
Genome-Wide Association Analysis of Salt-Tolerant Traits in Terrestrial Cotton at Seedling Stage

Abstract
:1. Introduction
2. Results
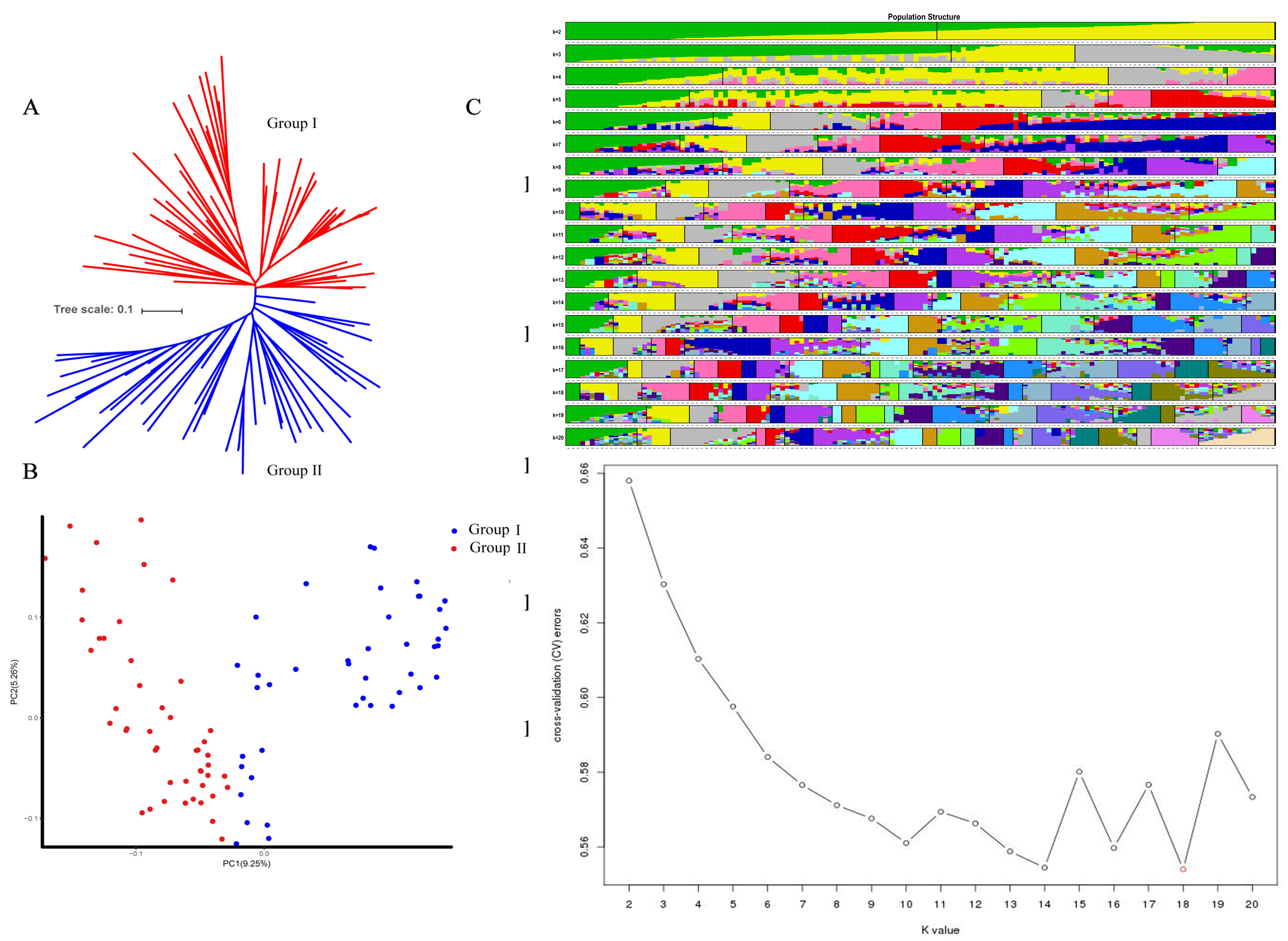
2.1. Genetic Diversity Analysis and Population Structure
2.1.1. Group Structure
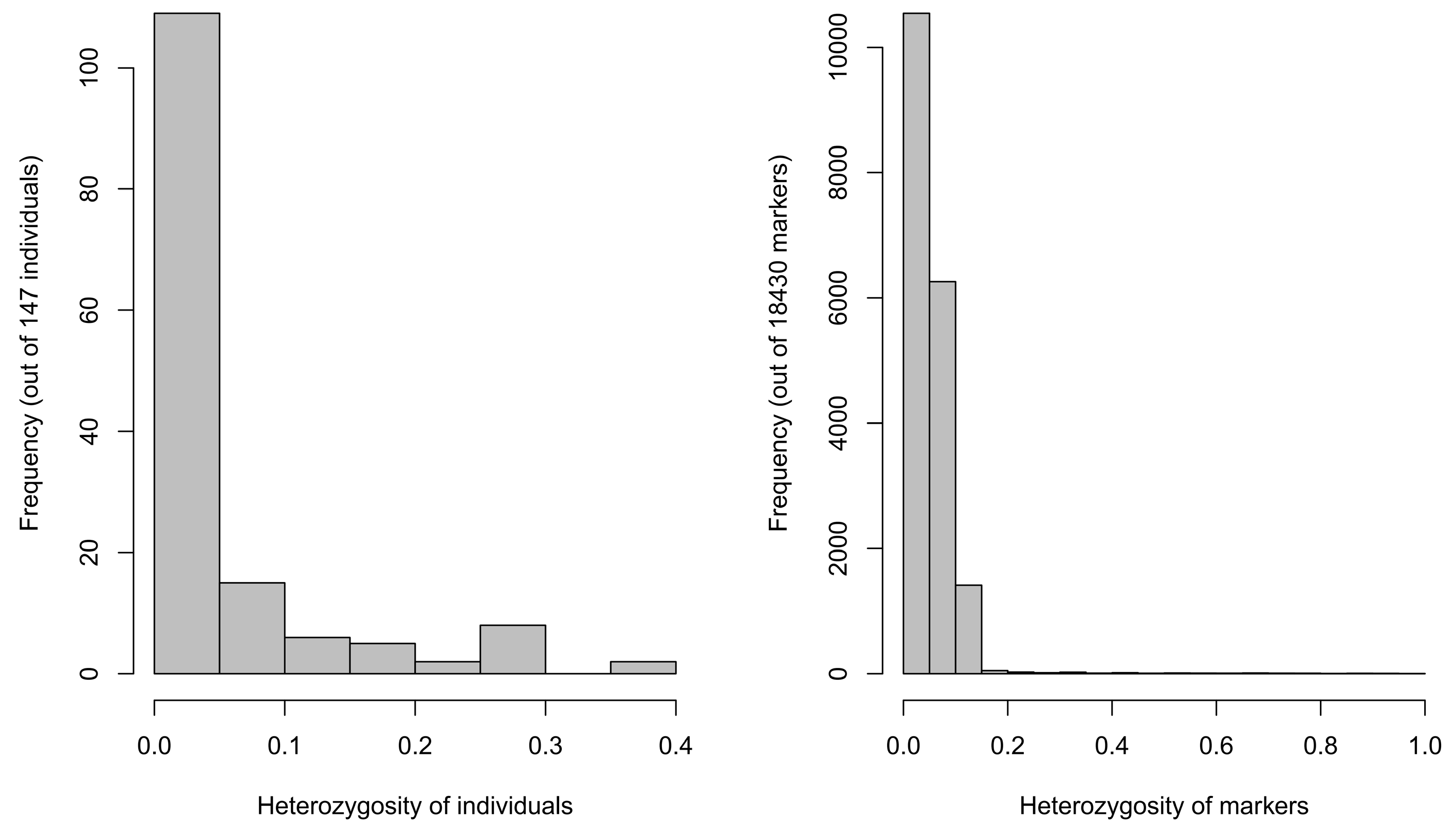
2.1.2. Material Heterozygosity
2.1.3. Kinship Distribution
2.1.4. Analysis of Linkage Disequilibrium
2.2. Phenotypic Statistical Analysis
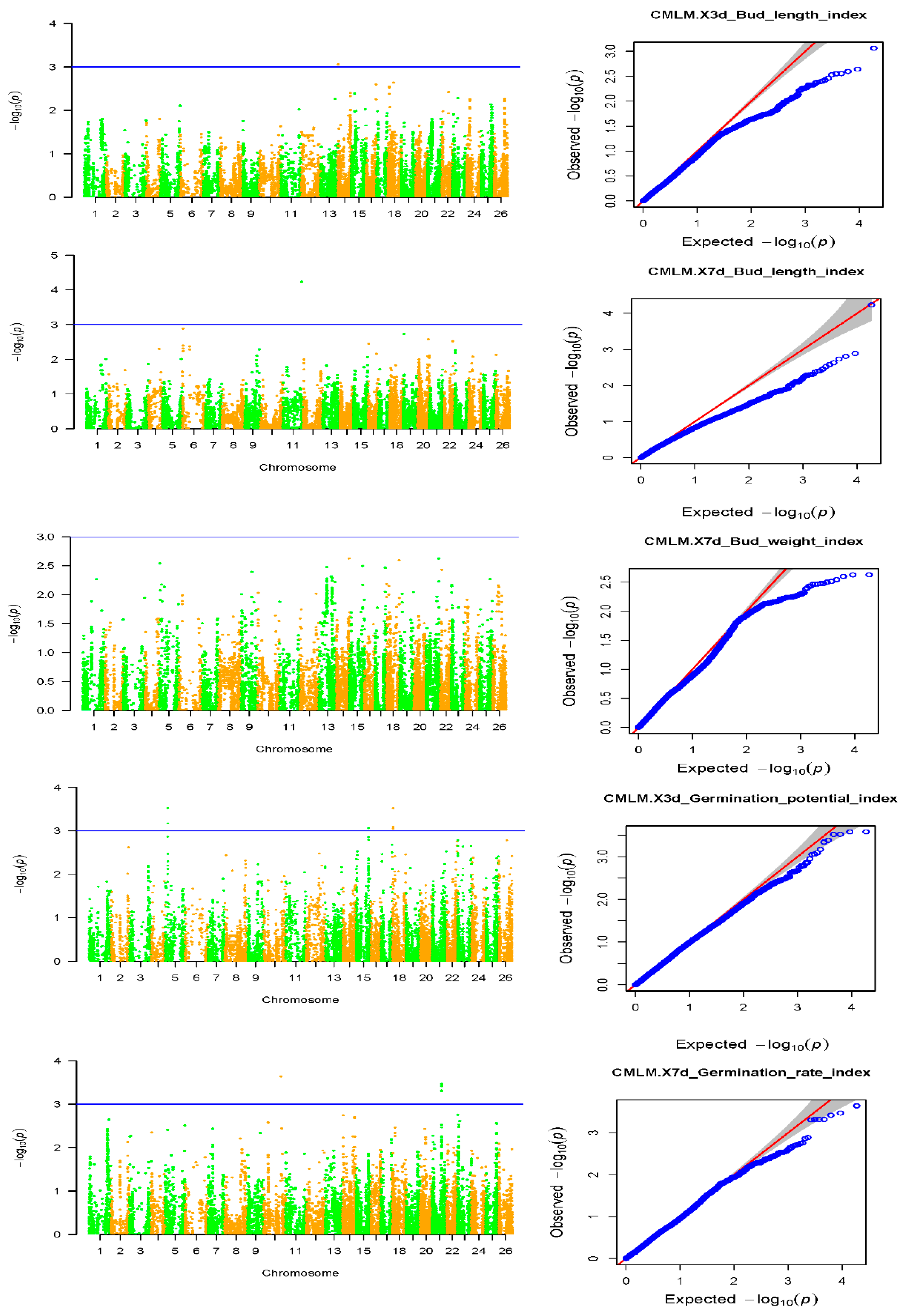
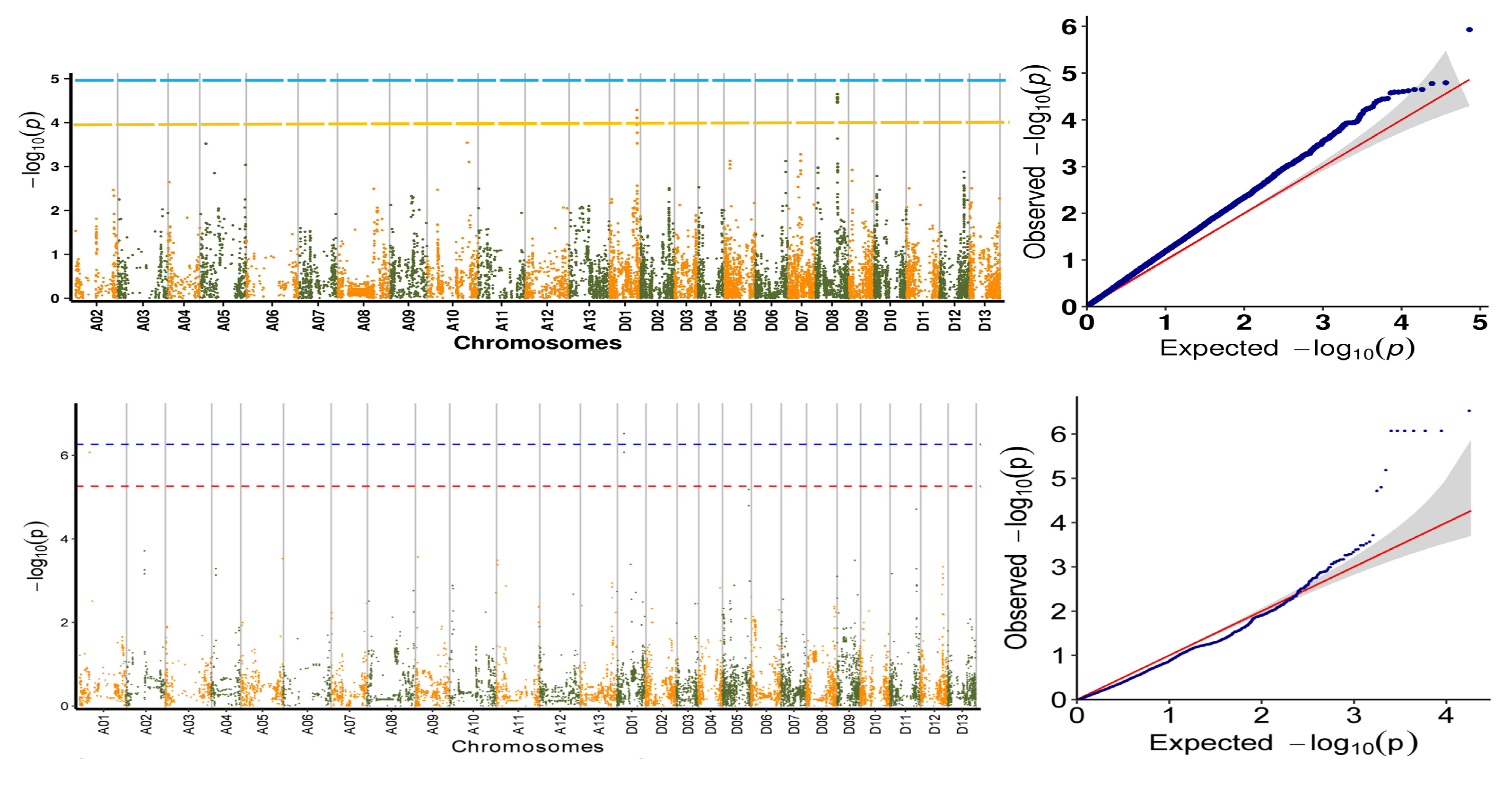
2.3. Association Analysis of Salt Tolerance Traits
2.4. Association Analysis of Salt Tolerance Index Traits among Cotton Accessions
