
Genetic Mapping by Sequencing More Precisely Detects Loci Responsible for Anaerobic Germination Tolerance in Rice

 , and
, and
Abstract
:1. Introduction
2. Results
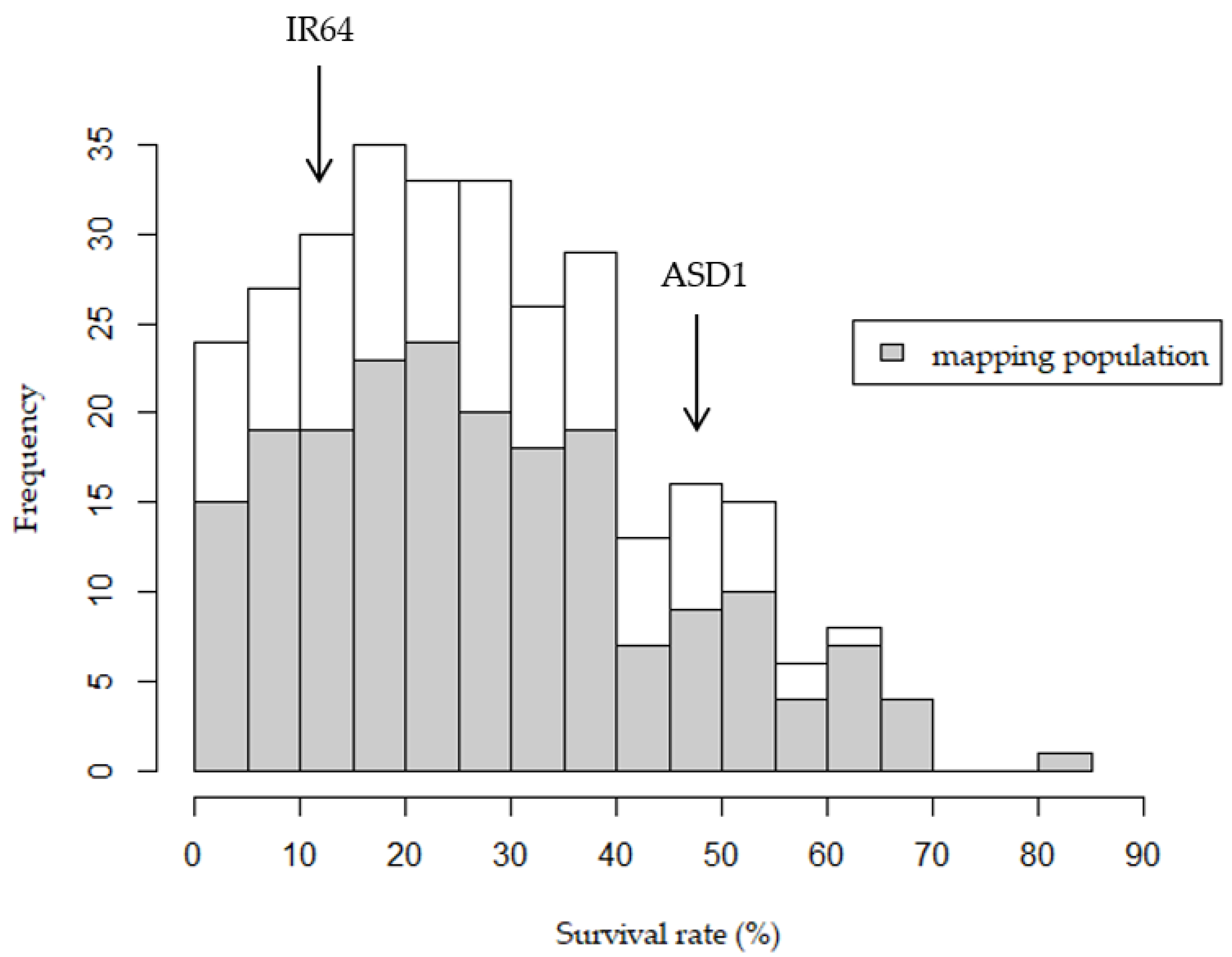
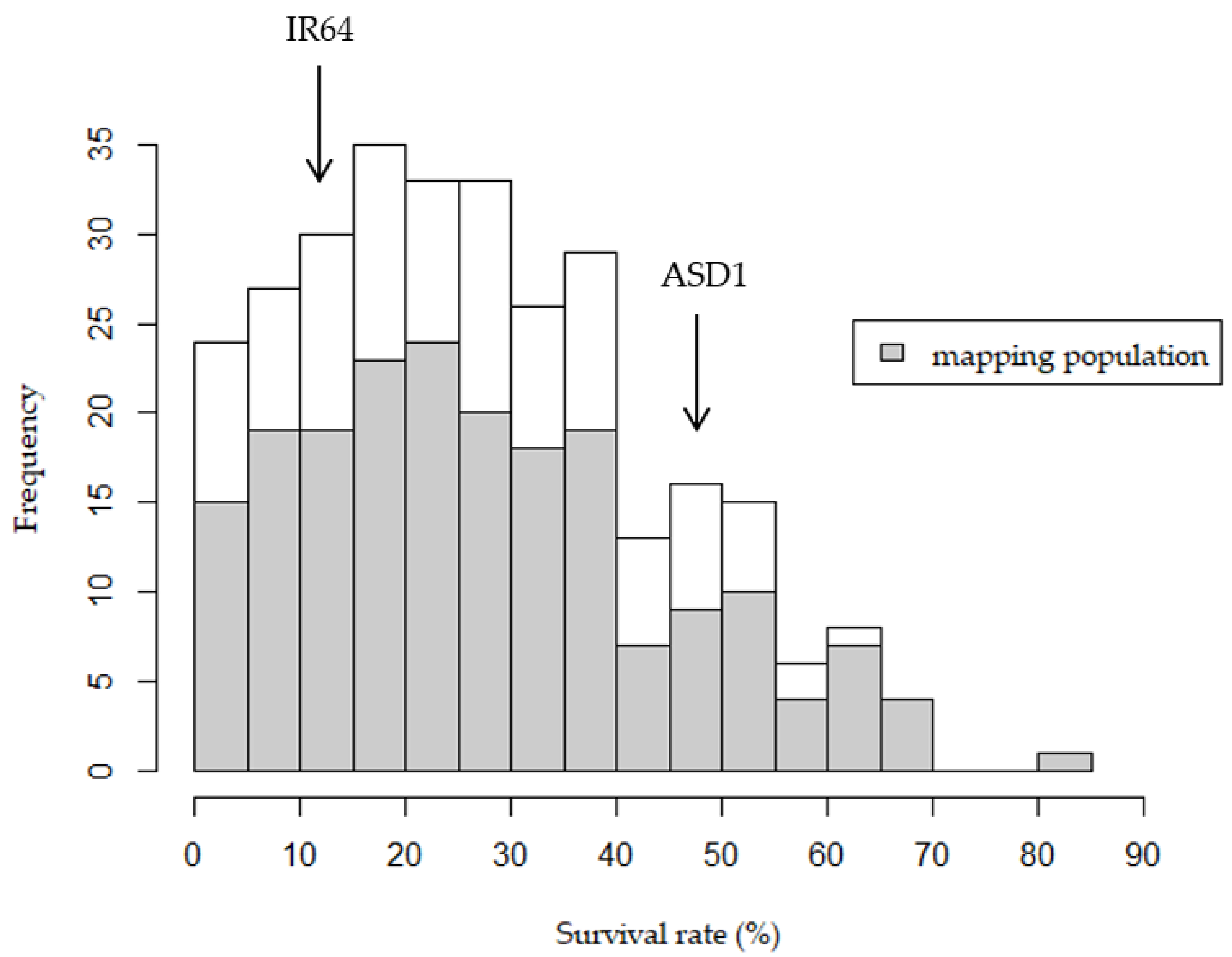
2.1. Phenotypic Performance
2.2. Genotyping
2.2.1. Library Preparation and Sequencing
2.2.2. Variant Detection
2.2.3. Variant and Sample Filtering
2.2.4. Adapting Genotyping Data for F2 Genetic Analysis
2.3. QTL Analysis
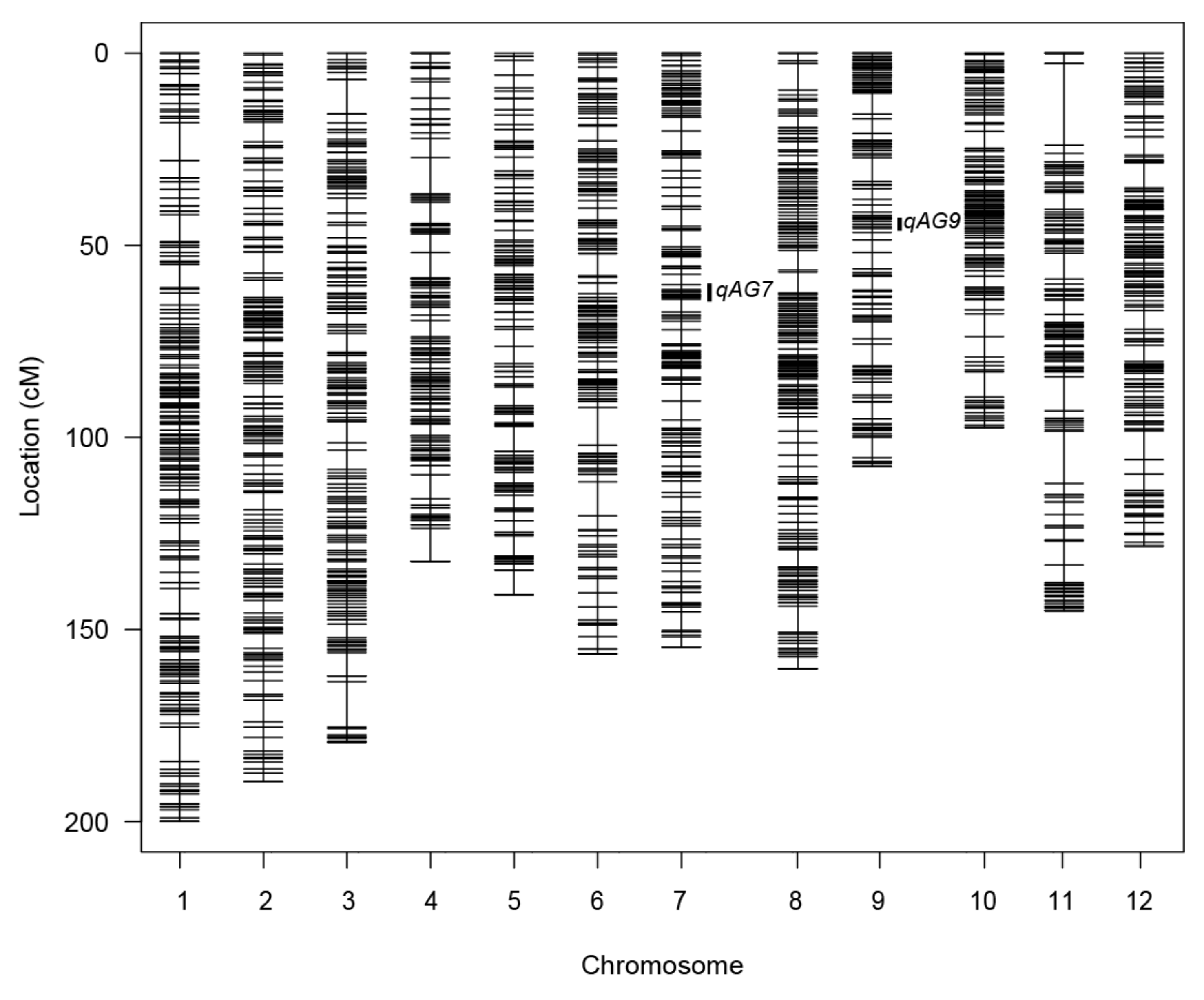
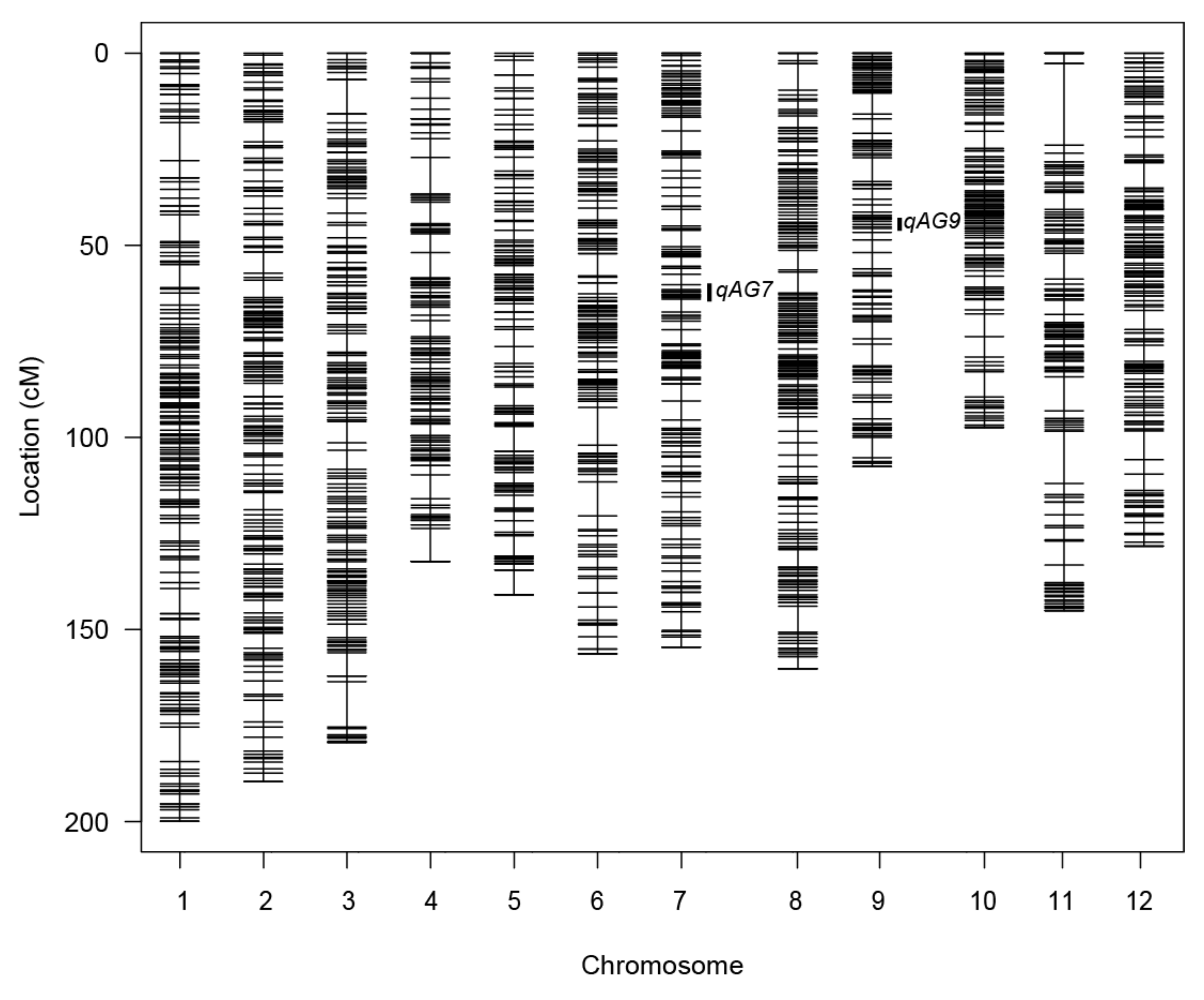
2.3.1. Genetic Map Estimation
2.3.2. QTL Detection
3. Discussion
4. Materials and Methods
4.1. Development of Mapping Population
4.2. Phenotyping
4.3. Genotyping
4.3.1. Library Preparation and Sequencing
4.3.2. Variant Detection
4.3.3. Variant and Sample Filtering
4.3.4. Adapting genotyping Data for F2 Genetics Analysis
4.4. QTL Analysis
4.4.1. Genetic Map Estimation
4.4.2. QTL Detection Using QGene Software
4.4.3. QTL Setection Using QTL Cartographer Software
4.5. QTL Comparisons
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pandey, S.; Mortimer, M.; Wade, L.; Tuong, T.; Lopez, K.; Hardy, B. Direct seeding: Research issues and opportunities. In Proceedings of the International Workshop on Direct Seeding in Asian Rice Systems: Strategic Research Issues and Opportunities, Bangkok, Thailand, 25–28 January 2000; International Rice Research Institute: Los Baños, Philippines, 2002; pp. 3–12. [Google Scholar]
- Angaji, S.A.; Septiningsih, E.M.; Mackill, D.J.; Ismail, A.M. QTLs associated with tolerance of flooding during germination in rice (Oryza sativa L.). Euphytica 2010, 172, 159–168. [Google Scholar] [CrossRef]
- Ismail, A.M.; Ella, E.S.; Vergara, G.V.; Mackill, D.J. Mechanisms associated with tolerance to flooding during germination and early seedling growth in rice (Oryza sativa). Ann. Bot. 2009, 103, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Ghosal, S.; Casal, C.; Quilloy, F.A.; Septiningsih, E.M.; Mendioro, M.S.; Dixit, S. Deciphering Genetics Underlying Stable Anaerobic Germination in Rice: Phenotyping, QTL Identification, and Interaction Analysis. Rice 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Alam, R.; Hummel, M.; Yeung, E.; Locke, A.M.; Ignacio, J.C.I.; Baltazar, M.D.; Jia, Z.; Ismail, A.M.; Septiningsih, E.M.; Bailey-Serres, J. Flood resilience loci SUBMERGENCE 1 and ANAEROBIC GERMINATION 1 interact in seedlings established underwater. Plant Direct 2020, 4, e00240. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Khan, M.I.R.; Entila, F.; Dixit, S.; Pompe, P.C.; Panna Ali, M.; Pittendrigh, B.; Septiningsih, E.M.; Ismail, A.M. Responses of AG1 and AG2 QTL introgression lines and seed pre-treatment on growth and physiological processes during anaerobic germination of rice under flooding. Sci. Rep. 2020, 10, 10214. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Khan, M.I.R.; Dixit, S.; Pompe, P.C.; Septiningsih, E.M.; Ismail, A.M. Growth, productivity and grain quality of AG1 and AG2 QTLs introgression lines under flooding in direct-seeded rice system. Field Crop. Res. 2020, 248, 107713. [Google Scholar] [CrossRef]
- Toledo, A.M.U.; Ignacio, J.C.I.; Casal, C.; Gonzaga, Z.J.; Mendioro, M.S.; Septiningsih, E.M. Development of Improved Ciherang-Sub1 Having Tolerance to Anaerobic Germination Conditions. Plant Breed. Biotechnol. 2015, 3, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Ghosal, S.; Quilloy, F.A.; Casal, C.; Septiningsih, E.M.; Mendioro, M.S.; Dixit, S. Trait-based mapping to identify the genetic factors underlying anaerobic germination of rice: Phenotyping, GXE, and QTL mapping. BMC Genet. 2020, 21, 6. [Google Scholar] [CrossRef]
- Miro, B.; Ismail, A.M. Tolerance of anaerobic conditions caused by flooding during germination and early growth in rice (Oryza sativa L.). Front. Plant Sci. 2013, 4, 269. [Google Scholar] [CrossRef] [Green Version]
- Septiningsih, E.M.; Ignacio, J.C.I.; Sendon, P.M.D.; Sanchez, D.L.; Ismail, A.M.; Mackill, D.J. QTL mapping and confirmation for tolerance of anaerobic conditions during germination derived from the rice landrace Ma-Zhan Red. Theor. Appl. Genet. 2013, 126, 1357–1366. [Google Scholar] [CrossRef]
- Baltazar, M.D.; Ignacio, J.C.I.; Thomson, M.J.; Ismail, A.M.; Mendioro, M.S.; Septiningsih, E.M. QTL mapping for tolerance of anaerobic germination from IR64 and the aus landrace Nanhi using SNP genotyping. Euphytica 2014, 197, 251–260. [Google Scholar] [CrossRef]
- Baltazar, M.D.; Ignacio, J.C.I.; Thomson, M.J.; Ismail, A.M.; Mendioro, M.S.; Septiningsih, E.M. QTL mapping for tolerance to anaerobic germination in rice from IR64 and the aus landrace Kharsu 80A. Breed. Sci. 2019, 69, 227–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kretzschmar, T.; Pelayo, M.A.F.; Trijatmiko, K.R.; Gabunada, L.F.M.; Alam, R.; Jimenez, R.; Mendioro, M.S.; Slamet-Loedin, I.H.; Sreenivasulu, N.; Bailey-Serres, J.; et al. A trehalose-6-phosphate phosphatase enhances anaerobic germination tolerance in rice. Nat. Plants 2015, 1. [Google Scholar] [CrossRef] [PubMed]
- Chamara, B.S.; Marambe, B.; Kumar, V.; Ismail, A.M.; Septiningsih, E.M.; Chauhan, B.S. Optimizing Sowing and Flooding Depth for Anaerobic Germination-Tolerant Genotypes to Enhance Crop Establishment, Early Growth, and Weed Management in Dry-Seeded Rice (Oryza sativa L.). Front. Plant Sci. 2018, 9, 1654. [Google Scholar] [CrossRef]
- Lal, B.; Gautam, P.; Nayak, A.K.; Raja, R.; Shahid, M.; Tripathi, R.; Singh, S.; Septiningsih, E.M.; Ismail, A.M. Agronomic manipulations can enhance the productivity of anaerobic tolerant rice sown in flooded soils in rainfed areas. Field Crop. Res. 2018, 220, 105–116. [Google Scholar] [CrossRef]
- Septiningsih, E.M.; Mackill, D.J. Genetics and breeding of flooding tolerance in rice. In Rice Genomics, Genetics and Breeding; Springer: Singapore, 2018; pp. 275–295. ISBN 9789811074615. [Google Scholar]
- Ling, J.; MingYu, H.; ChunMing, W.; JianMin, W. Quantitative trait loci and epistatic analysis of seed anoxia germinability in rice (Oryza sativa). Rice Sci. 2004, 11, 238–244. [Google Scholar]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 2008, 3. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [Green Version]
- Poland, J.A.; Rife, T.W. Genotyping-by-Sequencing for Plant Breeding and Genetics. Plant Genome 2012, 5, 92–102. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Wang, S.B.; Jian, J.; Geng, Q.C.; Wen, J.; Song, Q.; Wu, Z.; Li, G.J.; Liu, Y.Q.; Dunwell, J.M.; et al. Identification of domestication-related loci associated with flowering time and seed size in soybean with the RAD-seq genotyping method. Sci. Rep. 2015, 5, 9350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, N.; Li, M.; Hu, X.; Ma, Q.; Mu, Y.; Tan, Z.; Xia, Q.; Zhang, G.; Nian, H. Construction of high-density genetic map and QTL mapping of yield-related and two quality traits in soybean RILs population by RAD-sequencing. BMC Genom. 2017, 18, 466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Cheng, Y.; Yang, C.; Yang, C.; Mu, Y.; Xia, Q.; Ma, Q. QTL mapping for aluminum tolerance in RIL population of soybean (Glycine max L.) by RAD sequencing. PLoS ONE 2019, 14, e0223674. [Google Scholar] [CrossRef]
- Wang, L.; Conteh, B.; Fang, L.; Xia, Q.; Nian, H. QTL mapping for soybean (Glycine max L.) leaf chlorophyll-content traits in a genotyped RIL population by using RAD-seq based high-density linkage map. BMC Genom. 2020, 21, 739. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Baring, M.; Wang, S.; Septiningsih, E.M. Mapping QTLs for Leafspot Resistance in Peanut Using SNP-Based Next-Generation Sequencing Markers. Plant Breed. Biotechnol. 2017, 5, 115–122. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Baring, M.R.; Septiningsih, E.M. Mapping of quantitative trait loci for yield and grade related traits in peanut (Arachis hypogaea L.) using high-resolution SNP markers. Plant Breed. Biotechnol. 2018, 6, 454–462. [Google Scholar] [CrossRef]
- Liang, Y.; Cason, J.M.; Baring, M.R.; Septiningsih, E.M. Identification of QTLs associated with Sclerotinia blight resistance in peanut (Arachis hypogaea L.). Genet. Resour. Crop Evol. 2020, 68, 629–637. [Google Scholar] [CrossRef]
- Pan, L.; Wang, N.; Wu, Z.; Guo, R.; Yu, X.; Zheng, Y.; Xia, Q.; Gui, S.; Chen, C. A High Density Genetic Map Derived from RAD Sequencing and Its Application in QTL Analysis of Yield-Related Traits in Vigna unguiculata. Front. Plant Sci. 2017, 8, 1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumura, H.; Miyagi, N.; Taniai, N.; Fukushima, M.; Tarora, K.; Shudo, A.; Urasaki, N. Mapping of the Gynoecy in Bitter Gourd (Momordica charantia) Using RAD-Seq Analysis. PLoS ONE 2014, 9, e87138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, J.; Luo, S.; Niu, Y.; Huang, R.; Wen, Q.; Su, J.; Miao, N.; He, W.; Dong, Z.; Cheng, J.; et al. A RAD-Based Genetic Map for Anchoring Scaffold Sequences and Identifying QTLs in Bitter Gourd (Momordica charantia). Front. Plant Sci. 2018, 9, 477. [Google Scholar] [CrossRef] [PubMed]
- Laila, R.; Park, J.I.; Robin, A.H.K.; Natarajan, S.; Vijayakumar, H.; Shirasawa, K.; Isobe, S.; Kim, H.T.; Nou, I.S. Mapping of a novel clubroot resistance QTL using ddRAD-seq in Chinese cabbage (Brassica rapa L.). BMC Plant Biol. 2019, 19, 13. [Google Scholar] [CrossRef]
- Zhang, F.; Kang, J.; Long, R.; Yu, L.X.; Wang, Z.; Zhao, Z.; Zhang, T.; Yang, Q. High-density linkage map construction and mapping QTL for yield and yield components in autotetraploid alfalfa using RAD-seq. BMC Plant Biol. 2019, 19, 165. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Liu, H.; Yang, M.; Tao, Y.; Ma, H.; Wu, W.; Zuo, Y.; Zhao, Y. High-density genetic map construction and QTLs analysis of grain yield-related traits in Sesame (Sesamum indicum L.) based on RAD-Seq techonology. BMC Plant Biol. 2014, 14, 274. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, Z.; Du, X.; Yang, H.; Han, F.; Han, Y.; Yuan, F.; Zhang, L.; Peng, S.; Guo, E. A high-density genetic map and QTL analysis of agronomic traits in foxtail millet [Setaria italica (L.) P. Beauv.] using RAD-seq. PLoS ONE 2017, 12, e0179717. [Google Scholar] [CrossRef] [PubMed]
- Kajiya-Kanegae, H.; Takanashi, H.; Fujimoto, M.; Ishimori, M.; Ohnishi, N.; Wacera W, F.; Omollo, E.A.; Kobayashi, M.; Yano, K.; Nakano, M.; et al. RAD-seq-Based High-Density Linkage Map Construction and QTL Mapping of Biomass-Related Traits in Sorghum using the Japanese Landrace Takakibi NOG. Plant Cell Physiol. 2020, 61, 1262–1272. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Hu, Y.; Mao, B.; Xiang, H.; Shao, Y.; Pan, Y.; Sheng, X.; Li, Y.; Ni, X.; Xia, Y.; et al. Genetic analysis for rice grain quality traits in the YVB stable variant line using RAD-seq. Mol. Genet. Genom. 2016, 291, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.; Wu, D.; Chen, K. Mapping of QTLs controlling pre-harvest sprouting in the cross between japonica rice cultivars using RAD sequencing. Crop. Environ. Bioinform. 2014, 11, 11–25. [Google Scholar]
- Rebolledo, M.C.; Dingkuhn, M.; Clément-Vidal, A.; Rouan, L.; Luquet, D. Phenomics of rice early vigour and drought response: Are sugar related and morphogenetic traits relevant? Rice 2012, 5, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamauchi, M.; Winn, T. Rice seed vigor and seedling establishment in anaerobic soil. Crop Sci. 1996, 36, 680–686. [Google Scholar] [CrossRef]
- Furuta, T.; Ashikari, M.; Jena, K.K.; Doi, K.; Reuscher, S. Adapting genotyping-by-sequencing for rice F2 populations. G3 Genes Genomes Genet. 2017, 7, 881–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, J.C. QGENE: Software for marker-based genomic analysis and breeding. Mol. Breed. 1997, 3, 239–245. [Google Scholar] [CrossRef]
- Wang, S.; Basten, C.J.; Zeng, Z.-B. Windows QTL Cartographer 2.5; Department of Statistics, North Carolina State University: Raleigh, NC, USA, 2012; Available online: http://statgen.ncsu.edu/qtlcart/WQTLCart.htm (accessed on 12 December 2019).
- Kawahara, Y.; de la Bastide, M.; Hamilton, J.P.; Kanamori, H.; McCombie, W.R.; Ouyang, S.; Schwartz, D.C.; Tanaka, T.; Wu, J.; Zhou, S.; et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 2013, 6, 4. [Google Scholar] [CrossRef] [Green Version]
- Myint, O.; Hernandez, J.E.; Ismail, A.M.; Septiningsih, E.M. Mapping QTLs for tolerance to flooding during germination and early seedling growth in rice (Oryza sativa L.) using IR64 × TKM9 population. In Proceedings of the Seventh Agricultural Research Conference, Nay Pyi Taw, Myanmar, 10–11 January 2013; pp. 81–91. [Google Scholar]
- Biswas, J.K.; Yamauchi, M. Mechanism of seedling establishment of direct-seeded rice (Oryza sativa L.) under lowland conditions. Bot. Bull. Acad. Sin. 1997, 38, 29–32. [Google Scholar]
- Developer Core Team, R. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Coombes, N.E. DiGGer: DiGGer Design Generator under Correlation and Blocking; NSW DPI Biometrics: Sydney, Australia, 2011. [Google Scholar]
- Doyle, J.; Doyle, J.L. Genomic plant DNA preparation from fresh tissue-CTAB method. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner; LBNL Department of Energy Joint Genome Institute: Walnut Creek, CA, USA, 2014. [Google Scholar]
- Herten, K.; Hestand, M.S.; Vermeesch, J.R.; Van Houdt, J.K.J. GBSX: A toolkit for experimental design and demultiplexing genotyping by sequencing experiments. BMC Bioinf. 2015, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, T.; Wu, J.; Kanamori, H.; Katayose, Y.; Fujisawa, M.; Namiki, N.; Mizuno, H.; Yamamoto, K.; Antonio, B.A.; Baba, T.; et al. The map-based sequence of the rice genome. Nature 2005, 436, 793–800. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Broad Institute. Picard Toolkit. GitHub Repository. 2019. Available online: http://broadinstitute.github.io/picard/ (accessed on 12 December 2019).
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; Van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 2017, 201178. [Google Scholar] [CrossRef] [Green Version]
- Datta, K.; Gururaj, K.; Naik, M.; Narvaez, P.; Rutar, M. “GenomicsDB - Sparse Array Storage for Genomics.” 2019. Available online: https://www.genomicsdb.org/ (accessed on 12 December 2019).
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- RStudio Team. RStudio: Integrated Development Environment for R; RStudio PBC: Boston, MA, USA, 2019. [Google Scholar]
- Reuscher, S.; Furuta, T. ABHgenotypeR: Easy Visualization of ABH Genotypes. Available online: https://CRAN.R-project.org/package=ABHgenotypeR (accessed on 12 December 2019).
- Broman, K.W.; Wu, H.; Sen, S.; Churchill, G.A. R/qtl: {QTL} mapping in experimental crosses. Bioinformatics 2003, 19, 889–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Chr. | No. of Markers | Length (cM) | Ave. Interval (cM) | Max Interval (cM) |
|---|---|---|---|---|
| 1 | 202 | 199.9 | 1 | 9.9 |
| 2 | 193 | 189.6 | 1 | 5.7 |
| 3 | 191 | 179.4 | 0.9 | 11.8 |
| 4 | 128 | 132.3 | 1 | 9.6 |
| 5 | 136 | 141 | 1 | 6.5 |
| 6 | 166 | 156.3 | 0.9 | 9.8 |
| 7 | 161 | 154.6 | 1 | 5.2 |
| 8 | 212 | 160.2 | 0.8 | 6.9 |
| 9 | 116 | 107.6 | 0.9 | 6.2 |
| 10 | 134 | 97.5 | 0.7 | 6.6 |
| 11 | 127 | 145.1 | 1.2 | 21.2 |
| 12 | 155 | 128.3 | 0.8 | 7.4 |
| overall | 1921 | 1791.5 | 0.9 | 21.2 |
| QTL | Chr. | Markers | QGene IM | QGene CIM | QTL Cart IM | QTL Cart CIM | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Left flank | Peak | Right flank | LOD | R2 | Add | LOD | R2 | Add | LOD | R2 | Add | LOD | R2 | Add | ||
| qAG5 | 5 | S05_7886279 | S05_9596301 | S05_14907902 | - | - | - | 2.1 a | 5.4 | 2.2 | 2.1 | 0.0 | 0.0 | 2.9 | 0.0 | −0.5 |
| qAG6 | 6 | S06_10782720 | S06_11109090 | S06_11200565 | 2.3 | 5.1 | 5.1 | 2.2 | 5.0 | 2.4 | 2.6 | 2.7 | 3.9 | 2.6 | 0.3 | 1.3 |
| qAG7 | 7 | S07_6302957 | S07_7547815 | S07_7650364 | 5.6 | 12.1 | 8.4 | 7.1 | 15.1 | 8.0 | 5.5 | 11.7 | 8.0 | 7.5 | 10.7 | 8.0 |
| qAG9 | 9 | S09_12029625 | S09_12277535 | S09_12331208 | 12.0 | 24.3 | 13.0 | 15.0 | 29.4 | 13.0 | 12.0 | 30.2 | 14.0 | 14.5 | 27.6 | 15.0 |
| QTL | Source | Marker Position (Mb) | Range (Mb) | Marker | Software | IM | CIM | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Left Flank | Peak | Right Flank | LOD | R2 | LOD | R2 | |||||
| qAG-9-2 | Khao Hlan On | 11.75 | 12.31 | 12.55 | 0.80 | 136 SSR | QTLCart | 20.3 | 33.5 | 15.3 | 20.6 |
| qAG9a | ASD1 | 12.03 | 12.28 | 12.33 | 0.30 | RAD-seq | QGene | 12.0 | 24.3 | 15.0 | 29.4 |
| QTL | Source | Marker Position (Mb) | Range(Mb) | Marker | Software | IM | CIM | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Left Flank | Peak | Right Flank | LOD | R2 | LOD | R2 | |||||
| qAG7.1 | Ma-Zhan Red | 8.04 | 9.07 | 12.97 | 4.93 | 118 SSR, indel | QGene | 13.7 | 30.3 | 14.5 | 31.7 |
| qAGS-7 | TKM 9 | 7.61 | 11.09 | 12.78 | 5.17 | 91 SSR | QGene | 7.8 | 11.1 | 7.6 | 17.8 |
| qAG-7-1 | Khao Hlan On | 6.07 | 10.37 | 17.67 | 11.60 | 136 SSR | QTLCart | 5.1 | 9.9 | - | - |
| qAG7.1 | Kharsu 80A | 3.68 | 4.80 | 6.01 | 2.32 | BeadXpress | QGene | 5.5 | 17.5 | 4.8 | 15.4 |
| qAG7 | Nanhi | 4.80 | NAa | 18.48 | 13.68 | BeadXpress | QTLCart | 11.2 | 11.7 | 13.9 | 14.1 |
| qSUR7–1Rc238-SCR-21 | Kalarata | NA | 6.07 | NA | NA | KASP | IciM | 20.3 | 39.7 | NA | NA |
| qAG7b | ASD1 | 6.30 | 7.55 | 7.65 | 1.35 | RAD-seq | QGene | 5.6 | 12.1 | 7.1 | 15.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ignacio, J.C.I.; Zaidem, M.; Casal, C., Jr.; Dixit, S.; Kretzschmar, T.; Samaniego, J.M.; Mendioro, M.S.; Weigel, D.; Septiningsih, E.M. Genetic Mapping by Sequencing More Precisely Detects Loci Responsible for Anaerobic Germination Tolerance in Rice. Plants 2021, 10, 705. https://doi.org/10.3390/plants10040705
Ignacio JCI, Zaidem M, Casal C Jr., Dixit S, Kretzschmar T, Samaniego JM, Mendioro MS, Weigel D, Septiningsih EM. Genetic Mapping by Sequencing More Precisely Detects Loci Responsible for Anaerobic Germination Tolerance in Rice. Plants. 2021; 10(4):705. https://doi.org/10.3390/plants10040705
Chicago/Turabian StyleIgnacio, John Carlos I., Maricris Zaidem, Carlos Casal, Jr., Shalabh Dixit, Tobias Kretzschmar, Jaime M. Samaniego, Merlyn S. Mendioro, Detlef Weigel, and Endang M. Septiningsih. 2021. "Genetic Mapping by Sequencing More Precisely Detects Loci Responsible for Anaerobic Germination Tolerance in Rice" Plants 10, no. 4: 705. https://doi.org/10.3390/plants10040705
APA StyleIgnacio, J. C. I., Zaidem, M., Casal, C., Jr., Dixit, S., Kretzschmar, T., Samaniego, J. M., Mendioro, M. S., Weigel, D., & Septiningsih, E. M. (2021). Genetic Mapping by Sequencing More Precisely Detects Loci Responsible for Anaerobic Germination Tolerance in Rice. Plants, 10(4), 705. https://doi.org/10.3390/plants10040705