
A Traceable DNA-Replicon Derived Vector to Speed Up Gene Editing in Potato: Interrupting Genes Related to Undesirable Postharvest Tuber Traits as an Example

 ,
, 
Abstract
:1. Introduction
2. Results
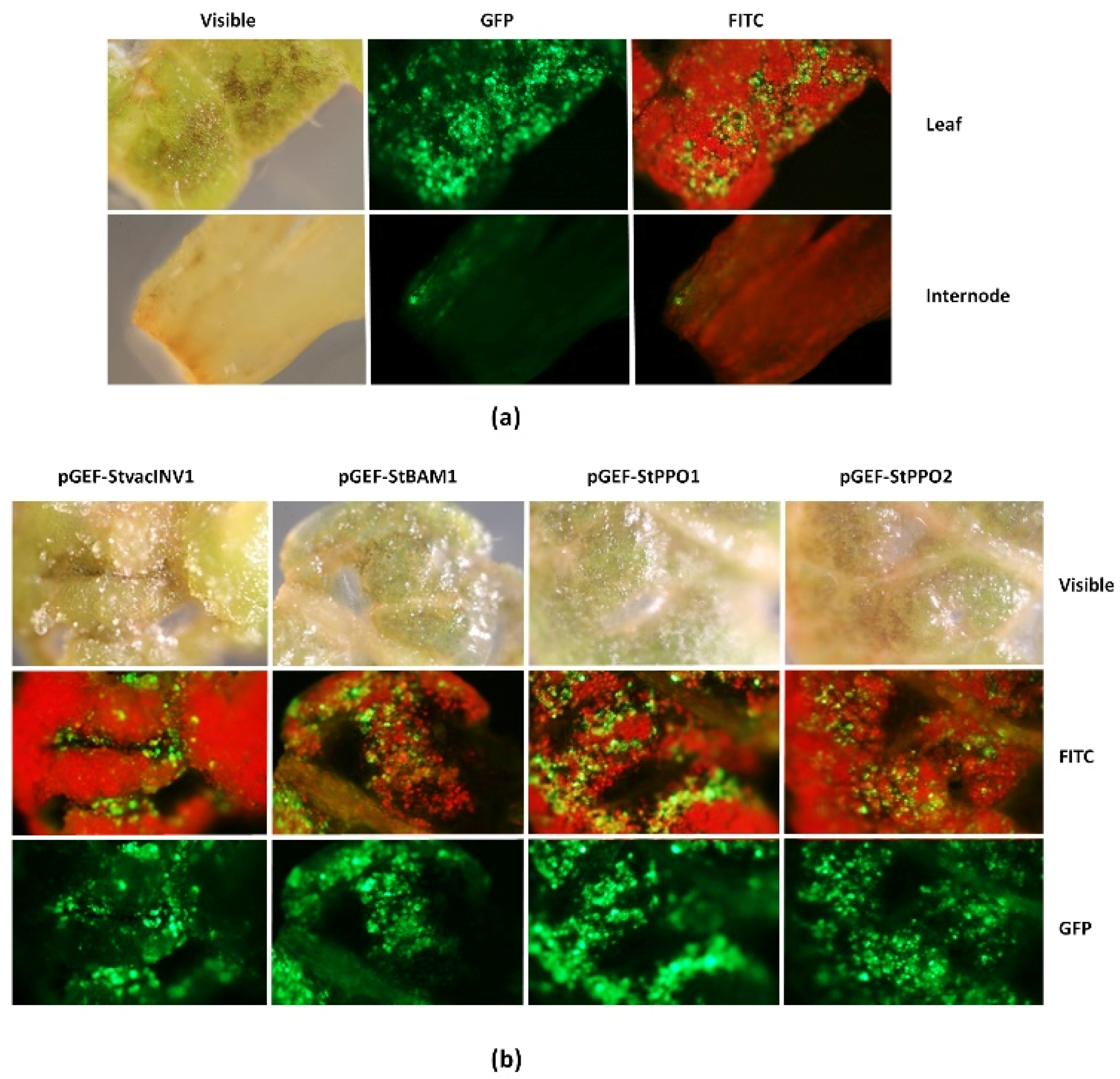
2.1. Functionality of the Universal Fluorescent Editor Geminivirus-Based Plasmid (pGEF-U) in Potato
2.2. Guide RNAs Design and Evaluation
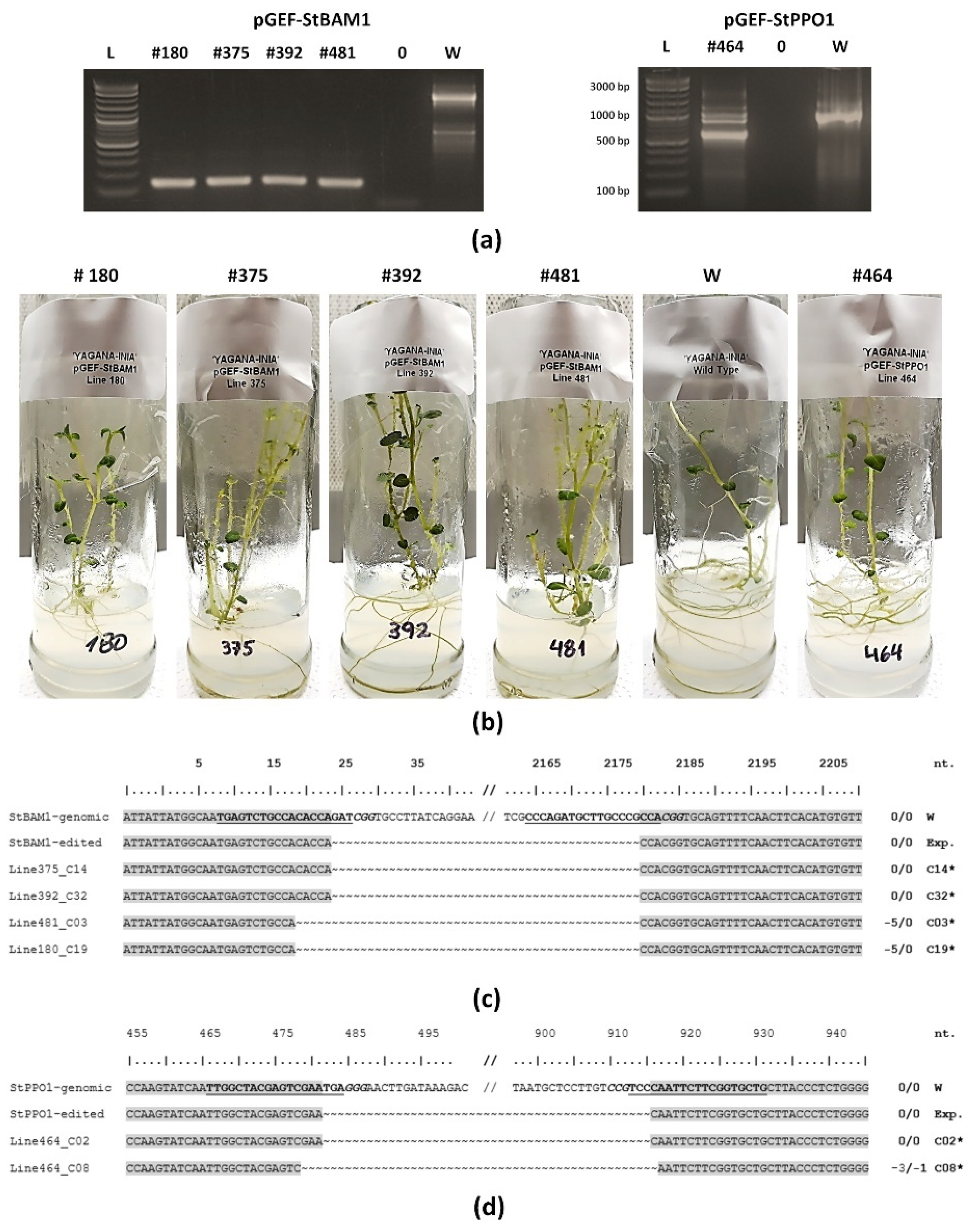
2.3. ‘Yagana-INIA’ Regeneration and Gene Editing
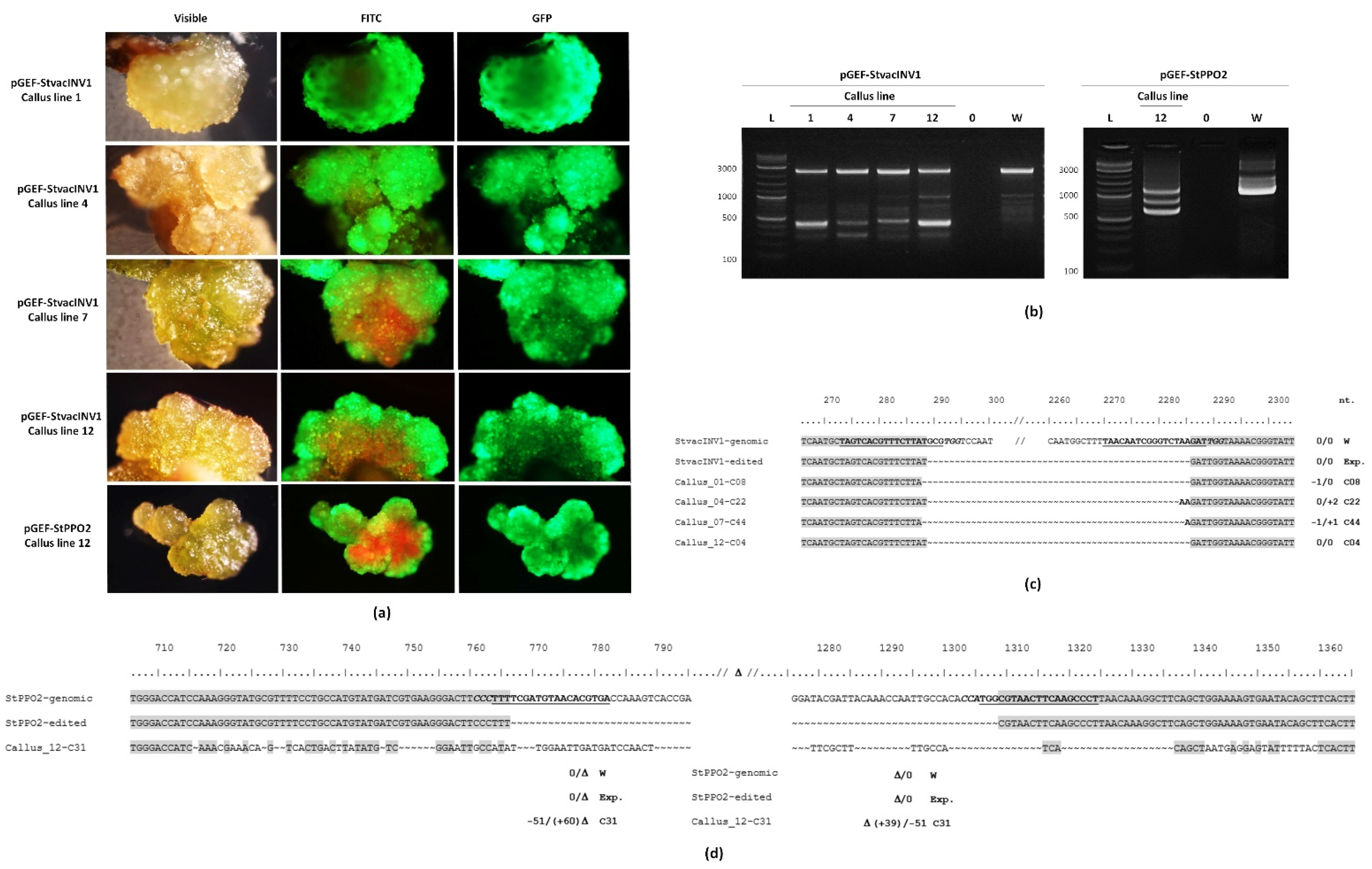
2.4. Gene Editing in Callus Lines
3. Discussion
4. Materials and Methods
4.1. Plant Material
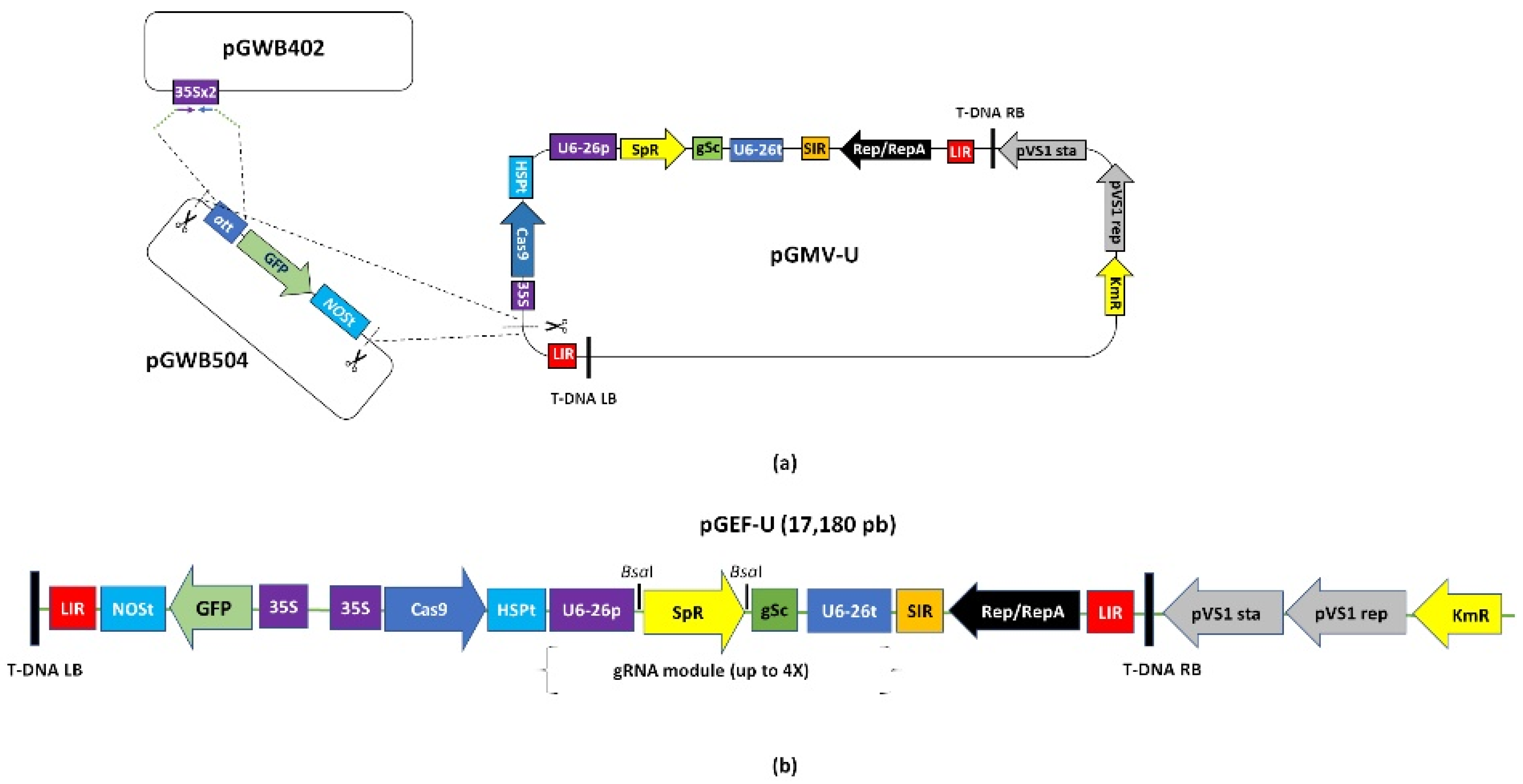
4.2. Construction of the Geminivirus Editor Fluorescent Universal Plasmid (pGEF-U) for Potato Gene Editing
4.3. Gene Transfer Experiments
4.3.1. Agrobacterium Preparation
4.3.2. Explants Pre-Culture
4.3.3. Explants’ Infection, Co-Culture, and Callus Induction
4.4. Plant Regeneration
4.5. Design and Selection of Guide RNAs (gRNA) for Potato Genome Editing
4.6. Gene Editing Vectors for Potato Genes
4.6.1. Paired-gRNA Module Cloning into pGEF-U
4.6.2. Validation of Guide RNAs
4.7. Genomic DNA Extraction
4.8. GFP Detection in Potato Explants
4.9. Verification of Edited Lines
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, S.; Zhang, S.; Wang, W.; Xiong, X.; Meng, F.; Cui, X. Efficient targeted mutagenesis in potato by the CRISPR/Cas9 system. Plant Cell Rep. 2015, 34, 1473–1476. [Google Scholar] [CrossRef] [Green Version]
- Ramírez Gonzales, L.; Shi, L.; Bergonzi Bergonzi, S.; Oortwijn, M.; Franco-Zorrilla, J.M.; Solano-Tavir, R.; Visser, R.G.F.; Abelenda, J.A.; Bachem, C.W.B. Potato CYCLING DOF FACTOR 1 and its lncRNA counterpart StFLORE link tuber development and drought response. Plant J. 2021, 105, 855–869. [Google Scholar] [CrossRef] [PubMed]
- Andersson, M.; Turesson, H.; Olsson, N.; Fält, A.-S.; Ohlsson, P.; Gonzalez, M.N.; Samuelsson, M.; Hofvander, P. Genome editing in potato via CRISPR-Cas9 ribonucleoprotein delivery. Physiol. Plant. 2018, 164, 378–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Jayarathna, S.; Turesson, H.; Fält, A.-S.; Nestor, G.; González, M.N.; Olsson, N.; Beganovic, M.; Hofvander, P.; Andersson, R.; et al. Amylose starch with no detectable branching developed through DNA-free CRISPR-Cas9 mediated mutagenesis of two starch branching enzymes in potato. Sci. Rep. 2021, 11, 4311–4323. [Google Scholar] [CrossRef] [PubMed]
- González, M.N.; Massa, G.A.; Andersson, M.; Turesson, H.; Olsson, N.; Fält, A.-S.; Storani, L.; Décima Oneto, C.A.; Hofvander, P.; Feingold, S.E. Reduced Enzymatic Browning in Potato Tubers by Specific Editing of a Polyphenol Oxidase Gene via Ribonucleoprotein Complexes Delivery of the CRISPR/Cas9 System. Front. Plant Sci. 2020, 10, 1649. [Google Scholar] [CrossRef] [PubMed]
- Maher, M.F.; Nasti, R.A.; Vollbrecht, M.; Starker, C.G.; Clark, M.D.; Voytas, D.F. Plant gene editing through de novo induction of meristems. Nat. Biotechnol. 2020, 38, 84–89. [Google Scholar] [CrossRef]
- Uranga, M.; Aragon, V.; Selma, S.; Vazquez-Vilar, M.; Orzaez, D.; Daros, J.A. Efficient Cas9 multiplex editing using unspaced sgRNA arrays engineering in a Potato virus X vector. Plant J. 2021, 106, 555–565. [Google Scholar] [CrossRef]
- Baltes, N.J.; Gil-Humanes, J.; Čermák, T.; Atkins, P.A.; Voytas, D.F. DNA replicons for plant genome engineering. Plant Cell 2014, 26, 151–163. [Google Scholar] [CrossRef] [Green Version]
- Nadakuduti, S.S.; Starker, C.G.; Voytas, D.F.; Buell, C.R.; Douches, D.S. Genome editing in potato with CRISPR/Cas9. In Plant Genome Editing with CRISPR Systems; Methods in Molecular Biology Volume 1917; Qi, Y., Ed.; Humana Press: New York, NY, USA, 2019. [Google Scholar] [CrossRef]
- Morris, B.A.M.; Richardson, K.A.; Haley, A.; Zhan, X.; Thomas, J.E. The nucleotide sequence of the infectious cloned DNA component of Tobacco yellow dwarf virus reveals features of geminiviruses infecting monocotyledonous plants. Virology 1992, 187, 633–642. [Google Scholar] [CrossRef]
- Sanz-Burgos, A.P.; Gutierrez, C. Organization of the cis-acting element required for wheat dwarf geminivirus DNA replication and visualization of a rep protein-DNA complex. Virology 1998, 243, 119–129. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Saunders, K.; Thomas, C.L.; Davies, J.W.; Stanley, J. Bean yellow dwarf virus RepA, but not Rep, binds to maize retinoblastoma protein, and the virus tolerates mutations in the consensus binding motif. Virology 1999, 256, 270–279. [Google Scholar] [CrossRef] [Green Version]
- Mor, T.S.; Moon, T.-S.; Palmer, K.E.; Mason, H.S. Geminivirus vectors for high-level expression of foreign proteins in plant cells. Biotechnol. Bioeng. 2003, 81, 430–437. [Google Scholar] [CrossRef]
- Čermák, T.; Baltes, N.J.; Čegan, R.; Zhang, Y.; Voytas, D.F. High-frequency, precise modification of the tomato genome. Genome Biol. 2015, 16, 232–245. [Google Scholar] [CrossRef] [Green Version]
