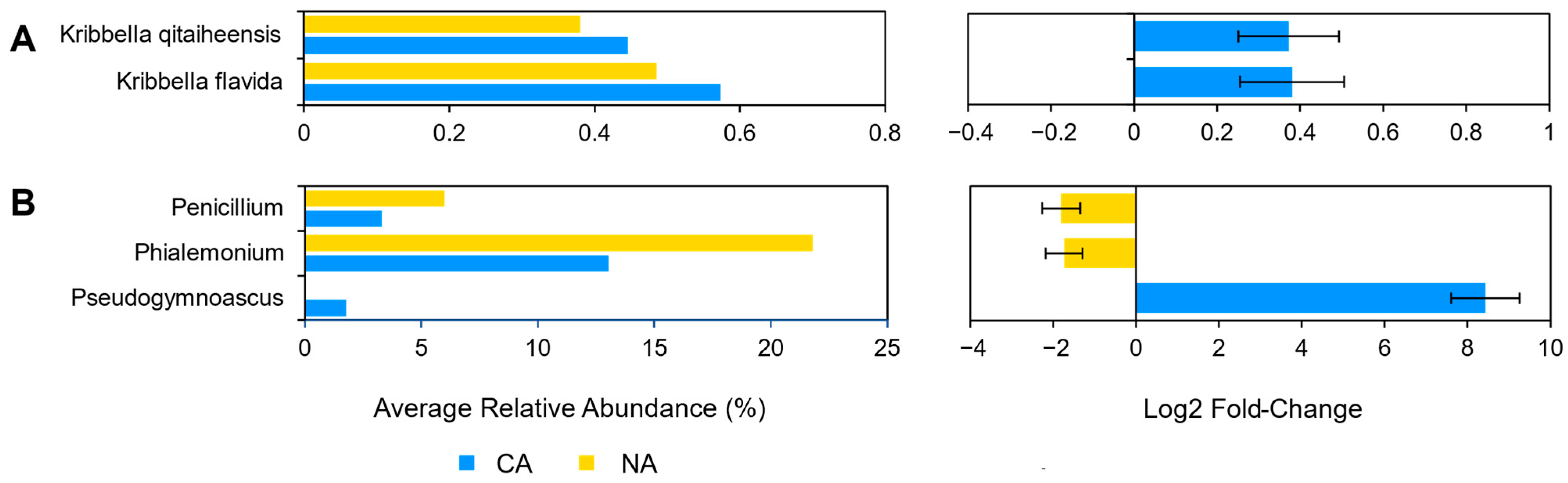
3.3. Cold Acclimation and the Rhizosphere Microbiome
3.4. Cold Acclimation and the Leaf Microbiome
3.5. Dissimilarity Comparisons and Core Microbiome
4. Discussion
4.1. Little Change in Rhizosphere Communities after Cold Acclimation
4.2. Shifts in Leaf Communities Accompany Cold Acclimation
4.3. Leaf Cold Acclimation Associated with Low Temperature and Pathogen Responses
4.4. Prospects and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Juurakko, C.L.; di Cenzo, G.C.; Walker, V.K. Cold acclimation and prospects for cold-resilient crops. Plant Stress 2021, 2, 100028. [Google Scholar] [CrossRef]
- Suzuki, N.; Mittler, R. Reactive oxygen species and temperature stresses: A delicate balance between signaling and destruction. Physiol. Plant. 2006, 126, 45–51. [Google Scholar] [CrossRef]
- Grady, K.L.; Sorensen, J.W.; Stopnisek, N.; Guittar, J.; Shade, A. Assembly and seasonality of core phyllosphere microbiota on perennial biofuel crops. Nat. Commun. 2019, 10, 4135. [Google Scholar] [CrossRef] [Green Version]
- Bei, Q.; Moser, G.; Müller, C.; Liesack, W. Seasonality affects function and complexity but not diversity of the rhizosphere microbiome in European temperate grassland. Sci. Total Environ. 2021, 784, 147036. [Google Scholar] [CrossRef]
- Chialva, M.; De Rose, S.; Novero, M.; Lanfranco, L.; Bonfante, P. Plant genotype and seasonality drive fine changes in olive root microbiota. Curr. Plant Biol. 2021, 28, 100219. [Google Scholar] [CrossRef]
- Juurakko, C.L.; Bredow, M.; Nakayama, T.; Imai, H.; Kawamura, Y.; di Cenzo, G.C.; Walker, V.K. The Brachypodium distachyon cold-acclimated plasma membrane proteome is primed for stress resistance. G3 2021, 11, jkab198. [Google Scholar] [CrossRef]
- Thomashow, M.F. Plant cold acclimation: Freezing tolerance genes and regulatory mechanisms. Annu. Rev. Plant Biol. 1999, 50, 571–599. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Liu, D.; Chong, K. Cold signaling in plants: Insights into mechanisms and regulation. J. Integr. Plant Biol. 2018, 60, 745–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acuña-Rodríguez, I.S.; Newsham, K.K.; Gundel, P.E.; Torres-Díaz, C.; Molina-Montenegro, M.A. Functional roles of microbial symbionts in plant cold tolerance. Ecol. Lett. 2020, 23, 1034–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Brettell, L.E.; Qiu, Z.; Singh, B.K. Microbiome-mediated stress resistance in plants. Trends Plant Sci. 2020, 25, 733–743. [Google Scholar] [CrossRef]