2.5. Candidate Gene Screening
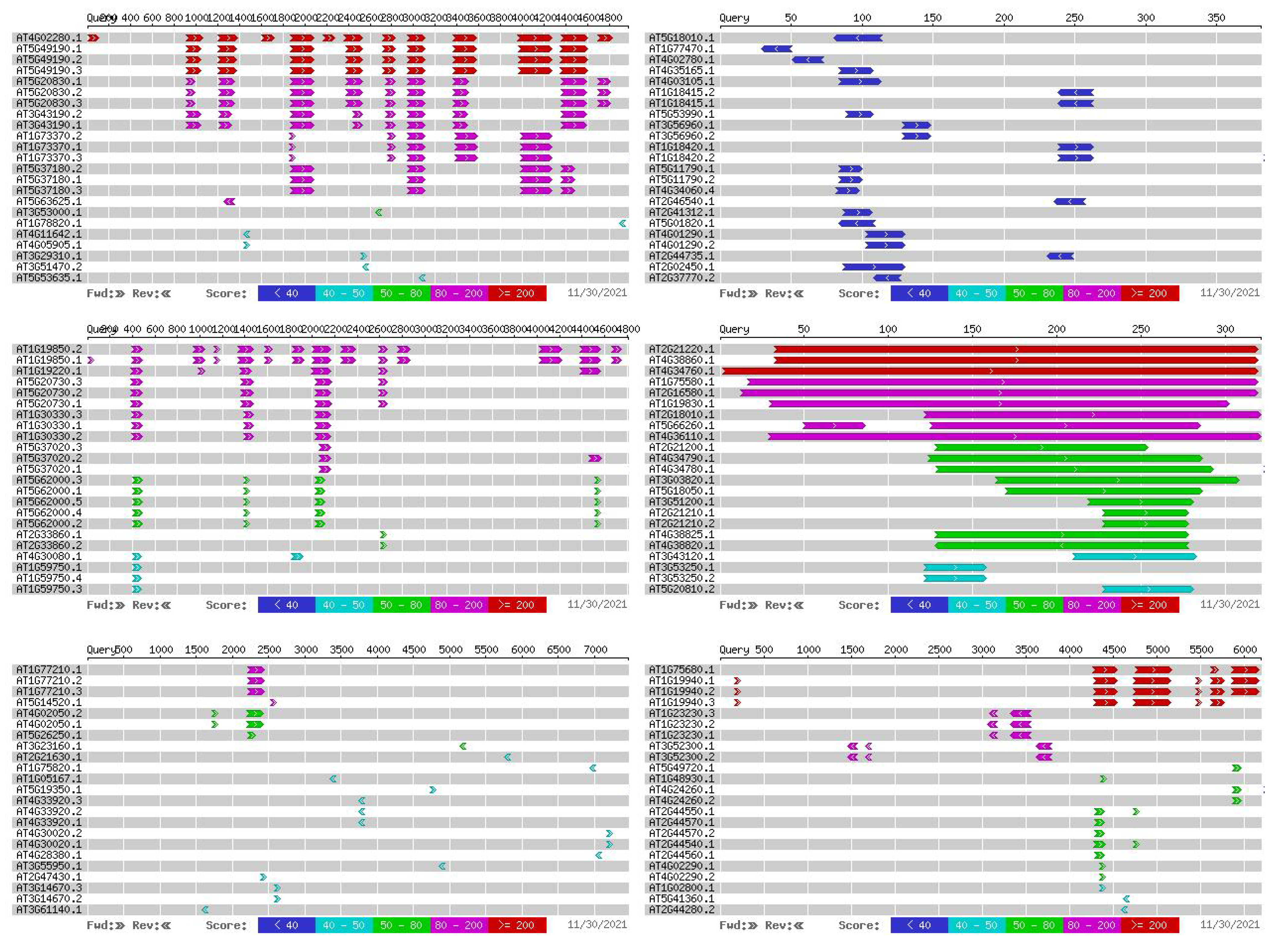
2.6. Alignment of Salt Resistance-Related Genes and Arabidopsis Homologous Sequences
3. Discussion
3.1. Target Gene Identification Based on GWAS
3.2. Functional Analysis of Candidate Genes
4. Conclusions
5. Materials and Methods
5.1. Test Materials
5.2. DNA Extraction and Genotyping
5.3. Molecular Genetic Diversity and Phylogenetic Analyses
5.4. Population Structure and Kinship Analysis
5.5. Linkage Disequilibrium Analysis
5.6. Association Analysis of Salt Tolerance Traits
5.7. Salt Stress Conditions and Salt-Tolerant Trait Collection