- Butler, N.M.; Atkins, P.A.; Voytas, D.F.; Douches, D.S. Generation and inheritance of targeted mutations in potato (Solanum tuberosum L.) using the CRISPR/Cas system. PLoS ONE 2015, 10, e0144591. [Google Scholar] [CrossRef] [Green Version]
- Butler, N.M.; Baltes, N.J.; Voytas, D.F.; Douches, D.S. Geminivirus-mediated genome editing in potato (Solanum tuberosum L.) using sequence-specific nucleases. Front. Plant Sci. 2016, 7, 1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil-Humanes, J.; Wang, Y.; Liang, Z.; Shan, Q.; Ozuna, C.V.; Sánchez-León, S.; Baltes, N.J.; Starker, C.; Barro, F.; Gao, C.; et al. High-efficiency gene targeting in hexaploid wheat using DNA replicons and CRISPR/Cas9. Plant J. 2017, 89, 1251–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, B.L.; Möller, S.R.; Mravec, J.; Jørgensen, B.; Christensen, M.; Liu, Y.; Wandall, H.H.; Bennett, E.P.; Yang, Z. Improved CRISPR/Cas9 gene editing by fluorescence activated cell sorting of green fluorescence protein tagged protoplasts. BMC Biotechnol. 2019, 19, 36. [Google Scholar] [CrossRef]
- Chi, M.; Bhagwat, B.; Lane, W.D.; Tang, G.; Su, Y.; Sun, R.; Oomah, B.D.; Wiersma, P.A.; Xiang, Y. Reduced polyphenol oxidase gene expression and enzymatic browning in potato (Solanum tuberosum L.) with artificial microRNAs. BMC Plant Biol. 2014, 14, 62. [Google Scholar] [CrossRef] [Green Version]
- Clasen, B.M.; Stoddard, T.J.; Luo, S.; Demorest, Z.L.; Li, J.; Cedrone, F.; Tibebu, R.; Davison, S.; Ray, E.E.; Daulhac, A.; et al. Improving cold storage and processing traits in potato through targeted gene knockout. Plant Biotechnol. J. 2016, 14, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Zhang, H.; Liu, J.; Reid, S.; Liu, T.; Xu, S.; Tian, Z.; Sonnewald, U.; Song, B.; Xie, C. Amylases StAmy23, StBAM1 and StBAM9 regulate cold-induced sweetening of potato tubers in distinct ways. J. Exp. Bot. 2017, 68, 2317–2331. [Google Scholar] [CrossRef] [Green Version]
- Prieto, H.; Miccono, M.; Aguirre, C.; Sánchez, E.; Castro, Á. Grape Biotechnology: Past, Present, and Future. In The Grape Genome. Compendium of Plant Genomes; Cantu, D., Walker, M., Eds.; Springer: Cham, Switzerland, 2019; pp. 349–367. [Google Scholar] [CrossRef]
- Craze, M.; Bates, R.; Bowden, S.; Wallington, E.J. Highly efficient Agrobacterium-mediated transformation of potato (Solanum tuberosum) and production of transgenic microtubers. Curr. Protoc. Plant Biol. 2018, 3, 3–41. [Google Scholar] [CrossRef] [PubMed]
- Hayes, R.J.; Petty, I.T.D.; Coutts, R.H.A.; Buck, K.W. Gene amplification and expression in plants by a replicating geminivirus vector. Nature 1988, 334, 179–182. [Google Scholar] [CrossRef]
- Lozano-Durán, R. Geminiviruses for biotechnology: The art of parasite taming. New Phytol. 2016, 210, 58–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergara, R.; Olivares, F.; Olmedo, B.; Toro, C.; Muñoz, M.; Zúñiga, C.; Mora, R.; Plantat, P.; Miccono, M.; Loyola, R.; et al. Gene Editing in Prunus Spp.: The Challenge of Adapting Regular Gene Transfer Procedures for Precision Breeding. In Prunus: Recent Advances; Küdenm, A., Ed.; IntechOpen: London, UK, 2021. [Google Scholar] [CrossRef]
- Chen, L.; Li, W.; Katin-Grazzini, L.; Ding, J.; Gu, X.; Li, Y.; Gu, T.; Lin, X.; Deng, Z.; McAvoy, R.J.; et al. A method for the production and expedient screening of CRISPR/Cas9 mediated non-transgenic mutant plants. Hort. Res. 2018, 5, 13. [Google Scholar] [CrossRef] [PubMed]