- Saikkonen, K.; Faeth, S.H.; Helander, M.; Sullivan, T.J. Fungal endophytes: A continuum of interactions with host plants. Annu. Rev. Ecol. Syst. 1998, 29, 319–343. [Google Scholar] [CrossRef]
- Porras-Alfaro, A.; Bayman, P. Hidden fungi, emergent properties: Endophytes and microbiomes. Annu. Rev. Phytopathol. 2011, 49, 291–315. [Google Scholar] [CrossRef] [Green Version]
- Rho, H.; Kim, S.H. Endophyte effects on photosynthesis and water use of plant hosts: A meta-analysis. In Functional Importance of the Plant Microbiome; Springer: Cham, Switzerland, 2017; pp. 43–69. [Google Scholar]
- Yadav, S.K. Cold stress tolerance mechanisms in plants. A review. Agron. Sustain. Dev. 2010, 30, 515–527. [Google Scholar] [CrossRef] [Green Version]
- Hardoim, P.R.; Van Overbeek, L.S.; Berg, G.; Pirttilä, A.M.; Compant, S.; Campisano, A.; Sessitsch, A. The hidden world within plants: Ecological and evolutionary considerations for defining functioning of microbial endophytes. Microbiol. Mol. Biol. Rev. 2015, 79, 293–320. [Google Scholar] [CrossRef] [Green Version]
- Rho, H.; Hsieh, M.; Kandel, S.L.; Cantillo, J.; Doty, S.L.; Kim, S.H. Do endophytes promote growth of host plants under stress? A meta-analysis on plant stress mitigation by endophytes. Microb. Ecol. 2018, 75, 407–418. [Google Scholar] [CrossRef]
- Beirinckx, S.; Viaene, T.; Haegeman, A.; Debode, J.; Amery, F.; Vandenabeele, S.; Goormachtig, S. Tapping into the maize root microbiome to identify bacteria that promote growth under chilling conditions. Microbiome 2020, 8, 54. [Google Scholar] [CrossRef]
- Theocharis, A.; Bordiec, S.; Fernandez, O.; Paquis, S.; Dhondt-Cordelier, S.; Baillieul, F.; Barka, E.A. Burkholderia phytofirmans PsJN primes Vitis vinifera L. and confers a better tolerance to low nonfreezing temperatures. Mol. Plant Microbe Interact. 2012, 25, 241–249. [Google Scholar] [CrossRef] [Green Version]
- Jeon, C.W.; Kim, D.R.; Bae, E.J.; Kwak, Y.S. Changes in bacterial community structure and enriched functional bacteria associated with turfgrass monoculture. Front. Bioeng. Biotechnol. 2021, 8, 1495. [Google Scholar] [CrossRef]
- Liu, X.M.; Xu, Q.L.; Li, Q.Q.; Zhang, H.; Xiao, J.X. Physiological responses of the two blueberry cultivars to inoculation with an arbuscular mycorrhizal fungus under low-temperature stress. J. Plant Nutr. 2017, 40, 2562–2570. [Google Scholar] [CrossRef]
- Rocha, I.; Ma, Y.; Souza-Alonso, P.; Vosátka, M.; Freitas, H.; Oliveira, R.S. Seed coating: A tool for delivering beneficial microbes to agricultural crops. Front. Plant Sci. 2019, 10, 1357. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, A.; Donn, S.; Ryan, P.R.; Mathesius, U.; Devilla, R.; Jones, A.; Watt, M. Microbiome and exudates of the root and rhizosphere of Brachypodium distachyon, a model for wheat. PLoS ONE 2016, 11, e0164533. [Google Scholar] [CrossRef] [Green Version]
