
Two Korean Endemic Clematis Chloroplast Genomes: Inversion, Reposition, Expansion of the Inverted Repeat Region, Phylogenetic Analysis, and Nucleotide Substitution Rates

 ,
,  ,
, 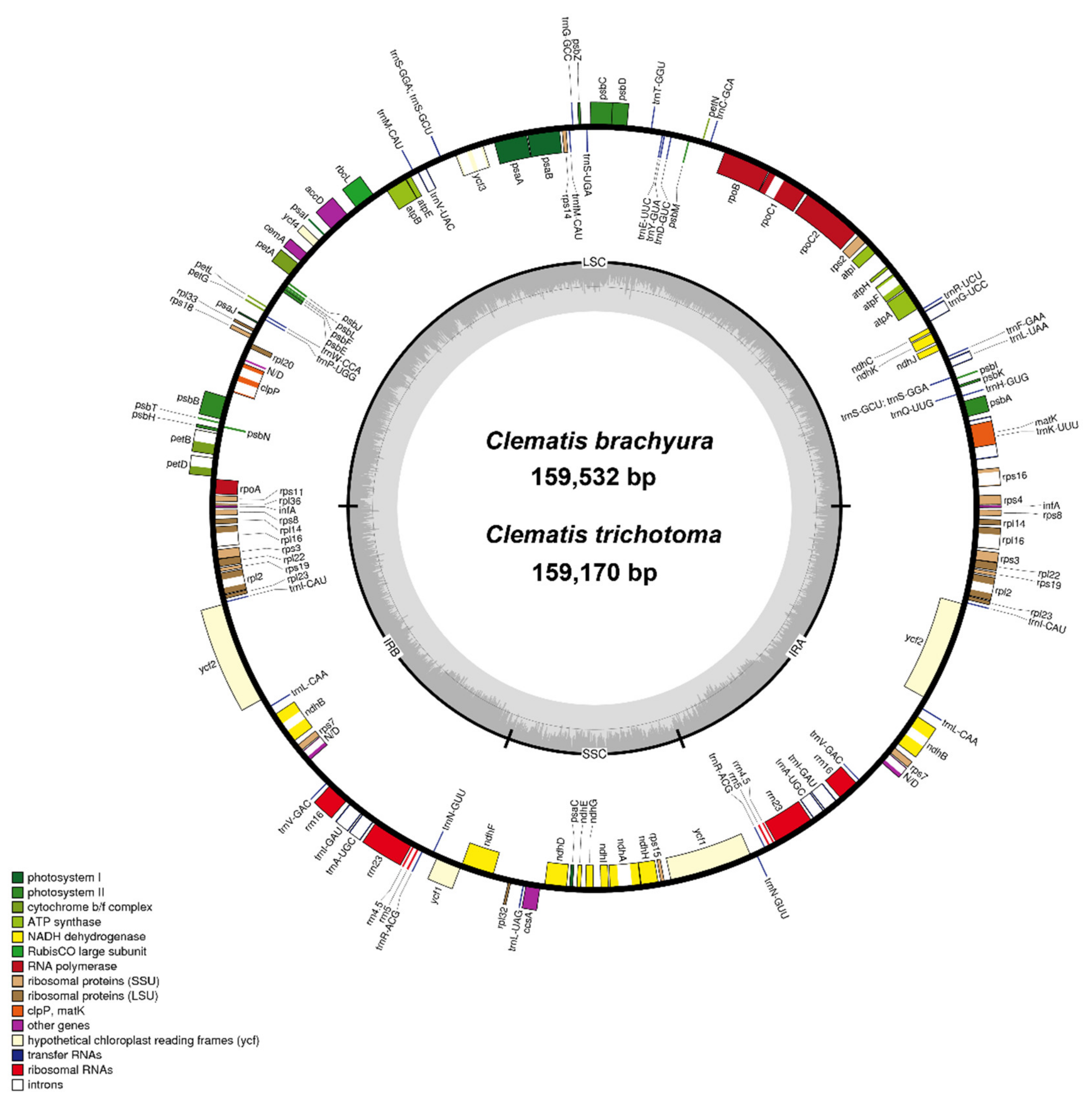{kind=link}
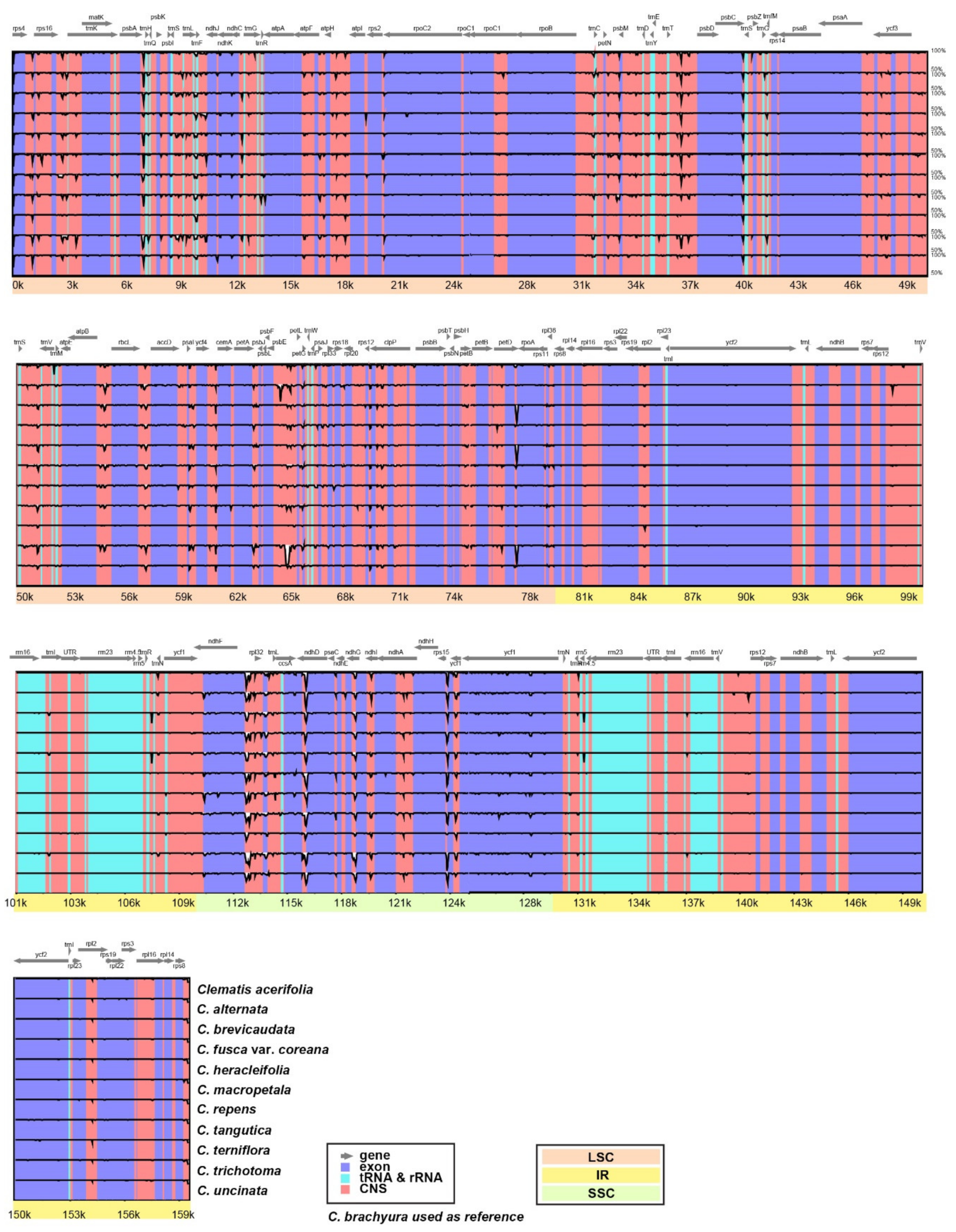{kind=link}
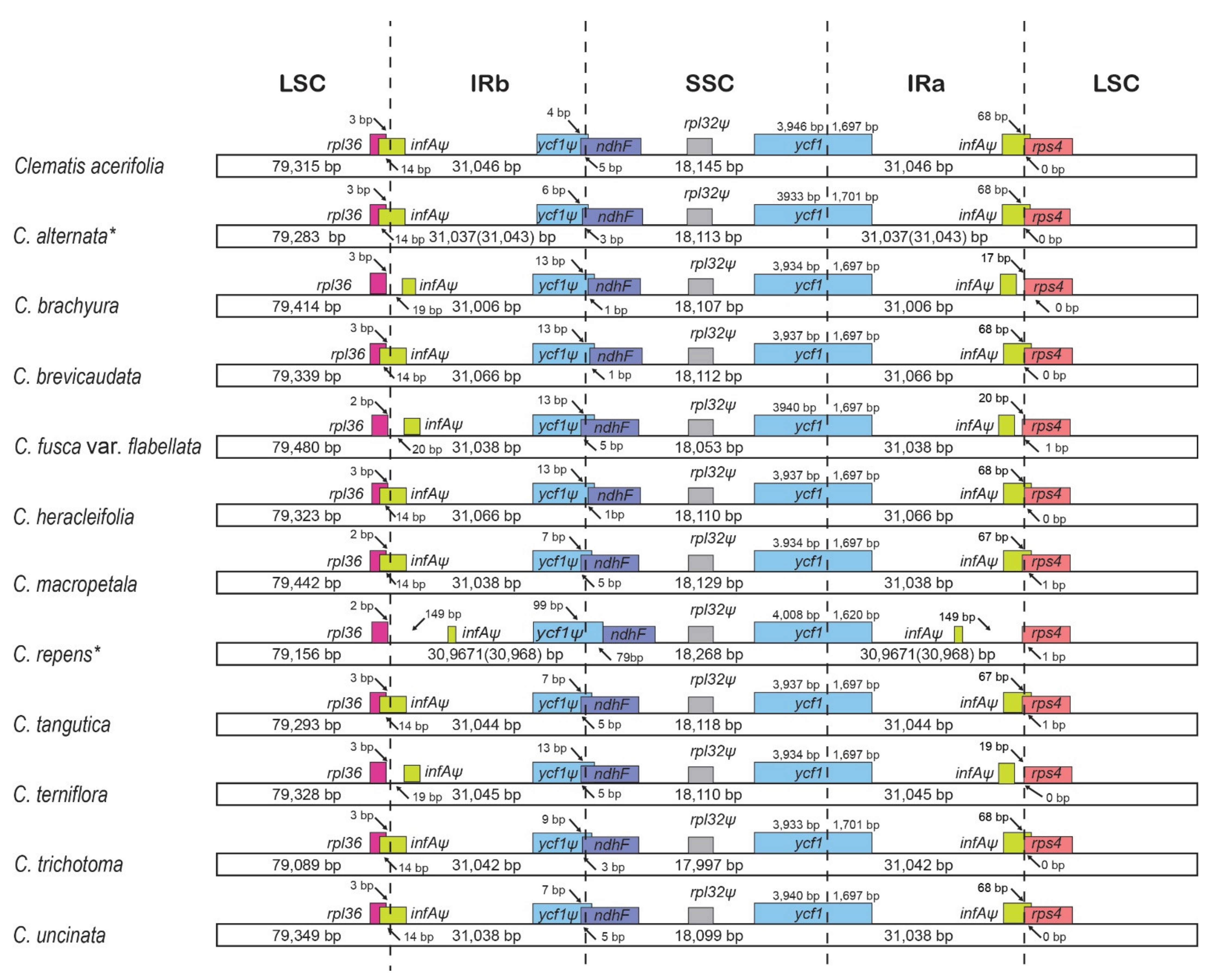{kind=link}
{kind=link}
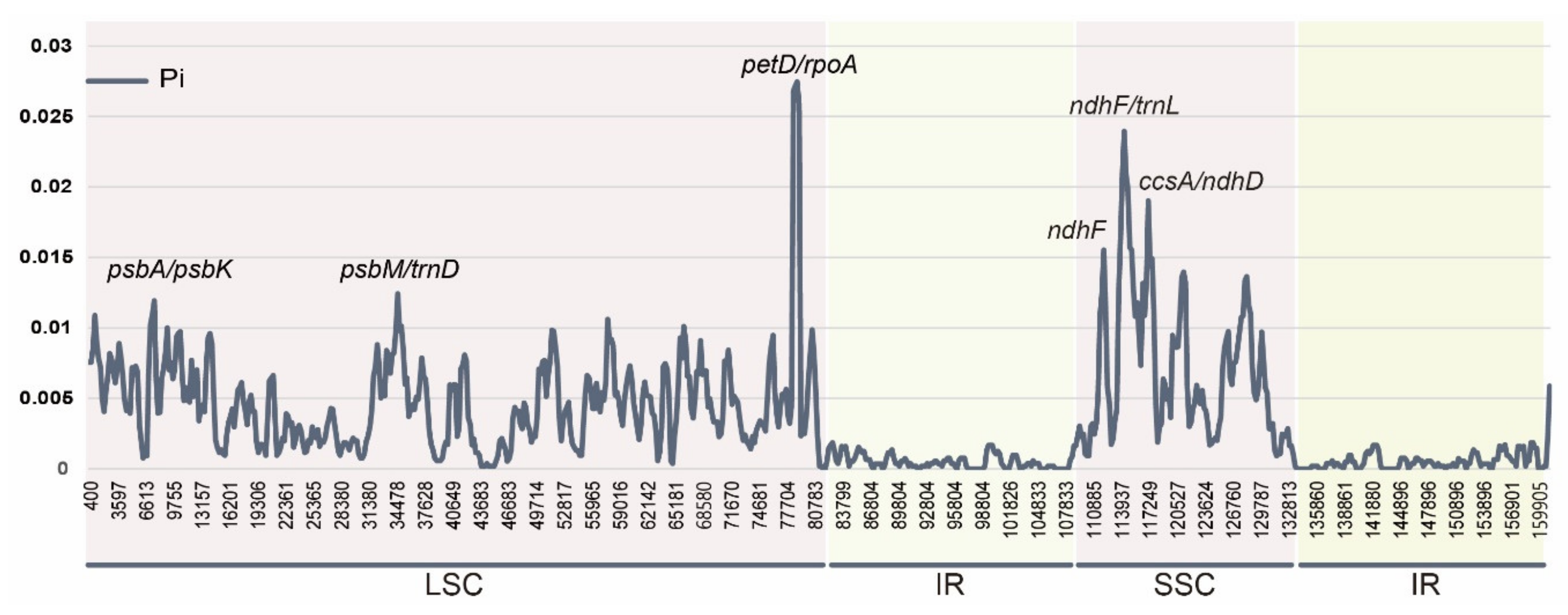{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. General Chloroplast Genome Features
2.2. Comparative Analyses
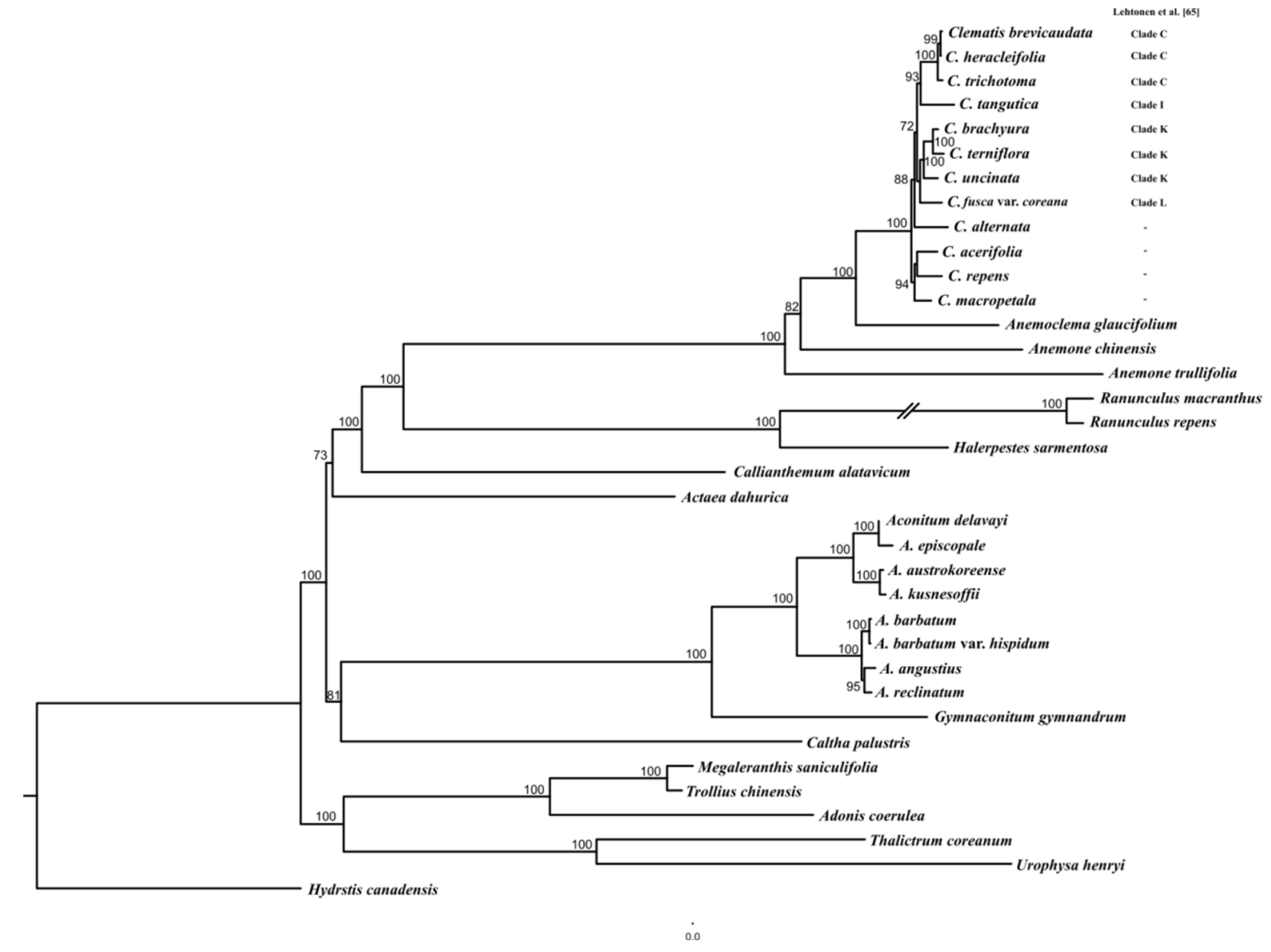
2.3. Phylogenetic Relationships Analysis
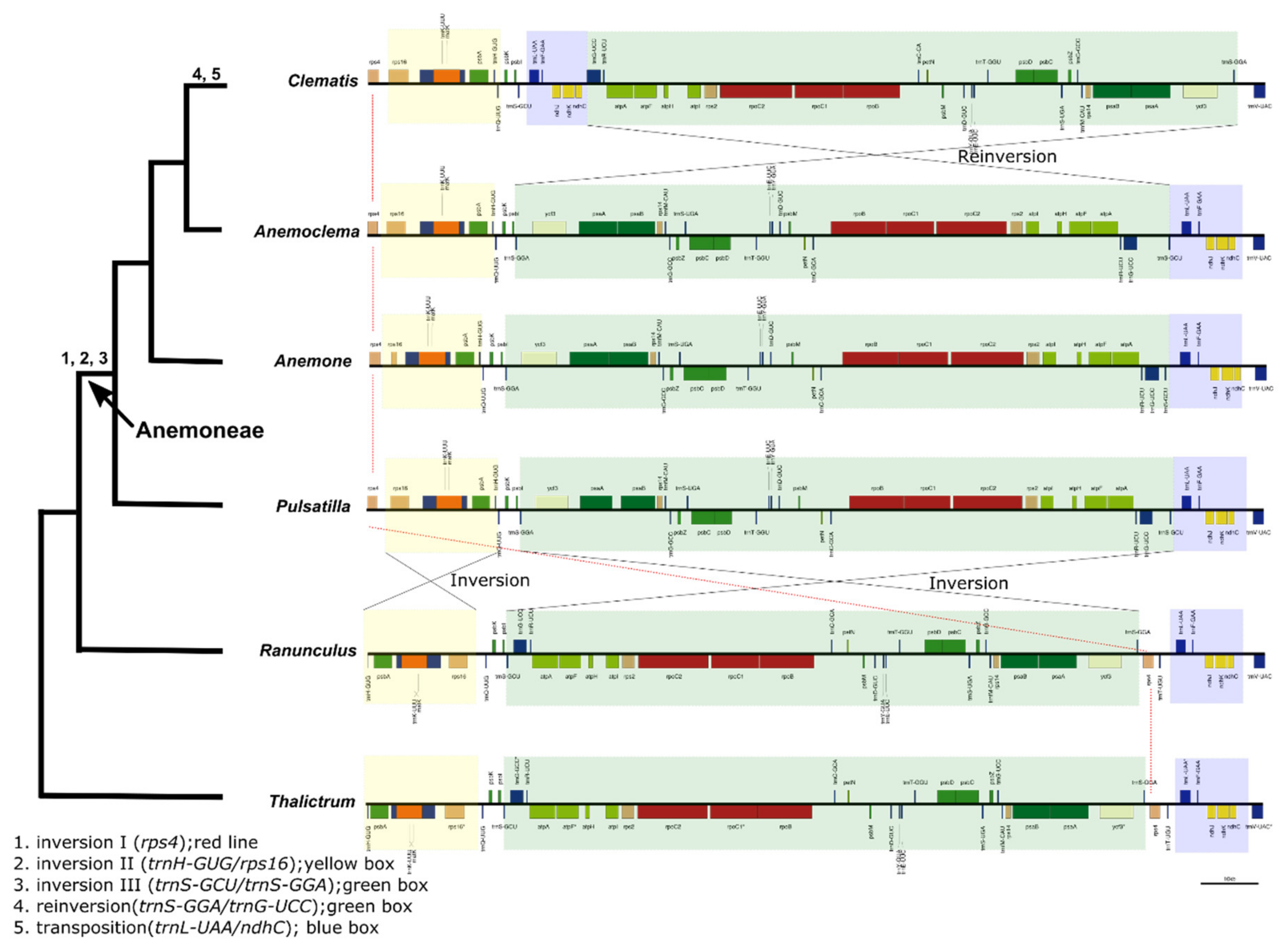
2.4. Intergeneric Genome Comparative Analyses
2.5. Comparison of Substituion Rates of Genes among Clematis, Anemoneae, and Ranunculaceae
3. Discussion
4. Materials and Methods
4.1. Taxon Sampling, DNA Extraction, Chloroplast Genome Sequencing, and Characterization
4.2. Interspecific Genome Comparative Analyses
4.3. Phylogenetic Analysis
4.4. Substitution Rate Estimation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mardis, E.R. DNA sequencing technologies: 2006–2016. Nat. Protoc. 2017, 12, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Wang, L.; You, F.M.; Rodriguez, J.C.; Deal, K.R.; Chen, L.; Li, J.; Chakraborty, S.; Balan, B.; Jiang, C.-Z. Sequencing a Juglans regia × J. microcarpa hybrid yields high-quality genome assemblies of parental species. Hortic. Res. 2019, 6, 55. [Google Scholar] [CrossRef]
- Ha, Y.-H.; Kim, C.; Choi, K.; Kim, J.-H. Molecular phylogeny and dating of Forsythieae (Oleaceae) provide insight into the Miocene history of Eurasian temperate shrubs. Front. Plant Sci. 2018, 9, 99. [Google Scholar] [CrossRef]
- Velasco, R.; Zharkikh, A.; Affourtit, J.; Dhingra, A.; Cestaro, A.; Kalyanaraman, A.; Fontana, P.; Bhatnagar, S.K.; Troggio, M.; Pruss, D. The genome of the domesticated apple (Malus × domestica Borkh.). Nat. Genet. 2010, 42, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; An, B.; Park, S. Recurrent gene duplication in the angiosperm tribe Delphinieae (Ranunculaceae) inferred from intracellular gene transfer events and heteroplasmic mutations in the plastid matK gene. Sci. Rep. 2020, 10, 2720. [Google Scholar] [CrossRef] [PubMed]
- Howe, C.J.; Barbrook, A.C.; Koumandou, V.L.; Nisbet, R.E.R.; Symington, H.A.; Wightman, T.F. Evolution of the chloroplast genome. Philos. Trans. R. Soc. London Ser. B Biol. Sci. 2003, 358, 99–107. [Google Scholar] [CrossRef]