- Veillet, F.; Perrot, L.; Chauvin, L.; Kermarrec, M.P.; Guyon-Debast, A.; Chauvin, J.E.; Nogué, F.; Mazier, M. Transgene-free genome editing in tomato and potato plants using Agrobacterium-mediated delivery of a CRISPR/Cas9 cytidine base editor. Int. J. Mol. Sci. 2019, 20, 402. [Google Scholar] [CrossRef] [Green Version]
- Bánfalvi, Z.; Csákvári, E.; Villányi, V.; Kondrák, M. Generation of transgene-free PDS mutants in potato by Agrobacterium-mediated transformation. BMC Biotech. 2020, 20, 25–35. [Google Scholar] [CrossRef]
- Nocarova, E.; Opatrny, Z.; Fischer, L. Successive silencing of tandem reporter genes in potato (Solanum tuberosum) over 5 years of vegetative propagation. Ann. Bot. 2010, 106, 565–572. [Google Scholar] [CrossRef] [Green Version]
- Shen, B.; Zhang, W.; Zhang, J.; Zhou, J.; Wang, J.; Chen, L.; Wang, L.; Hodgkins, A.; Iyer, V.; Huang, X.; et al. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat. Methods 2014, 11, 399–402. [Google Scholar] [CrossRef]
- Pulido-Quetglas, C.; Aparicio-Prat, E.; Arnan, C.; Polidori, T.; Hermoso, T.; Palumbo, E.; Ponomarenko, E.; Guigo, R.; Johnson, R. Scalable design of paired CRISPR guide RNAs for genomic deletion. PLoS Comput. Biol. 2017, 13, e1005341. [Google Scholar] [CrossRef]
- Canver, M.C.; Bauer, D.E.; Dass, A.; Yien, Y.Y.; Chung, J.; Masuda, T.; Maeda, T.; Paw, B.H.; Orkin, S.H. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. J. Biol. Chem. 2014, 289, 21312–21324. [Google Scholar] [CrossRef] [Green Version]
- Potato Pedigree Database. Available online: https://www.plantbreeding.wur.nl/PotatoPedigree/index.html (accessed on 27 July 2021).
- Modrzejewski, D.; Hartung, F.; Lehnert, H.; Sprink, T.; Kohl, C.; Keilwagen, J.; Wilhelm, R. Which factors affect the occurrence of off-target effects caused by the use of CRISPR/Cas: A systematic review in plants. Front. Plant Sci. 2020, 11, 574959. [Google Scholar] [CrossRef] [PubMed]
- Sowokinos, J. Biochemical and molecular control of cold-induced sweetening in potatoes. Am. J. Potato Res. 2001, 78, 221–236. [Google Scholar] [CrossRef]
- Roitsch, T.; Gonzalez, M.C. Function and regulation of plant invertases: Sweet sensations. Trends Plant Sci. 2004, 9, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Steentoft, C.; Hauge, C.; Hansen, L.; Lind Thomsen, A.; Niola, F.; Vester-Christensen, M.B.; Frödin, M.; Clausen, H.; Wandall, H.H.; et al. Fast and sensitive detection of indels induced by precise gene targeting. Nucleic Acids Res. 2015, 43, e59. [Google Scholar] [CrossRef] [Green Version]
- McCormac, A.C.; Elliott, M.C.; Chen, D.F. A simple method for the production of highly competent cells of Agrobacterium for transformation via electroporation. Mol. Biotechnol. 1998, 9, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Visser, R.G.F.; Jacobsen, E.; Hesseling-Meinders, A.; Schans, M.J.; Witholt, B.; Feenstra, W.J. Transformation of homozygous diploid potato with an Agrobacterium tumefaciens binary vector system by adventitious shoot regeneration on leaf and stem segments. Plant Mol. Biol. 1989, 12, 329–337. [Google Scholar] [CrossRef]
- Xu, X.; Pan, S.; Cheng, S.; Zhang, B.; Mu, D.; Ni, P.; Zhang, G.; Yang, S.; Li, R.; Wang, J.; et al. Potato Genome Sequencing Consortium. Genome sequence and analysis of the tuber crop potato. Nature 2011, 475, 189–195. [Google Scholar] [CrossRef] [Green Version]
- Doench, J.G.; Hartenian, E.; Graham, D.B.; Tothova, Z.; Hegde, M.; Smith, I.; Sullender, M.; Ebert, B.L.; Xavier, R.J.; Root, D.E. Rational design of highly active sgRNAs for CRISPR-Cas9–mediated gene inactivation. Nat. Biotechnol. 2014, 32, 1262–1267. [Google Scholar] [CrossRef] [Green Version]
- Skinner, M.E.; Uzilov, A.V.; Stein, L.D.; Mungall, C.J.; Holmes, I.H. JBrowse: A next-generation genome browser. Genome Res. 2009, 19, 1630–1638. [Google Scholar] [CrossRef] [Green Version]
- Priyam, A.; Woodcroft, B.J.; Rai, V.; Munagala, A.; Moghul, I.; Ter, F.; Gibbins, M.A.; Moon, H.; Leonard, G.; Rumpf, W.; et al. Sequenceserver: A modern graphical user interface for custom BLAST databases. Mol. Biol. Evol. 2019, 36, 2922–2924. [Google Scholar] [CrossRef]
- Xing, H.L.; Dong, L.; Wang, Z.P.; Zhang, H.Y.; Han, C.Y.; Liu, B.; Wang, X.C.; Chen, Q.J. A CRISPR/Cas9 toolkit for multiplex genome editing in plants. BMC Plant Biol. 2014, 14, 327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engler, C.; Gruetzner, R.; Kandzia, R.; Marillonnet, S. Golden gate shuffling: A one-pot DNA shuffling method based on type IIs restriction enzymes. PLoS ONE 2009, 4, e5553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steenkamp, J.; Wild, I.; Lourens, A.; Van Helden, P. Improved method for DNA extraction from Vitis vinifera. Am. J. Enol. Vitic. 1994, 45, 102–106. [Google Scholar]
- Gibson, D. Enzymatic assembly of overlapping DNA fragments. Methods Enzymol. 2011, 98, 349–361. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| gRNA Name | gRNA Sequence | gRNA Position | Size of the Expected Deletion |
|---|---|---|---|
| gRNA1-StvacINV1B | TAGTCACGTTTCTTATGCG | 273–291 | 1997 bp |
| gRNA2-StvacINV1B | TAACAATCGGGTCTAAGAT | 2270–2288 | |
| gRNA1-StBAM1A | TGAGTCTGCCACACCAGAT | 8–26 | 2156 bp |
| gRNA2-StBAM1A | CCCAGATGCTTGCCCGCCA | 2164–2182 | |
| gRNA1-StPPO1B | TTGGCTACGAGTCGAATGA | 466–484 | 434 bp |
| gRNA2-StPPO1C | CAGCACCGAAGAATTGGGA | 913–931 | |
| gRNA1-StPPO2C | TCACGTGTTACATCGAAAA | 764–782 | 542 bp |
| gRNA2-StPPO2C | AGGGCTTGAAGTTACGCCA | 1306–1324 |
| pGEF-X X: | Initial Explants | GFP+ Leaves (%) | GFP+ Leaf Explants in PSM400cc Day 7 (pi) * (%) | Individualized Shoots | Shoots per Callus |
|---|---|---|---|---|---|
| StvacINV1 | 23 | 18 (78.3) | 4 (17.4) | 15 | 3.8 |
| 35 | 16 (45.7) | 10 (28.6) | 61 | 6.1 | |
| 80 | 31 (38.8) | 23 (28,8) | 48 | 2.1 | |
| StBAM1 | 65 | 33 (50.8) | 22 (33.8) | 74 | 3.4 |
| 101 | 54 (53.5) | 26 (25.7) | 52 | 2.0 | |
| 61 | 12 (19.7) | 5 (8.2) | 2 | 0.4 | |
| StPPO1 | 58 | 45 (77,6) | 39 (67.2) | 86 | 2.2 |
| 200 | 126 (63.0) | 84 (42.0) | 52 | 0.6 | |
| 81 | 57 (70.4) | 36 (44.4) | 3 | 0.1 | |
| StPPO2 | 26 | 18 (69.2) | 14 (53.8) | 37 | 2.6 |
| 263 | 152 (57.8) | 79 (30.0) | 122 | 1.5 | |
| 47 | 18 (38.3) | 15 (31.9) | 11 | 0.7 |
| Gene Editor | Initial Explants | GFP Positive | PSM400cc Day 7 (pi) | PSM200cc Day 82 (pi) | PSM200cc Day 271 (pi) | Edited TG Calli * |
|---|---|---|---|---|---|---|
| pGEF-StvacINV1 | 93 | 51 | 32 | 12 | 6 | 4 |
| pGEF-StPPO2 | 62 | 29 | 19 | 14 | 3 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Acha, G.; Vergara, R.; Muñoz, M.; Mora, R.; Aguirre, C.; Muñoz, M.; Kalazich, J.; Prieto, H. A Traceable DNA-Replicon Derived Vector to Speed Up Gene Editing in Potato: Interrupting Genes Related to Undesirable Postharvest Tuber Traits as an Example. Plants 2021, 10, 1882. https://doi.org/10.3390/plants10091882
Acha G, Vergara R, Muñoz M, Mora R, Aguirre C, Muñoz M, Kalazich J, Prieto H. A Traceable DNA-Replicon Derived Vector to Speed Up Gene Editing in Potato: Interrupting Genes Related to Undesirable Postharvest Tuber Traits as an Example. Plants. 2021; 10(9):1882. https://doi.org/10.3390/plants10091882
Chicago/Turabian StyleAcha, Giovana, Ricardo Vergara, Marisol Muñoz, Roxana Mora, Carlos Aguirre, Manuel Muñoz, Julio Kalazich, and Humberto Prieto. 2021. "A Traceable DNA-Replicon Derived Vector to Speed Up Gene Editing in Potato: Interrupting Genes Related to Undesirable Postharvest Tuber Traits as an Example" Plants 10, no. 9: 1882. https://doi.org/10.3390/plants10091882
APA StyleAcha, G., Vergara, R., Muñoz, M., Mora, R., Aguirre, C., Muñoz, M., Kalazich, J., & Prieto, H. (2021). A Traceable DNA-Replicon Derived Vector to Speed Up Gene Editing in Potato: Interrupting Genes Related to Undesirable Postharvest Tuber Traits as an Example. Plants, 10(9), 1882. https://doi.org/10.3390/plants10091882