- Feehery, G.R.; Yigit, E.; Oyola, S.O.; Langhorst, B.W.; Schmidt, V.T.; Stewart, F.J.; Pradhan, S. A method for selectively enriching microbial DNA from contaminating vertebrate host DNA. PLoS ONE 2013, 8, e76096. [Google Scholar]
- Clarke, E.L.; Taylor, L.J.; Zhao, C.; Connell, A.; Lee, J.J.; Fett, B.; Bittinger, K. Sunbeam: An extensible pipeline for analyzing metagenomic sequencing experiments. Microbiome 2019, 7, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 20 August 2021).
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [Green Version]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Pruitt, K.D. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Breitwieser, F.P.; Thielen, P.; Salzberg, S.L. Bracken: Estimating species abundance in metagenomics data. PeerJ Comput. Sci. 2017, 3, e104. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D. Vegan: Community Ecology Package. R Package Version 2.5-7. 2020. Available online: https://rdrr.io/cran/vegan/ (accessed on 15 December 2021).
- Hammer, Ø.; Harper, D.A.; Ryan, P.D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 9. [Google Scholar]
- Clarke, K.R. Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 1993, 18, 117–143. [Google Scholar] [CrossRef]
- Lazcano, C.; Boyd, E.; Holmes, G.; Hewavitharana, S.; Pasulka, A.; Ivors, K. The rhizosphere microbiome plays a role in the resistance to soil-borne pathogens and nutrient uptake of strawberry cultivars under field conditions. Sci. Rep. 2021, 11, 3188. [Google Scholar] [CrossRef] [PubMed]
- Marsh, R.; Gavillet, H.; Hanson, L.H.; Ng, C.; Mitchell-Whyte, M.; Major, G.; van der Gast, C. Intestinal function and transit associate with gut microbiota dysbiosis in cystic fibrosis. MedRxiv 2021. Available online: https://www.medrxiv.org/content/10.1101/2021.08.24.21262265v2 (accessed on 20 October 2021). [CrossRef]
- Beghini, F.; McIver, L.J.; Blanco-Míguez, A.; Dubois, L.; Asnicar, F.; Maharjan, S.; Segata, N. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. Elife 2021, 10, e65088. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Li, H. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Mallick, H.; Tickle, T.L.; McIver, L.J.; Rahnavard, G.; Nguyen, L.H.; Weingart, G.; Subramanian, A. Multivariable Association in Population-Scale Meta’omic Surveys. Submission. 2020. Available online: https://huttenhower.sph.harvard.edu/maaslin2/ (accessed on 6 September 2021).
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 2016, 18, 1403–1414. [Google Scholar] [CrossRef]
- Apprill, A.; McNally, S.; Parsons, R.; Weber, L. Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 2015, 75, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Gardes, M.; Bruns, T.D. ITS primers with enhanced specificity for basidiomycetes-application to the identification of mycorrhizae and rusts. Mol. Ecol. 1993, 2, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Tannenbaum, I.; Kaur, J.; Mann, R.; Sawbridge, T.; Rodoni, B.; Spangenberg, G. Profiling the Lolium perenne microbiome: From seed to seed. Phytobiomes J. 2020, 4, 281–289. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Caporaso, J.G. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Glöckner, F.O. The SILVA and “all-species living tree project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef] [Green Version]
- Glöckner, F.O.; Yilmaz, P.; Quast, C.; Gerken, J.; Beccati, A.; Ciuprina, A.; Ludwig, W. 25 years of serving the community with ribosomal RNA gene reference databases and tools. J. Biotechnol. 2017, 261, 169–176. [Google Scholar] [CrossRef]
- Nilsson, R.H.; Larsson, K.H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Abarenkov, K. The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2019, 47, D259–D264. [Google Scholar] [CrossRef] [PubMed]
- Abarenkov, K.; Zirk, A.; Piirmann, T.; Pöhönen, R.; Ivanov, F.; Nilsson, R.H.; Kõljalg, U. UNITE General FASTA Release for Fungi. Version 04.02. 2020, UNITE Community. Available online: https://search.datacite.org/works/10.15156/bio/786385 (accessed on 15 December 2021).
- Kõljalg, U.; Nilsson, H.R.; Schigel, D.; Tedersoo, L.; Larsson, K.H.; May, T.W.; Abarenkov, K. The taxon hypothesis paradigm—On the unambiguous detection and communication of taxa. Microorganisms 2020, 8, 1910. [Google Scholar] [CrossRef]
- Lin, H.; Peddada, S.D. Analysis of compositions of microbiomes with bias correction. Nat. Commun. 2020, 11, 3514. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Brader, G.; Sessitsch, A.; Mäki, A.; van Elsas, J.D.; Nissinen, R. Plants assemble species specific bacterial communities from common core taxa in three arcto-alpine climate zones. Front. Microbiol. 2017, 8, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos-Medellín, C.; Liechty, Z.; Edwards, J.; Nguyen, B.; Huang, B.; Weimer, B.C.; Sundaresan, V. Prolonged drought imparts lasting compositional changes to the rice root microbiome. Nat. Plants 2021, 7, 1065–1077. [Google Scholar] [CrossRef]
- Song, Y.; Haney, C.H. Drought dampens microbiome development. Nat. Plants 2021, 7, 994–995. [Google Scholar] [CrossRef] [PubMed]
- Castanheira, N.; Dourado, A.C.; Kruz, S.; Alves, P.I.L.; Delgado-Rodríguez, A.I.; Pais, I.; Fareleira, P. Plant growth-promoting Burkholderia species isolated from annual ryegrass in Portuguese soils. J. Appl. Microbiol. 2016, 120, 724–739. [Google Scholar] [CrossRef] [Green Version]
- Challacombe, J.F.; Hesse, C.N.; Bramer, L.M.; McCue, L.A.; Lipton, M.; Purvine, S.; Kuske, C.R. Genomes and secretomes of Ascomycota fungi reveal diverse functions in plant biomass decomposition and pathogenesis. BMC Genom. 2019, 20, 976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Kim, J.; Lee, J. Colonization of toxic cyanobacteria on the surface and inside of leafy green: A hidden source of cyanotoxin production and exposure. Food Microbiol. 2021, 94, 103655. [Google Scholar] [CrossRef]
- Worsley, S.F.; Newitt, J.; Rassbach, J.; Batey, S.F.; Holmes, N.A.; Murrell, J.C.; Hutchings, M.I. Streptomyces endophytes promote host health and enhance growth across plant species. Appl. Environ. Microbiol. 2020, 86, e01053-20. [Google Scholar] [CrossRef]
- Hutchings, M.I.; Truman, A.W.; Wilkinson, B. Antibiotics: Past, present and future. Curr. Opin. Microbiol. 2019, 51, 72–80. [Google Scholar] [CrossRef]
- Gupta, S.; Pandey, S. ACC deaminase producing bacteria with multifarious plant growth promoting traits alleviates salinity stress in French bean (Phaseolus vulgaris) plants. Front. Microbiol. 2019, 10, 1506. [Google Scholar] [CrossRef]
- Rinu, K.; Malviya, M.K.; Sati, P.; Tiwari, S.C.; Pandey, A. Response of cold-tolerant Aspergillus spp. to solubilization of Fe and Al phosphate in presence of different nutritional sources. Int. Sch. Res. Not. 2013, 2013, 598541. [Google Scholar] [CrossRef] [Green Version]
- Granzow, S.; Kaiser, K.; Wemheuer, B.; Pfeiffer, B.; Daniel, R.; Vidal, S.; Wemheuer, F. The effects of cropping regimes on fungal and bacterial communities of wheat and faba bean in a greenhouse pot experiment differ between plant species and compartment. Front. Microbiol. 2017, 8, 902. [Google Scholar] [CrossRef] [Green Version]
- Pearce, R.S. Plant freezing and damage. Ann. Bot. 2001, 87, 417–424. [Google Scholar] [CrossRef]
- Daffonchio, D.; Zanardini, E.; Vatta, P.; Sorlini, C. Cometabolic degradation of thiocarbamate herbicides by Streptomyces sp. strain M2 and effects on the cell metabolism. Ann. Microbiol. Enzimol. 1999, 49, 13–22. [Google Scholar]
- Singh, S.P.; Gaur, R. Endophytic Streptomyces spp. underscore induction of defense regulatory genes and confers resistance against Sclerotium rolfsii in chickpea. Biol. Control. 2017, 104, 44–56. [Google Scholar] [CrossRef]
- Taechowisan, T.; Peberdy, J.F.; Lumyong, S. Isolation of endophytic actinomycetes from selected plants and their antifungal activity. World J. Microbiol. Biotechnol. 2003, 19, 381–385. [Google Scholar] [CrossRef]
- Human, Z.R.; Moon, K.; Bae, M.; De Beer, Z.W.; Cha, S.; Wingfield, M.J.; Venter, S.N. Antifungal Streptomyces spp. associated with the infructescences of Protea spp. in South Africa. Front. Microbiol. 2016, 7, 1657. [Google Scholar] [CrossRef] [Green Version]
- Beales, N. Adaptation of microorganisms to cold temperatures, weak acid preservatives, low pH, and osmotic stress: A review. Compr. Rev. Food Sci. Food Saf. 2004, 3, 1–20. [Google Scholar] [CrossRef]
- Kim, M.J.; Lee, Y.K.; Lee, H.K.; Im, H. Characterization of cold-shock protein A of Antarctic Streptomyces sp. AA8321. Protein J. 2007, 26, 51–59. [Google Scholar] [CrossRef]
- Manullang, W.; Chuang, H.W. Streptomyces sp. mitigates abiotic stress response and promotes plant growth. J. Plant Prot. Res. 2020, 263–274. [Google Scholar]
- Warrad, M.; Hassan, Y.M.; Mohamed, M.S.; Hagagy, N.; Al-Maghrabi, O.A.; Selim, S.; Abd-Elgawad, H. A bioactive fraction from Streptomyces sp. enhances maize tolerance against drought stress. J. Microbiol. Biotechnol. 2020, 30, 1156–1168. [Google Scholar] [CrossRef]
- Zou, P.; Schrempf, H. The heme-independent manganese-peroxidase activity depends on the presence of the C-terminal domain within the Streptomyces reticuli catalase-peroxidase CpeB. Eur. J. Biochem. 2000, 267, 2840–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folcher, M.; Gaillard, H.; Nguyen, L.T.; Nguyen, K.T.; Lacroix, P.; Bamas-Jacques, N.; Thompson, C.J. Pleiotropic functions of a Streptomyces pristinaespiralis autoregulator receptor in development, antibiotic biosynthesis, and expression of a superoxide dismutase. J. Biol. Chem. 2001, 276, 44297–44306. [Google Scholar] [CrossRef] [Green Version]
- Akanuma, G.; Hara, H.; Ohnishi, Y.; Horinouchi, S. Dynamic changes in the extracellular proteome caused by absence of a pleiotropic regulator AdpA in Streptomyces griseus. Mol. Microbiol. 2009, 73, 898–912. [Google Scholar] [CrossRef]
- Killham, K.; Firestone, M.K. Proline transport increases growth efficiency in salt-stressed Streptomyces griseus. Appl. Environ. Microbiol. 1984, 48, 239–241. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Solanki, M.K.; Yu, Z.X.; Yang, L.T.; An, Q.L.; Dong, D.F.; Li, Y.R. Draft genome analysis offers insights into the mechanism by which Streptomyces chartreusis WZS021 increases drought tolerance in sugarcane. Front. Microbiol. 2019, 9, 3262. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lai, R.; Li, W.J. The Family Solimonadaceae. In The Prokaryotes; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Panoff, J.M.; Thammavongs, B.; Guéguen, M.; Boutibonnes, P. Cold stress responses in mesophilic bacteria. Cryobiology 1998, 36, 75–83. [Google Scholar] [CrossRef]
- Stokes, J.M.; French, S.; Ovchinnikova, O.G.; Bouwman, C.; Whitfield, C.; Brown, E.D. Cold stress makes Escherichia coli susceptible to glycopeptide antibiotics by altering outer membrane integrity. Cell Chem. Biol. 2016, 23, 267–277. [Google Scholar] [CrossRef] [Green Version]
- Tiryaki, D.; Aydın, İ.; Atıcı, Ö. Psychrotolerant bacteria isolated from the leaf apoplast of cold-adapted wild plants improve the cold resistance of bean (Phaseolus vulgaris L.) under low temperature. Cryobiology 2019, 86, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.M.; Muñoz, C.A.; Kao-Kniffin, J.; Kessler, A. Soil microbiomes from fallow fields have species-specific effects on crop growth and pest resistance. Front. Plant Sci. 2020, 11, 1171. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxa | NA (%) | CA (%) | Average Dissimilarity | Contribution (%) | Cumulative (%) |
|---|---|---|---|---|---|
| Shotgun Rhizo (Overall Average Dissimilarity 7.1%) | |||||
| Streptomyces sp. M2 | 32.5 | 32.8 | 0.9 | 12.8 | 12.8 |
| Actinocatenispora sera | 5.7 | 7.3 | 0.8 | 11.2 | 24.0 |
| Actinocatenispora thailandica | 3.6 | 4.9 | 0.6 | 9.1 | 33.1 |
| 16S Rhizo (Overall Average Dissimilarity 10.7%) | |||||
| Actinocatenispora | 8.1 | 11.4 | 1. 7 | 15.7 | 15.7 |
| Streptomyces | 24.2 | 26.6 | 1.4 | 13.4 | 29.2 |
| ITS Rhizo (Overall Average Dissimilarity 19.0%) | |||||
| Phialemonium | 21.3 | 13.4 | 4.0 | 21.1 | 21.1 |
| Apiotrichum | 29.9 | 30.8 | 4.0 | 21.1 | 42.2 |
| Shotgun Leaf (Overall Average Dissimilarity 52. 6%) | |||||
| Microcystis aeruginosa | 12.1 | 27.4 | 9.6 | 18.2 | 18.2 |
| Streptomyces sp. M2 | 4.4 | 15.1 | 5.4 | 10.2 | 28.4 |
| 16S Leaf (Overall Average Dissimilarity 60.9%) | |||||
| Pseudomonas | 19.7 | 1.0 | 9.4 | 15.4 | 15.4 |
| Rhodococcus | 15.0 | 0.4 | 7.3 | 12.0 | 27.4 |
| ITS Leaf (Overall Average Dissimilarity 80.3%) | |||||
| Aspergillus | 15.9 | 47.5 | 16.6 | 20.7 | 20.7 |
| Goidanichiella | 25.1 | 0.0 | 12.6 | 15.7 | 36.4 |
| Phyla | Class | Order | Family | Genus | Species |
|---|---|---|---|---|---|
| Core rhizosphere species (shotgun) | |||||
| Actinobacteria | Actinomycetia | Streptomycetales | Streptomycetaceae | Streptomyces | Streptomyces sp. M2 |
| Actinobacteria | Actinomycetia | Micromonosporales | Micromonosporaceae | Actinocatenispora | Actinocatenispora sera |
| Core rhizosphere genera (16S) | |||||
| Actinobacteria | Actinomycetia | Streptomycetales | Streptomycetaceae | Streptomyces | |
| Actinobacteria | Actinomycetia | Micromonosporales | Micromonosporaceae | Actinocatenispora | |
| Proteobacteria | Gammaproteobacteria | Xanthomonadales | Rhodanobacteraceae | Rhodanobacter | |
| Core rhizosphere genera (ITS) | |||||
| Ascomycota | |||||
| Basidiomycota | Tremellomycetes | Trichosporonales | Trichosporonaceae | Apiotrichum | |
| Ascomycota | Sordariomycetes | Sordariales | Cephalothecaceae | Phialemonium | |
| Ascomycota | Saccharomycetes | Saccharomycetales | Saccharomycetaceae | Candida | |
| Core leaf species (shotgun) | |||||
| Actinobacteria | Actinomycetia | Streptomycetales | Streptomycetaceae | Streptomyces | Streptomyces sp. M2 |
| Proteobacteria | Alphaproteobacteria | Hyphomicrobiales | Rhizobiaceae | Liberibacter | * ‘Candidatus L. a.’ |
| Core leaf genera (16S) | |||||
| Actinobacteria | Actinomycetia | Streptomycetales | Streptomycetaceae | Streptomyces | |
| Proteobacteria | Gammaproteobacteria | Xanthomonadales | Rhodanobacteraceae | Rhodanobacter | |
| Proteobacteria | Gammaproteobacteria | Salinisphaerales | Solimonadaceae | Solimonas | |
| Core leaf genera (ITS) | |||||
| Unidentified Fungi | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juurakko, C.L.; diCenzo, G.C.; Walker, V.K. Cold Acclimation in Brachypodium Is Accompanied by Changes in Above-Ground Bacterial and Fungal Communities. Plants 2021, 10, 2824. https://doi.org/10.3390/plants10122824
Juurakko CL, diCenzo GC, Walker VK. Cold Acclimation in Brachypodium Is Accompanied by Changes in Above-Ground Bacterial and Fungal Communities. Plants. 2021; 10(12):2824. https://doi.org/10.3390/plants10122824
Chicago/Turabian StyleJuurakko, Collin L., George C. diCenzo, and Virginia K. Walker. 2021. "Cold Acclimation in Brachypodium Is Accompanied by Changes in Above-Ground Bacterial and Fungal Communities" Plants 10, no. 12: 2824. https://doi.org/10.3390/plants10122824
APA StyleJuurakko, C. L., diCenzo, G. C., & Walker, V. K. (2021). Cold Acclimation in Brachypodium Is Accompanied by Changes in Above-Ground Bacterial and Fungal Communities. Plants, 10(12), 2824. https://doi.org/10.3390/plants10122824