- Sugiura, M. The chloroplast genome. In 10 Years Plant Molecular Biology; Schilperoort, R.A., Dure, L., Eds.; Springer: Dordrecht, the Netherlands, 1992; pp. 149–168. [Google Scholar]
- Wicke, S.; Schneeweiss, G.M.; Depamphilis, C.W.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Funk, H.T.; Berg, S.; Krupinska, K.; Maier, U.G.; Krause, K. Complete DNA sequences of the plastid genomes of two parasitic flowering plant species, Cuscuta reflexa and Cuscuta gronovii. BMC Plant Biol. 2007, 7, 45. [Google Scholar] [CrossRef] [PubMed]
- Wicke, S.; Müller, K.F.; de Pamphilis, C.W.; Quandt, D.; Wickett, N.J.; Zhang, Y.; Renner, S.S.; Schneeweiss, G.M. Mechanisms of functional and physical genome reduction in photosynthetic and nonphotosynthetic parasitic plants of the broomrape family. Plant Cell 2013, 25, 3711–3725. [Google Scholar] [CrossRef]
- Saski, C.; Lee, S.-B.; Daniell, H.; Wood, T.C.; Tomkins, J.; Kim, H.-G.; Jansen, R.K. Complete chloroplast genome sequence of Glycine max and comparative analyses with other legume genomes. Plant Mol. Biol. 2005, 59, 309–322. [Google Scholar] [CrossRef]
- Schwarz, E.N.; Ruhlman, T.A.; Sabir, J.S.M.; Hajrah, N.H.; Alharbi, N.S.; Al-Malki, A.L.; Bailey, C.D.; Jansen, R.K. Plastid genome sequences of legumes reveal parallel inversions and multiple losses of rps16 in papilionoids. J. Syst. Evol. 2015, 53, 458–468. [Google Scholar] [CrossRef]
- Dugas, D.V.; Hernandez, D.; Koenen, E.J.M.; Schwarz, E.; Straub, S.; Hughes, C.E.; Jansen, R.K.; Nageswara-Rao, M.; Staats, M.; Trujillo, J.T. Mimosoid legume plastome evolution: IR expansion, tandem repeat expansions, and accelerated rate of evolution in clpP. Sci. Rep. 2015, 5, 16958. [Google Scholar] [CrossRef]
- Zhu, A.; Guo, W.; Gupta, S.; Fan, W.; Mower, J.P. Evolutionary dynamics of the plastid inverted repeat: The effects of expansion, contraction, and loss on substitution rates. New Phytol. 2016, 209, 1747–1756. [Google Scholar] [CrossRef]
- Weng, M.L.; Ruhlman, T.A.; Jansen, R.K. Expansion of inverted repeat does not decrease substitution rates in Pelargonium plastid genomes. New Phytol. 2017, 214, 842–851. [Google Scholar] [CrossRef]
- Magee, A.M.; Aspinall, S.; Rice, D.W.; Cusack, B.P.; Sémon, M.; Perry, A.S.; Stefanović, S.; Milbourne, D.; Barth, S.; Palmer, J.D. Localized hypermutation and associated gene losses in legume chloroplast genomes. Genome Res. 2010, 20, 1700–1710. [Google Scholar] [CrossRef] [PubMed]