5.8. Prediction and Functional Annotation of Salt-Tolerant Candidate Genes
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liang, W.; Cui, W.; Ma, X.; Wang, G.; Huang, Z. Function of wheat Ta-UnP gene in enhancing salt tolerance in transgenic Arabidopsis and rice. Biochem. Biophys. Res. Commun. 2014, 450, 794–801. [Google Scholar] [CrossRef] [PubMed]
- Ullah, A.; Sun, H.; Yang, X.; Zhang, X. Drought coping strategies in cotton: Increased crop per drop. Plant Biotechnol. J. 2017, 15, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Long Enecker, D.E. The influence of soil so linity upon fruiting and shedding, boll characteristics, fibre properties and yields of two cotton species. Soil Sci. 1973, 115, 294–302. [Google Scholar] [CrossRef]
- Long Enecker, D.E. The influence of high so dium in soil upon fruiting and shedding, boll characteristics, fibre properties and yields of two cotton species. Soil Sci. 1974, 118, 387–396. [Google Scholar] [CrossRef]
- Razzouk, S.; Whittington, W. Effects of salinity on cotton yield and quality. Field Crop Res. 1991, 26, 305–314. [Google Scholar] [CrossRef]
- Xu, P.; Guo, Q.; Meng, S.; Zhang, X.; Xu, Z.; Guo, W.; Shen, X. Genome-wide association analysis reveals genetic variations and candidate genes associated with salt tolerance related traits in Gossypium hirsutum. BMC Genom. 2021, 22, 26. [Google Scholar] [CrossRef]
- Si, L.; Chen, J.; Huang, X.; Gong, H.; Luo, J.; Hou, Q.; Zhou, T.; Lu, T.; Zhu, J.; Shangguan, Y.; et al. OsSPL13 controls grain size in cultivated rice. Nat. Genet. 2016, 48, 447–456. [Google Scholar] [CrossRef]
- Yasir, M.; He, S.; Sun, G.; Geng, X.; Pan, Z.; Gong, W.; Jia, Y.; Du, X. A Genome-Wide Association Study Revealed Key SNPs/Genes Associated With Salinity Stress Tolerance In Upland Cotton. Genes 2019, 10, 829. [Google Scholar]
- Cai, C.; Zhu, G.; Zhang, T.; Guo, W. High-density 80 K SNP array is a powerful tool for genotypng G hirsutum accessions and genome analysis. BMC Genom. 2017, 18, 654. [Google Scholar]
- Sun, H.; Meng, M.; Yan, Z.; Lin, Z.; Nie, X.; Yang, X. Genome-wide association mapping of stress-tolerance traits in cotton. Crop J. 2018, 7, 77–88. [Google Scholar] [CrossRef]
- Huang, C.; Nie, X.; Shen, C.; You, C.; Li, W.; Zhao, W.; Zhang, X.; Lin, Z. Population structure and genetic basis of the agronomic traits of upland cotton China revealed by a genome-wide association study using high-density SNPs. Plant Biotechnol. J. 2017, 15, 1374–1386. [Google Scholar] [CrossRef]
- Paterson, A.H.; Wendel, J.F.; Gundlach, H.; