
Transcriptome Sequence Reveals Candidate Genes Involving in the Post-Harvest Hardening of Trifoliate Yam Dioscorea dumetorum


Abstract
:1. Introduction
2. Results
2.1. Descriptive Statistics of RNA-Seq Data
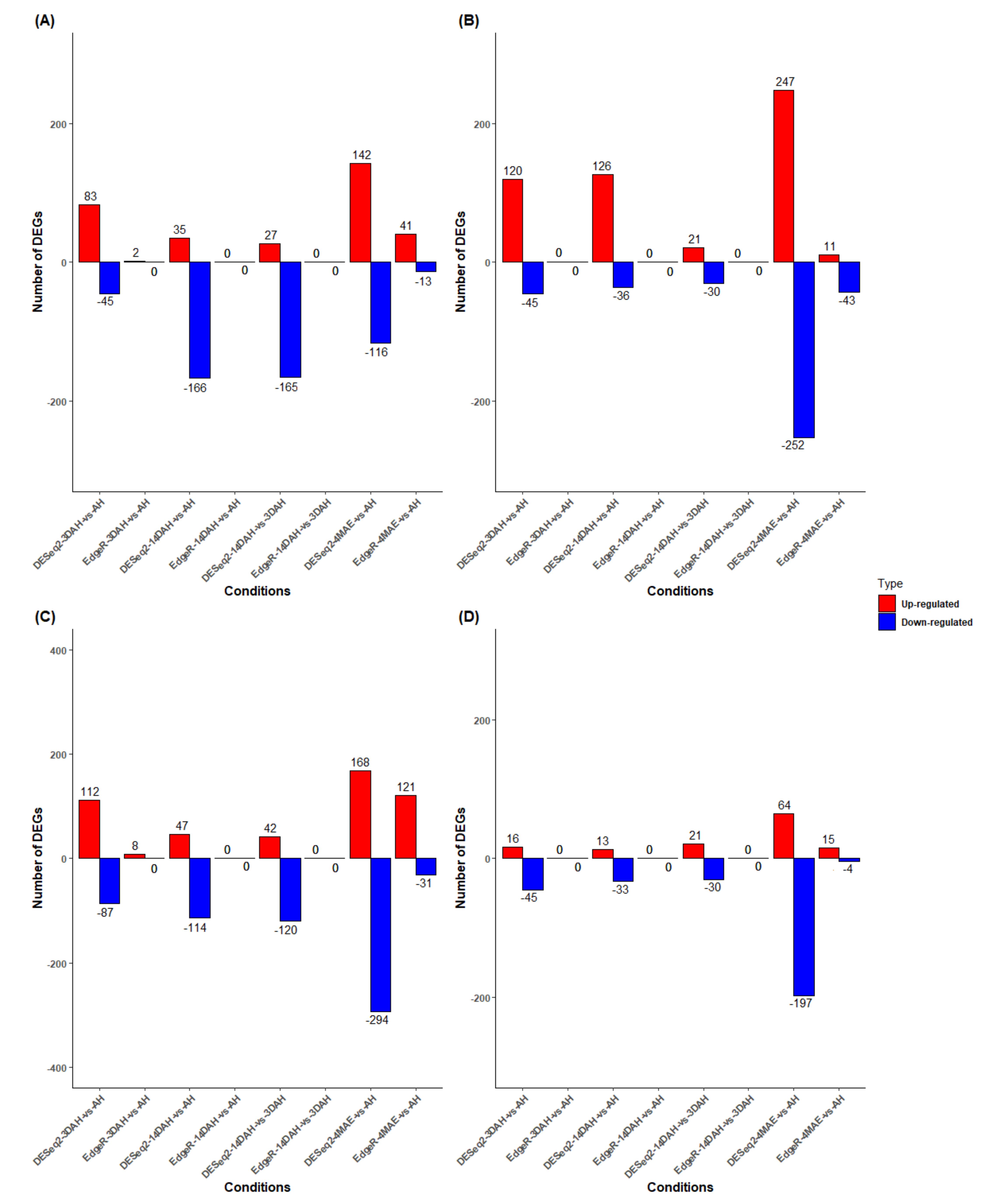
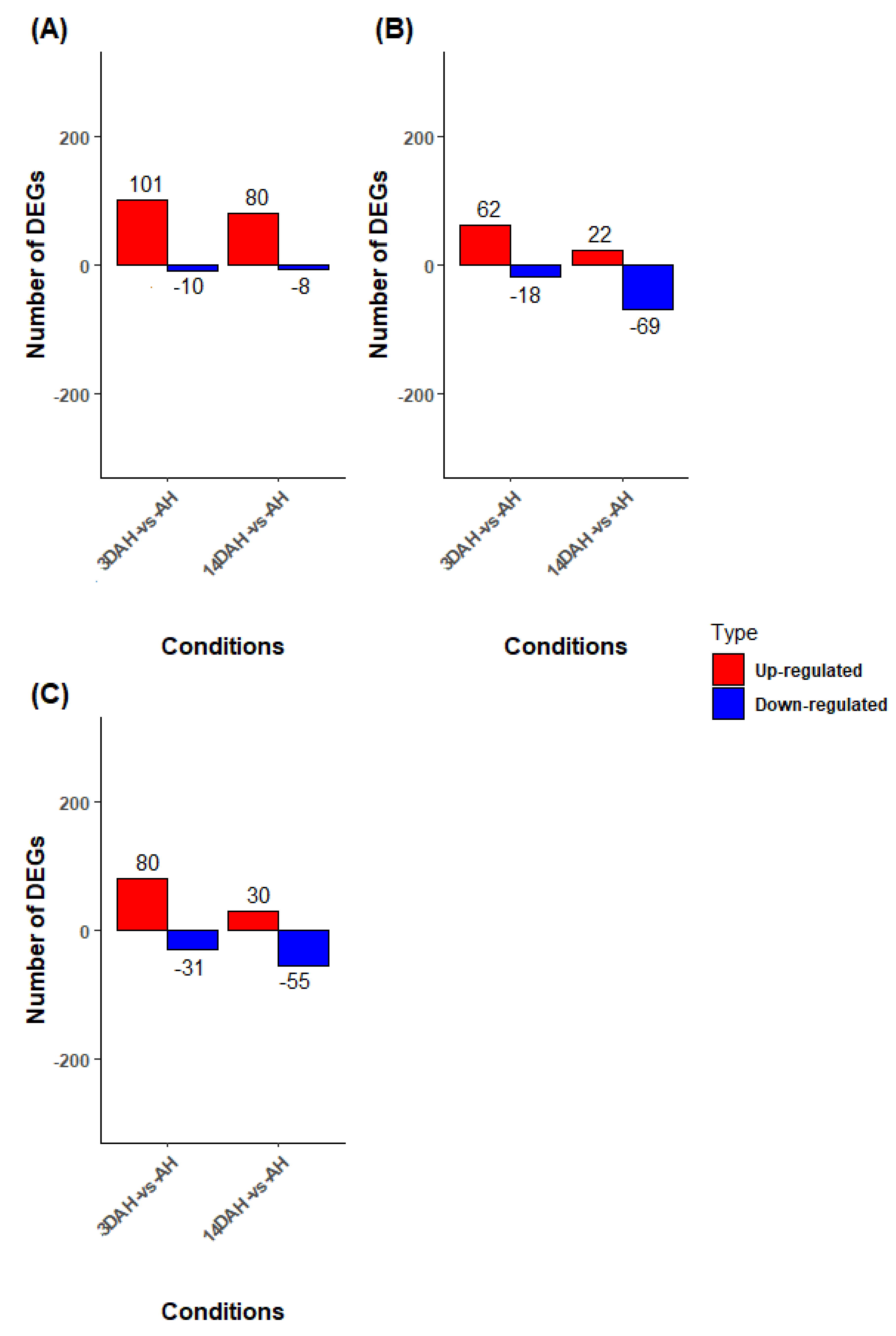
2.2. Differential Expression Analysis
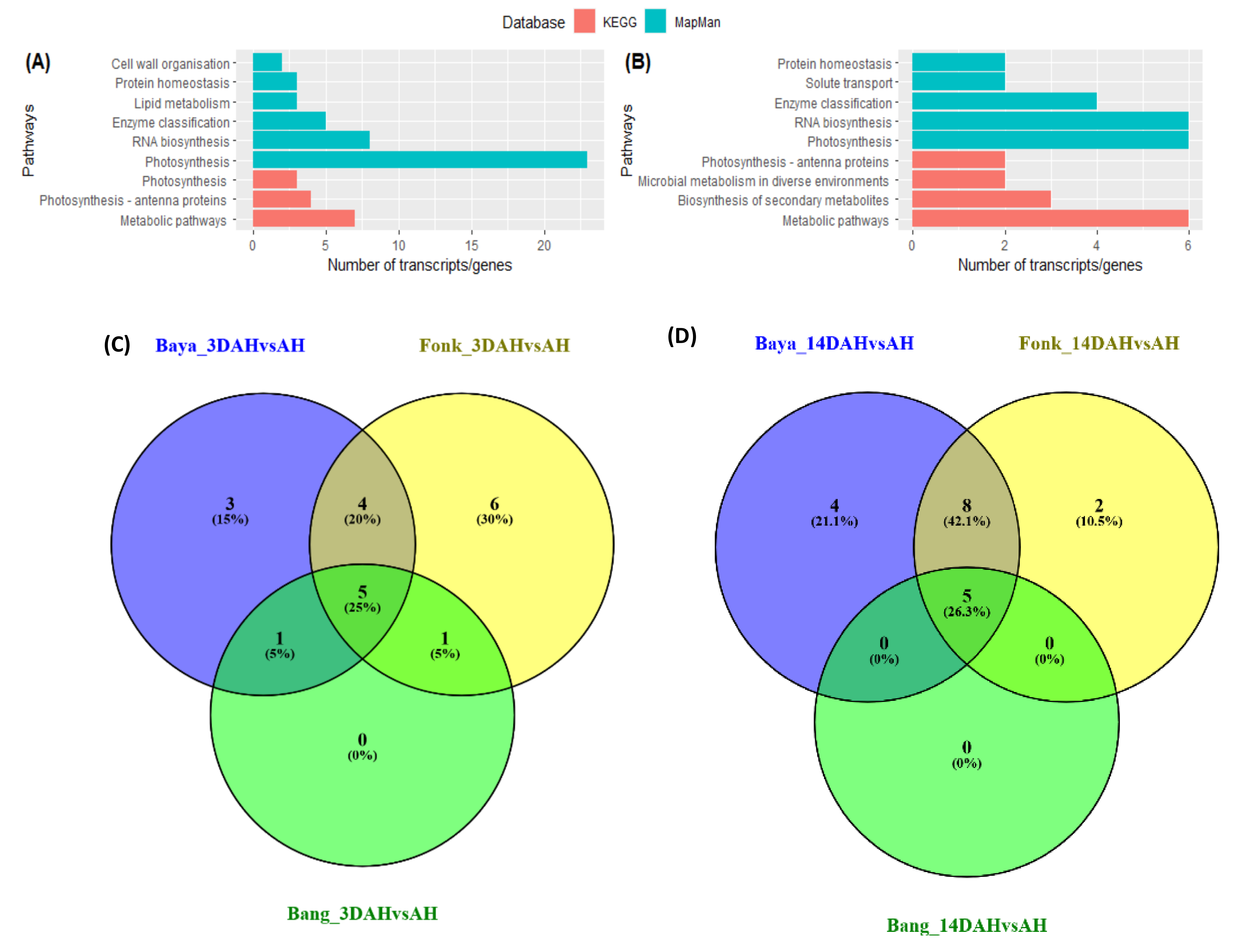
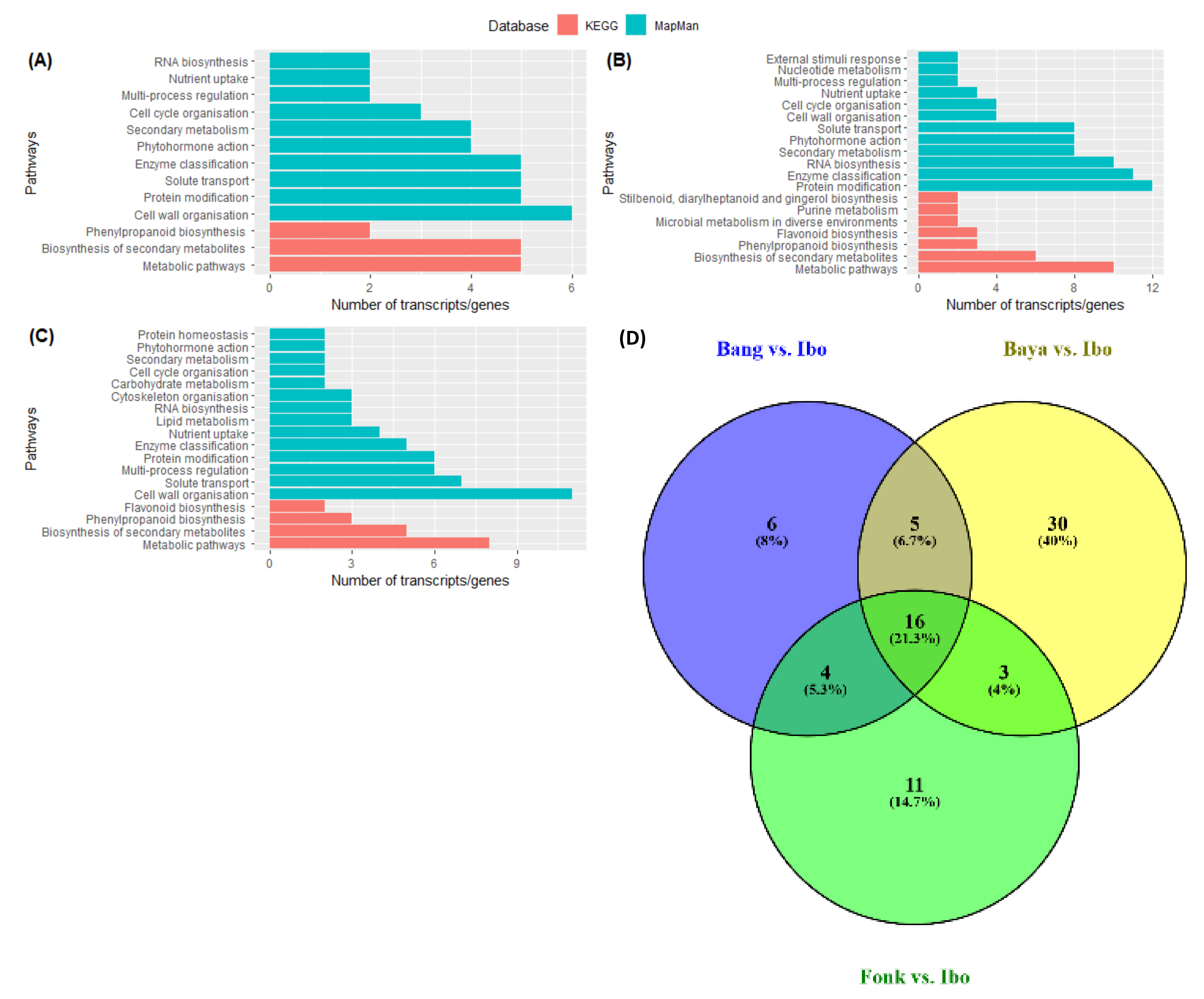
2.3. GO Enrichment and Functional Classification of DEGs with KEGG and Mapman
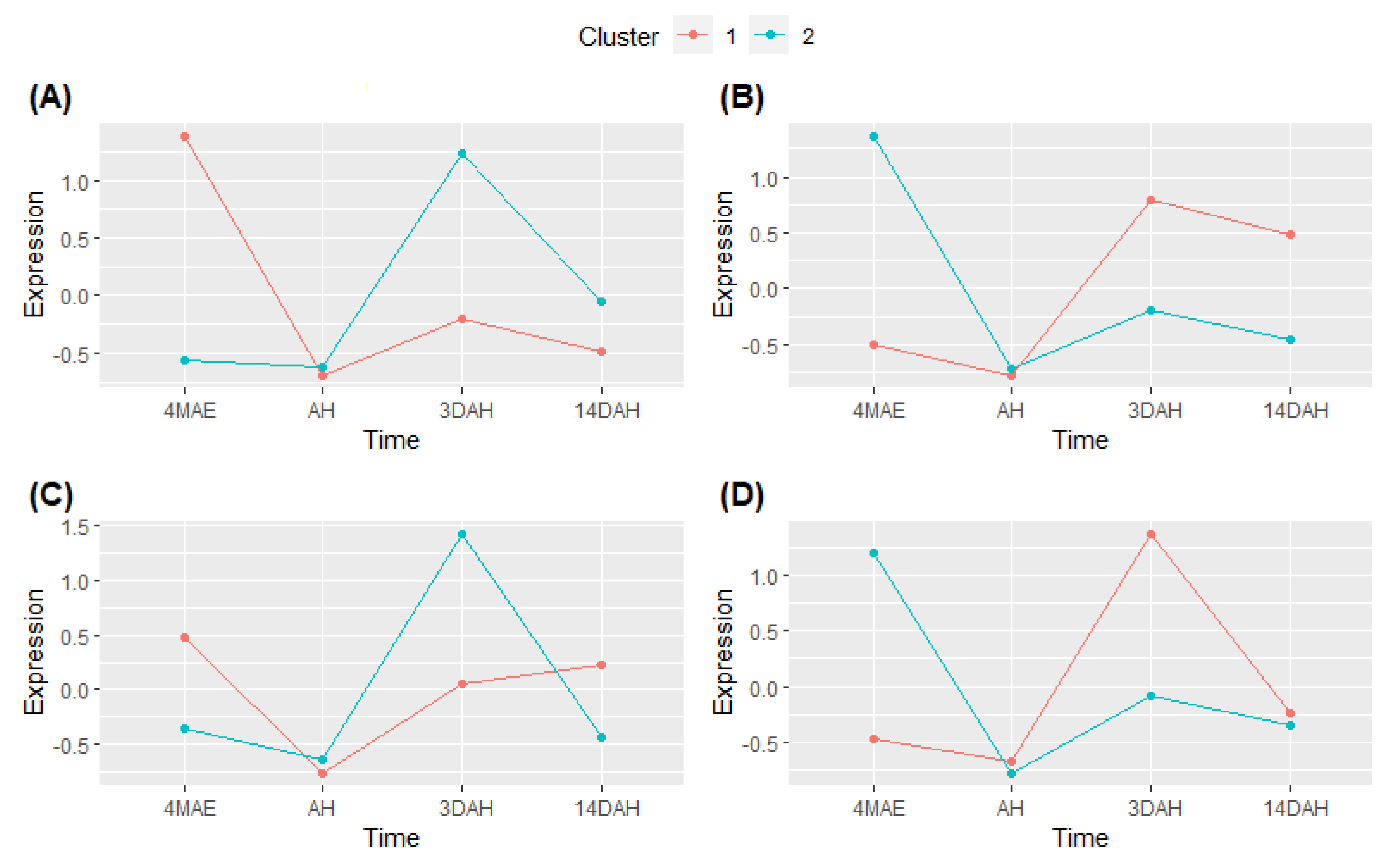
2.4. Cluster Expression Analysis
2.5. Comprehensive Analysis of Expression of Genes Potentially Involved in PHH
2.6. Comprehensive Difference between Harden and Non-Harden Accessions
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Sample Preparation
4.3. RNA-Seq Extraction
4.4. Library Construction and Illumina Sequencing
4.5. Data Processing and Functional Analysis
4.6. Cluster Differential Expressed Genes Analysis
4.7. qRT-PCR Validation of Targeted Gene
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sefa-Dedeh, S.; Afoakwa, E.O. Biochemical and textural changes in trifoliate yam Dioscorea dumetorum tubers after harvest. Food Chem. 2002, 79, 27–40. [Google Scholar] [CrossRef]
- Mbome Lape, I.; Treche, S. Nutritional quality of yam (Dioscorea dumetorum and D rotundata) flours for growing rats. J. Sci. Food Agric. 1994, 66, 447–455. [Google Scholar] [CrossRef]
- Afoakwa, E.O.; Sefa-Dedeh, S. Chemical composition and quality changes occurring in Dioscorea dumetorum Pax tubers after harvest. Food Chem. 2001, 75, 85–91. [Google Scholar] [CrossRef]
- Iwu, M.M.; Okunji, C.; Akah, P.; Corley, D.; Tempesta, S. Hypoglycaemic Activity of Dioscoretine from Tubers of Dioscorea dumetorum in Normal and Alloxan Diabetic Rabbits. Planta Med. 1990, 56, 264–267. [Google Scholar] [CrossRef]
- Nimenibo-Uadia, R.; Oriakhi, A. Proximate, Mineral and Phytochemical Composition of Dioscorea dumetorum Pax. J. Appl. Sci. Environ. Manag. 2017, 21, 771. [Google Scholar] [CrossRef] [Green Version]
- Medoua, G.N.; Mbofung, C.M.F. Hard-to-cook defect in trifoliate yam Dioscorea dumetorum tubers after harvest. Food Res. Int. 2006, 39, 513–518. [Google Scholar] [CrossRef]
- Medoua, G.N. Potentiels Nutritionnel et Technologique des Tubercules Durcis de l ’igname Dioscorea dumetorum (Kunth). Ph.D. Thesis, Ngaoundere University, Ngaoundere, Cameroon, 2005. [Google Scholar]
- Siadjeu, C.; Panyoo, E.A.; Mahbou Somo Toukam, G.; Bell, M.; Nono, B.; Medoua, G.N. Influence of cultivar on the post-harvest hardening of trifoliate yam (Dioscorea dumetorum) tubers. Adv. Agric. 2016, 2016, 2658983. [Google Scholar]
- Siadjeu, C.; Mayland-quellhorst, E.; Albach, D.C. Genetic diversity and population structure of trifoliate yam (Dioscorea dumetorum Kunth) in Cameroon revealed by genotyping-by-sequencing (GBS). BMC Plant Biol. 2018, 18, 359. [Google Scholar] [CrossRef] [Green Version]
- Siadjeu, C.; Pucker, B.; Viehöver, P.; Albach, D.C.; Weisshaar, B. High contiguity de novo genome sequence assembly of trifoliate yam (Dioscorea dumetorum) using long read sequencing. Genes 2020, 11, 274. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wang, Z.; Chen, J.; Liu, C.; Zhu, W.; Wang, L.; Meng, L. De Novo Transcriptome Assembly and Development of Novel Microsatellite Markers for the Traditional Chinese Medicinal Herb, Veratrilla Baillonii Franch (Gentianaceae). Evol. Bioinform. 2015, 11, 39–45. [Google Scholar] [CrossRef]
- Alves-Carvalho, S.; Aubert, G.; Carrère, S.; Cruaud, C.; Brochot, A.L.; Jacquin, F.; Klein, A.; Martin, C.; Boucherot, K.; Kreplak, J.; et al. Full-length de novo assembly of RNA-seq data in pea (Pisum sativum L.) provides a gene expression atlas and gives insights into root nodulation in this species. Plant J. 2015, 84, 1–19. [Google Scholar] [CrossRef]
- Becerra-Moreno, A.; Redondo-Gil, M.; Benavides, J.; Nair, V.; Cisneros-Zevallos, L.; Jacobo-Velázquez, D.A. Combined effect of water loss and wounding stress on gene activation of metabolic pathways associated with phenolic biosynthesis in carrot. Front. Plant Sci. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.-G.; Jiang, W.; Mantri, N.; Bao, X.-Q.; Chen, S.-L.; Tao, Z.-M. Transciptome analysis reveals flavonoid biosynthesis regulation and simple sequence repeats in yam (Dioscorea alata L.) tubers. BMC Genom. 2015, 16, 346. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, R.; Gedil, M.; Sartie, A.; Otoo, E.; Dumet, D.; Kikuno, H.; Kumar, P.L.; Asiedu, R. Dioscrorea. In Wild Crop Relatives: Genomic Breeding Resources; Kole, C., Ed.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 71–96. [Google Scholar] [CrossRef]
- Tamiru, M.; Natsume, S.; Takagi, H.; White, B.; Yaegashi, H.; Shimizu, M.; Yoshida, K.; Uemura, A.; Oikawa, K.; Abe, A.; et al. Genome sequencing of the staple food crop white Guinea yam enables the development of a molecular marker for sex determination. BMC Biol. 2017, 15, 86. [Google Scholar] [CrossRef] [Green Version]
- Medoua, G.N.; Mbome, I.L.; Agbor-Egbe, T.; Mbofung, C.M.F. Physicochemical changes occurring during post-harvest hardening of trifoliate yam (Dioscorea dumetorum) tubers. Food Chem. 2005, 90, 597–601. [Google Scholar] [CrossRef]
- Medoua, G.N.; Mbome, I.L.; Agbor-Egbe, T.; Mbofung, C.M.F. Study of the hard-to-cook property of stored yam tubers (Dioscorea dumetorum) and some determining biochemical factors. Food Res. Int. 2005, 38, 143–149. [Google Scholar] [CrossRef]
- Le Gall, H.; Philippe, F.; Domon, J.M.; Gillet, F.; Pelloux, J.; Rayon, C. Cell wall metabolism in response to abiotic stress. Plants 2015, 4, 112–166. [Google Scholar] [CrossRef]
- Speicher, T.L.; Li, P.Z.; Wallace, I.S. Phosphoregulation of the plant cellulose synthase complex and cellulose synthase-like proteins. Plants 2018, 7, 52. [Google Scholar] [CrossRef] [Green Version]
- Polko, J.K.; Kieber, J.J. The regulation of cellulose biosynthesis in plants. Plant Cell 2019, 31, 282–296. [Google Scholar] [CrossRef]
- Ambawat, S.; Sharma, P.; Yadav, N.R.; Yadav, R.C. MYB transcription factor genes as regulators for plant responses: An overview. Physiol. Mol. Biol. Plants 2013, 19, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Wang, Y.; Wang, L.; Hu, P.; Wang, Y.; Jia, Y.; Zhang, C.; Zhang, Y.; Zhang, Y.; Wang, C.; et al. Expression of the MYB transcription factor gene BplMYB46 affects abiotic stress tolerance and secondary cell wall deposition in Betula platyphylla. Plant Biotechnol. J. 2017, 15, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Medoua, G.N.; Lape Mbome, I.; Agbor-Egbe, T.; Mbofung, C.M.F. Antinutritional factors changes occurring in trifoliate yam (Dioscorea dumetorum) tubers after harvest. Food Chem. 2007, 102, 716–720. [Google Scholar] [CrossRef]
- Dao, T.T.H.; Linthorst, H.J.M.; Verpoorte, R. Chalcone synthase and its functions in plant resistance. Phytochem. Rev. 2011, 10, 397–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arcuri, M.L.C.; Fialho, L.C.; Vasconcellos Nunes-Laitz, A.; Fuchs-Ferraz, M.C.P.; Wolf, I.R.; Valente, G.T.; Marino, C.L.; Maia, I.G. Genome-wide identification of multifunctional laccase gene family in Eucalyptus grandis: Potential targets for lignin engineering and stress tolerance. Trees Struct. Funct. 2020, 34, 745–758. [Google Scholar] [CrossRef]
- Wang, Y.; Suzuki, H.; Xie, J.; Tomita, O.; Martin, D.J.; Higashi, M.; Kong, D.; Abe, R.; Tang, J. Mimicking Natural Photosynthesis: Solar to Renewable H2 Fuel Synthesis by Z-Scheme Water Splitting Systems. Chem. Rev. 2018, 118, 5201–5241. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.; Huber, W.; Pagès, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for Computing and Annotating Genomic Ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic. Acids Res. 2007, 35, 182–185. [Google Scholar] [CrossRef] [Green Version]
- Schwacke, R.; Ponce-Soto, G.Y.; Krause, K.; Bolger, A.M.; Arsova, B.; Hallab, A.; Gruden, K.; Stitt, M.; Bolger, M.E.; Usadel, B. MapMan4: A Refined Protein Classification and Annotation Framework Applicable to Multi-Omics Data Analysis. Mol. Plant 2019, 12, 879–892. [Google Scholar] [CrossRef]
- Stracke, R.; Werber, M.; Weisshaar, B. The R2R3-MYB gene family in Arabidopsis thaliana. Curr. Opin. Plant Biol. 2001, 4, 447–456. [Google Scholar] [CrossRef]
- Stracke, R.; Holtgräwe, D.; Schneider, J.; Pucker, B.; Rosleff Sörensen, T.; Weisshaar, B. Genome-wide identification and characterisation of R2R3-MYB genes in sugar beet (Beta vulgaris). BMC Plant Biol. 2014, 14, 249. [Google Scholar] [CrossRef] [Green Version]
- Pucker, B.; Pandey, A.; Weisshaar, B.; Stracke, R. The R2R3-MYB gene family in banana (Musa acuminata): Genome-wide identification, classification and expression patterns. PLoS ONE 2020, 15, e0239275. [Google Scholar] [CrossRef]
- Pucker, B.; Reiher, F.; Schilbert, H.M. Automatic identification of players in the flavonoid biosynthesis with application on the biomedicinal plant croton tiglium. Plants 2020, 9, 1103. [Google Scholar] [CrossRef]
- Götz, S.; García-Gómez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic. Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef]
- Yeung, K.Y.; Medvedovic, M.; Bumgarner, R.E. From co-expression to co-regulation: How many microarray experiments do we need? Genome Biol. 2004, 5, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 18, 134. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Contig | LF2C | Padj | Bin/KO | Gene\Name | Description |
|---|---|---|---|---|---|
| contig544.g2040 | 6.91 | 0.04740 | 21.4.1.1.3 | FLA | fasciclin-type AGP |
| contig278.g50 | 8.89 | 0.02720 | 21.1.2.2 | COB | regulatory protein |
| contig760.g29 | 18.35 | 0.03609 | 1.2.3/K05298 | GAPA | glyceraldehyde-3-phosphate dehydrogenase |
| contig119.g125 | 8.07 | 0.00170 | 1.1.6.1.1 | PGR5/PGRL1 | complex.component PGR5-like |
| contig678.g379 | 7.72 | 0.00000 | 1.1.4.1.4/K08910 | LHCA4 | chlorophyll a/b binding protein 4 |
| contig679.g24 | 7.98 | 0.00000 | 1.1.4.1.4/K08910 | LHCA4 | chlorophyll a/b binding protein 4 |
| contig549.g218 | 6.53 | 0.00000 | K02694 | psaF | photosystem I subunit III |
| contig222.g1555 | 5.27 | 0.00626 | 1.1.4.2.8/K02695 | psaH | photosystem I subunit VI |
| contig206.g10 | 5.55 | 0.00042 | 1.1.1.1.1/K08912 | LHCB1 | chlorophyll a/b binding protein 1 |
| contig206.g11 | 7.98 | 0.00000 | 1.1.1.1.1/K08912 | LHCB1 | chlorophyll a/b binding protein 1 |
| contig206.g6 | 7.12 | 0.00000 | 1.1.1.1.1/K08912 | LHCB1 | chlorophyll a/b binding protein 1 |
| contig206.g8 | 7.58 | 0.00000 | 1.1.1.1.1/K08912 | LHCB1 | chlorophyll a/b binding protein 1 |
| contig267.g402 | 5.81 | 0.00836 | 1.1.1.1.1/K08913 | LHCB2 | chlorophyll a/b binding protein 2 |
| contig355.g38 | 5.82 | 0.01516 | 1.1.1.1.1/K08913 | LHCB2 | chlorophyll a/b binding protein 2 |
| contig391.g20 | 6.24 | 0.00012 | 1.1.1.1.1/K08912 | LHCB1 | chlorophyll a/b binding protein 1 |
| contig391.g26 | 6.94 | 0.00000 | 1.1.1.1.1/K08912 | LHCB1 | chlorophyll a/b binding protein 1 |
| contig391.g28 | 5.72 | 0.00038 | 1.1.1.1.1/K08912 | LHCB1 | chlorophyll a/b binding protein 1 |
| contig391.g29 | 7.65 | 0.00000 | 1.1.1.1.1/K08912 | LHCB1 | chlorophyll a/b binding protein 1 |
| contig553.g402 | 4.31 | 0.04740 | 1.1.1.1.1/K08914 | LHCB3 | chlorophyll a/b binding protein 3 |
| contig565.g52 | 7.56 | 0.02366 | 1.1.1.1.1/K08912 | LHCB1 | chlorophyll a/b binding protein 1 |
| contig89.g1873 | 5.94 | 0.01452 | 1.1.1.6.2.1 | ELIP | LHC-related protein group.protein |
| contig544.g1881 | 5.17 | 0.04740 | 1.1.1.2.13 | 1.1.1.2.13/>PsbX | PS-II complex.component |
| contig544.g1970 | 5.05 | 0.00905 | 1.1.1.2.13 | 1.1.1.2.13/PsbX | PS-II complex.component |
| contig267.g494 | 20.89 | 0.00000 | 15.5.2.1/K09422 | MYB | transcription factor |
| Contig | LF2C | Padj | Bin/Ko | Gene\Name | Description |
|---|---|---|---|---|---|
| contig557.g748 | 9.02 | 3.15 × 10−9 | 21.1.1.1.1/K10999 | CESA | cellulose synthase A |
| contig60.g53 | 8.86 | 3.44 × 10−9 | 21.1.1.1.1/K10999 | CESA | cellulose synthase A |
| contig73.g5 | 8.94 | 3.78 × 10−9 | 21.1.1.1.1/K10999 | CESA | cellulose synthase A |
| contig267.g188 | 23.39 | 5.99 × 10−6 | 21.1.2.2 | COB | regulatory protein |
| contig278.g50 | 14.51 | 2.99 × 10−3 | 21.1.2.2 | COB | regulatory protein |
| contig143.g88 | 17.83 | 2.27 × 10−3 | 21.2.2.2.2 | XOAT | xylan O-acetyltransferase |
| contig145.g17 | 17.90 | 2.01 × 10−3 | 21.2.2.2.2 | XOAT | xylan O-acetyltransferase |
| contig199.g1435 | 12.17 | 1.46 × 10−4 | 21.2.2.2.2 | XOAT | xylan O-acetyltransferase |
| contig920.g250 | 17.89 | 1.76 × 10−4 | 21.2.2.2.2 | XOAT | xylan O-acetyltransferase |
| contig922.g12 | 11.49 | 7.50 × 10−3 | 21.2.2.2.2 | XOAT | xylan O-acetyltransferase |
| contig750.g97 | 6.45 | 8.83 × 10−4 | 21.6.1.7/K13066 | COMT | caffeic acid 3-O-methyltransferase |
| contig646.g19 | 5.52 | 1.68 × 10−2 | K18368 | CSE | caffeoylshikimate esterase |
| contig552.g180 | 5.18 | 1.60 × 10−2 | K00588 | E2.1.1.104 | caffeoyl-CoA O-methyltransferase |
| contig3.g487 | 5.66 | 4.55 × 10−2 | 21.6.1.2/K09754 | CYP98A | 5-O-(4-coumaroyl)-d-quinate 3′-monooxygenase |
| contig199.g1672 | 10.14 | 3.21 × 10−3 | 21.6.2.2/K05909 | E1.10.3.2 | Laccase |
| contig559.g139 | 26.23 | 4.72 × 10−8 | 21.6.2.1 | PMT | p-coumaroyl-CoA |
| contig119.g106 | 14.35 | 9.30 × 10−3 | K05350 | bglB | beta-glucosidase |
| contig390.g181 | 6.08 | 3.53 × 10−2 | 21.3.2.2.2 | BGAL | beta-galactosidase |
| contig678.g379 | 7.74 | 2.36 × 10−4 | 1.1.4.1.4/K08910 | LHCA4 | chlorophyll a/b binding protein 4 |
| contig679.g24 | 11.17 | 4.28 × 10−6 | 1.1.4.1.4/K08910 | LHCA4 | chlorophyll a/b binding protein 4 |
| contig206.g11 | 7.52 | 4.19 × 10−7 | 1.1.1.1.1/K08912 | LHCB1 | chlorophyll a/b binding protein 1 |
| contig391.g29 | 6.83 | 5.77 × 10−4 | 1.1.1.1.1/K08912 | LHCB1 | chlorophyll a/b binding protein 1 |
| contig546.g79 | 20.36 | 6.88 × 10−4 | 15.5.7.2 | DREB | transcription factor |
| contig771.g2 | 25.08 | 4.05 × 10−5 | 15.5.7.2 | DREB | transcription factor |
| contig267.g494 | 25.57 | 3.54 × 10−2 | 15.5.2.1/K09422 | MYB | transcription factor |
| contig678.g290 | 16.94 | 1.44 × 10−2 | K09422 | MYB | transcription factor |
| contig693.g10 | 6.77 | 4.76 × 10−2 | K09422 | MYB | transcription factor |
| contig750.g121 | 25.14 | 2.61 × 10−7 | K09422 | MYB | transcription factor |
| contig158.g23 | 37.78 | 5.01 × 10−6 | 15.5.17 | NAC | transcription factor |
| contig556.g459 | 37.78 | 5.01 × 10−6 | 15.5.17 | NAC | transcription factor |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siadjeu, C.; Mayland-Quellhorst, E.; Pande, S.; Laubinger, S.; Albach, D.C. Transcriptome Sequence Reveals Candidate Genes Involving in the Post-Harvest Hardening of Trifoliate Yam Dioscorea dumetorum. Plants 2021, 10, 787. https://doi.org/10.3390/plants10040787
Siadjeu C, Mayland-Quellhorst E, Pande S, Laubinger S, Albach DC. Transcriptome Sequence Reveals Candidate Genes Involving in the Post-Harvest Hardening of Trifoliate Yam Dioscorea dumetorum. Plants. 2021; 10(4):787. https://doi.org/10.3390/plants10040787
Chicago/Turabian StyleSiadjeu, Christian, Eike Mayland-Quellhorst, Shruti Pande, Sascha Laubinger, and Dirk C. Albach. 2021. "Transcriptome Sequence Reveals Candidate Genes Involving in the Post-Harvest Hardening of Trifoliate Yam Dioscorea dumetorum" Plants 10, no. 4: 787. https://doi.org/10.3390/plants10040787
APA StyleSiadjeu, C., Mayland-Quellhorst, E., Pande, S., Laubinger, S., & Albach, D. C. (2021). Transcriptome Sequence Reveals Candidate Genes Involving in the Post-Harvest Hardening of Trifoliate Yam Dioscorea dumetorum. Plants, 10(4), 787. https://doi.org/10.3390/plants10040787