- Knox, E.B. The dynamic history of plastid genomes in the Campanulaceae sensu lato is unique among angiosperms. Proc. Natl. Acad. Sci. USA 2014, 111, 11097–11102. [Google Scholar] [CrossRef] [PubMed]
- Yi, D.-K.; Lee, H.-L.; Sun, B.-Y.; Chung, M.Y.; Kim, K.-J. The complete chloroplast DNA sequence of Eleutherococcus senticosus (Araliaceae); comparative evolutionary analyses with other three asterids. Mol. Cells 2012, 33, 497–508. [Google Scholar] [CrossRef]
- Hirao, T.; Watanabe, A.; Kurita, M.; Kondo, T.; Takata, K. Complete nucleotide sequence of the Cryptomeria japonica D. Don. chloroplast genome and comparative chloroplast genomics: Diversified genomic structure of coniferous species. BMC Plant Biol. 2008, 8, 70. [Google Scholar] [CrossRef]
- Chumley, T.W.; Palmer, J.D.; Mower, J.P.; Fourcade, H.M.; Calie, P.J.; Boore, J.L.; Jansen, R.K. The complete chloroplast genome sequence of Pelargonium × hortorum: Organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol. Biol. Evol. 2006, 23, 2175–2190. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Yang, B.; Zhu, W.; Sun, L.; Tian, J.; Wang, X. The complete chloroplast genome sequence of Mahonia bealei (Berberidaceae) reveals a significant expansion of the inverted repeat and phylogenetic relationship with other angiosperms. Gene 2013, 528, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Sinn, B.T.; Sedmak, D.D.; Kelly, L.M.; Freudenstein, J.V. Total duplication of the small single copy region in the angiosperm plastome: Rearrangement and inverted repeat instability in Asarum. Am. J. Bot. 2018, 105, 71–84. [Google Scholar] [CrossRef]
- Downie, S.R.; Palmer, J.D. Use of chloroplast DNA rearrangements in reconstructing plant phylogeny. In Molecular Systematics of Plants; Soltis, P.S., Soltis, D.E., Doyle, J.J., Eds.; Springer: Boston, MA, USA, 1992; pp. 14–35. [Google Scholar]
- Blazier, J.C.; Jansen, R.K.; Mower, J.P.; Govindu, M.; Zhang, J.; Weng, M.-L.; Ruhlman, T.A. Variable presence of the inverted repeat and plastome stability in Erodium. Ann. Bot. 2016, 117, 1209–1220. [Google Scholar] [CrossRef]
- Zhai, W.; Duan, X.; Zhang, R.; Guo, C.; Li, L.; Xu, G.; Shan, H.; Kong, H.; Ren, Y. Chloroplast genomic data provide new and robust insights into the phylogeny and evolution of the Ranunculaceae. Mol. Phylogenet. Evol. 2019, 135, 12–21. [Google Scholar] [CrossRef]
- Tamura, M. Angiospermae: Ordnung Ranunculales, Fam. Ranunculaceae. In Die Natürlichen Pflanzenfamilien; Hiepko, P., Ed.; Duncker and Humblot: Berlin, Germany, 1995; Volume 17, pp. 324–349. [Google Scholar]
- Emadzade, K.; Lehnebach, C.; Lockhart, P.; Hörandl, E. A molecular phylogeny, morphology and classification of genera of Ranunculeae (Ranunculaceae). Taxon 2010, 59, 809–828. [Google Scholar] [CrossRef]
- Cossard, G.; Sannier, J.; Sauquet, H.; Damerval, C.; de Craene, L.R.; Jabbour, F.; Nadot, S. Subfamilial and tribal relationships of Ranunculaceae: Evidence from eight molecular markers. Plant Syst. Evol. 2016, 302, 419–431. [Google Scholar] [CrossRef]
- Gregory, W.C. Phylogenetic and cytological studies in the Ranunculaceae Juss. Trans. Am. Philos. Soc. 1941, 31, 443–521. [Google Scholar] [CrossRef]
- Hutchinson, J. Contributions towards a phylogenetic classification of flowering plants. Bull. Misc. Inf. 1923, 9, 65–89. [Google Scholar]
- Xie, L.; Wen, J.; Li, L.; Xie, L.; Wen, J.; Li, L. Phylogenetic analyses of Clematis (Ranunculaceae) based on sequences of nuclear ribosomal ITS and three plastid regions. Syst. Bot. 2011, 36, 907–921. [Google Scholar] [CrossRef]
- Park, S.; Jansen, R.K.; Park, S. Complete plastome sequence of Thalictrum coreanum (Ranunculaceae) and transfer of the rpl32 gene to the nucleus in the ancestor of the subfamily Thalictroideae. BMC Plant Biol. 2015, 15, 40. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, Q.; Li, Y.; Qian, J.; Han, J. Chloroplast genome of Aconitum barbatum var. puberulum (Ranunculaceae) derived from CCS reads using the PacBio RS platform. Front. Plant Sci. 2015, 6, 42. [Google Scholar] [CrossRef]
- Kong, H.; Liu, W.; Yao, G.; Gong, W. A comparison of chloroplast genome sequences in Aconitum (Ranunculaceae): A traditional herbal medicinal genus. Peer J. 2017, 5, e4018. [Google Scholar] [CrossRef][Green Version]
- Park, S.; Park, S. Large-scale phylogenomics reveals ancient introgression in Asian Hepatica and new insights into the origin of the insular endemic Hepatica maxima. Sci. Rep. 2020, 10, 16288. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Xing, Y.; Xu, L.; Bao, G.; Zhan, Z.; Yang, Y.; Wang, J. Comparative analysis of the complete chloroplast genome sequences of six species of Pulsatilla Miller, Ranunculaceae. Chin. Med. 2019, 14, 53. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yang, B.; Chen, Q.; Zhu, W.; Ma, J.; Tian, J. The complete chloroplast genome sequence of Clematis terniflora DC. (Ranunculaceae). Mitochondr. DNA A 2016, 27, 2470–2472. [Google Scholar] [CrossRef] [PubMed]
- Park, K.T.; Park, S. Complete chloroplast genome of Clematis fusca var. coreana (Ranunculaceae). Mitochondr. DNA A 2016, 27, 4056–4058. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; He, J.; Ding, C.; Lyu, R.; Pei, L.; Cheng, J.; Xie, L. Comparative analysis of complete chloroplast genomes of Anemoclema, Anemone, Pulsatilla, and Hepatica revealing structural variations among genera in tribe Anemoneae (Ranunculaceae). Front. Plant Sci. 2018, 9, 1097. [Google Scholar] [CrossRef]
- He, J.; Yao, M.; Lyu, R.-D.; Lin, L.-L.; Liu, H.-J.; Pei, L.-Y.; Yan, S.-X.; Xie, L.; Cheng, J. Structural variation of the complete chloroplast genome and plastid phylogenomics of the genus Asteropyrum (Ranunculaceae). Sci. Rep. 2019, 9, 15285. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Chen, L.; Song, L.; Fei, W.; He, M.; He, C.; Yin, Z. Diterpenoid alkaloids from the root of Aconitum sinchiangense WT Wang with their antitumor and antibacterial activities. Nat. Prod. Res. 2017, 31, 2016–2023. [Google Scholar] [CrossRef]
- Park, I.; Kim, W.; Yang, S.; Yeo, S.-M.; Li, H.; Moon, B.C. The complete chloroplast genome sequence of Aconitum coreanum and Aconitum carmichaelii and comparative analysis with other Aconitum species. PLoS ONE 2017, 12, e0184257. [Google Scholar] [CrossRef]
- Chaudhary, M.I.; Qing, H.; Xiao, P.G.; Cheng, Y. Clematis huchouensis T AMURA: A traditional Chinese herbal medicine and its quality control using a high performance liquid chromatography technique. Biol. Pharm. Bull. 2007, 30, 165–168. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yan, S.; Wang, P.; He, S.; Liu, B.; Li, Y. Ornamental characters of plants of Clematis L. from Henan and their utilization. Henan Sci. 2005, 23, 218–220. [Google Scholar]
- Bender, S. Clematis. In The Southern Living Garden Book; Bender, S., Ed.; Southern Living Magazine: Birmingham, AL, USA, 2004; pp. 0–250. [Google Scholar]
- Wen-Tsai, W.; Lei, X.I.E. A revision of Clematis sect. Tubulosae (Ranunculaceae). J. Syst. Evol. 2006, 45, 425–457. [Google Scholar]
- Jiang, N.; Zhou, Z.; Yang, J.-B.; Zhang, S.-D.; Guan, K.-Y.; Tan, Y.-H.; Yu, W.-B. Phylogenetic reassessment of tribe Anemoneae (Ranunculaceae): Non-monophyly of Anemone sl revealed by plastid datasets. PLoS ONE 2017, 12, e0174792. [Google Scholar] [CrossRef]
- Johansson, J.T. A revised chloroplast DNA phylogeny of the Ranunculaceae. In Systematics and Evolution of the Ranunculiflorae; Jensen, U., Kadereit, J.W., Eds.; Springer: Vienna, Austria, 1995; pp. 253–261. [Google Scholar]
- Wang, W.; Lu, A.-M.; Ren, Y.; Endress, M.E.; Chen, Z.-D. Phylogeny and classification of Ranunculales: Evidence from four molecular loci and morphological data. Perspect. Plant Ecol. Evol. Syst. 2009, 11, 81–110. [Google Scholar] [CrossRef]
- Liu, H.-J.; Ding, C.-H.; He, J.; Cheng, J.; Pei, L.Y.; Xie, L. Complete chloroplast genomes of Archiclematis, Naravelia and Clematis (Ranunculaceae), and their phylogenetic implications. Phytotaxa 2018, 343, 214–226. [Google Scholar] [CrossRef]
- Korea National Arboretum. Checklist of Vascular Plants in Korea; Korea National Arboretum: Pocheon, Korea, 2017; pp. 156–159. [Google Scholar]
- Oh, H.S. An anatomical study of Clematis in Korea. Korean J. Taxon. 1971, 3, 1–2. [Google Scholar] [CrossRef]
- Lehtonen, S.; Christenhusz, M.J..; Falck, D. Sensitive phylogenetics of Clematis and its position in Ranunculaceae. Bot. J. Linn. Soc. 2016, 182, 825–867. [Google Scholar] [CrossRef]
- Kubitzki, K.; Rohwer, J.G.; Bittrich, V. The Families and Genera of Vascular Plants: Volume II Flowering Plants: Dicotyledons, Magnoliid, Hamamelid and Caryophyllid Families; Springer: Berlin/Heidelberg, Germany, 1993. [Google Scholar]
- Wu, Z.Y.; Raven, P.H.; Hong, D.Y. Flora of China, Aconitum Linnaeus; Science Press: Beijing, China; Missouri Botanical Garden Press: St. Louis, MI, USA, 2001; Volume 6, pp. 333–386. [Google Scholar]
- Iwatsuki, K.; Yamazaki, T.; Boufford, D.E.; Ohba, H. Flora of Japan, Vol. IIa: Angiospermae, Dicotyledoneae, Archichlamydeae (a); Kodansha: Tokyo, Japan, 2006. [Google Scholar]
- Chung, G.Y.; Chang, K.S.; Chung, J.-M.; Choi, H.J.; Paik, W.-K.; Hyun, J.-O. A checklist of endemic plants on the Korean Peninsula. Korean J. Plant Taxon. 2017, 47, 264–288. [Google Scholar] [CrossRef]
- Lee, H.-L.; Jansen, R.K.; Chumley, T.W.; Kim, K.-J. Gene relocations within chloroplast genomes of Jasminum and Menodora (Oleaceae) are due to multiple, overlapping inversions. Mol. Biol. Evol. 2007, 24, 1161–1180. [Google Scholar] [CrossRef]
- Hirose, T.; Ideue, T.; Wakasugi, T.; Sugiura, M. The chloroplast infA gene with a functional UUG initiation codon. FEBS Lett. 1999, 445, 169–172. [Google Scholar] [CrossRef]
- Millen, R.S.; Olmstead, R.G.; Adams, K.L.; Palmer, J.D.; Lao, N.T.; Heggie, L.; Kavanagh, T.A.; Hibberd, J.M.; Gray, J.C.; Morden, C.W. Many parallel losses of infA from chloroplast DNA during angiosperm evolution with multiple independent transfers to the nucleus. Plant Cell 2001, 13, 645–658. [Google Scholar] [CrossRef]
- Lee, S.R.; Kim, K.; Lee, B.Y.; Lim, C.E. Complete chloroplast genomes of all six Hosta species occurring in Korea: Molecular structures, comparative, and phylogenetic analyses. BMC Genom. 2019, 20, 833. [Google Scholar] [CrossRef]
- Goulding, S.E.; Wolfe, K.H.; Olmstead, R.G.; Morden, C.W. Ebb and flow of the chloroplast inverted repeat. Mol. Gen. Genet. MGG 1996, 252, 195–206. [Google Scholar] [CrossRef]
- Perry, A.S.; Wolfe, K.H. Nucleotide substitution rates in legume chloroplast DNA depend on the presence of the inverted repeat. J. Mol. Evol. 2002, 55, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, K.H.; Li, W.-H.; Sharp, P.M. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci. USA 1987, 84, 9054–9058. [Google Scholar] [CrossRef] [PubMed]
- Yi, D.-K.; Kim, K.-J. Complete chloroplast genome sequences of important oilseed crop Sesamum indicum L. PLoS ONE 2012, 7, e35872. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, D.; Sakaguchi, S.; Isagi, Y.; Setoguchi, H. Comparative chloroplast genomics of series Sakawanum in genus Asarum (Aristolochiaceae) to develop single nucleotide polymorphisms (SNPs) and simple sequence repeat (SSR) markers. J. For. Res. 2018, 23, 387–392. [Google Scholar] [CrossRef]
- Palmer, J.D.; Nugent, J.M.; Herbon, L.A. Unusual structure of geranium chloroplast DNA: A triple-sized inverted repeat, extensive gene duplications, multiple inversions, and two repeat families. Proc. Natl. Acad. Sci. USA 1987, 84, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Weng, M.-L.; Blazier, J.C.; Govindu, M.; Jansen, R.K. Reconstruction of the ancestral plastid genome in Geraniaceae reveals a correlation between genome rearrangements, repeats, and nucleotide substitution rates. Mol. Biol. Evol. 2014, 31, 645–659. [Google Scholar] [CrossRef]
- Sun, Y.; Moore, M.J.; Meng, A.; Soltis, P.S.; Soltis, D.E.; Li, J.; Wang, H. Complete plastid genome sequencing of Trochodendraceae reveals a significant expansion of the inverted repeat and suggests a Paleogene divergence between the two extant species. PLoS ONE 2013, 8, e60429. [Google Scholar] [CrossRef]
- Sloan, D.B.; Alverson, A.J.; Wu, M.; Palmer, J.D.; Taylor, D.R. Recent acceleration of plastid sequence and structural evolution coincides with extreme mitochondrial divergence in the angiosperm genus. Silene Genome Biol. Evol. 2012, 4, 294–306. [Google Scholar]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, W686–W689. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef]
- Mayor, C.; Brudno, M.; Schwartz, J.R.; Poliakov, A.; Rubin, E.M.; Frazer, K.A.; Pachter, L.S.; Dubchak, I. VISTA: Visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 2000, 16, 1046. [Google Scholar] [CrossRef] [PubMed]
- Brudno, M.; Do, C.B.; Cooper, G.M.; Kim, M.F.; Davydov, E.; Green, E.D.; Sidow, A.; Batzoglou, S. NISC comparative sequencing program. LAGAN and Multi-LAGAN: Efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 2003, 13, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Santorum, J.M.; Darriba, D.; Taboada, G.L.; Posada, D. jmodeltest. org: Selection of nucleotide substitution models on the cloud. Bioinformatics 2014, 30, 1310–1311. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, K.S.; Ha, Y.-H.; Gil, H.-Y.; Choi, K.; Kim, D.-K.; Oh, S.-H. Two Korean Endemic Clematis Chloroplast Genomes: Inversion, Reposition, Expansion of the Inverted Repeat Region, Phylogenetic Analysis, and Nucleotide Substitution Rates. Plants 2021, 10, 397. https://doi.org/10.3390/plants10020397
Choi KS, Ha Y-H, Gil H-Y, Choi K, Kim D-K, Oh S-H. Two Korean Endemic Clematis Chloroplast Genomes: Inversion, Reposition, Expansion of the Inverted Repeat Region, Phylogenetic Analysis, and Nucleotide Substitution Rates. Plants. 2021; 10(2):397. https://doi.org/10.3390/plants10020397
Chicago/Turabian StyleChoi, Kyoung Su, Young-Ho Ha, Hee-Young Gil, Kyung Choi, Dong-Kap Kim, and Seung-Hwan Oh. 2021. "Two Korean Endemic Clematis Chloroplast Genomes: Inversion, Reposition, Expansion of the Inverted Repeat Region, Phylogenetic Analysis, and Nucleotide Substitution Rates" Plants 10, no. 2: 397. https://doi.org/10.3390/plants10020397
APA StyleChoi, K. S., Ha, Y.-H., Gil, H.-Y., Choi, K., Kim, D.-K., & Oh, S.-H. (2021). Two Korean Endemic Clematis Chloroplast Genomes: Inversion, Reposition, Expansion of the Inverted Repeat Region, Phylogenetic Analysis, and Nucleotide Substitution Rates. Plants, 10(2), 397. https://doi.org/10.3390/plants10020397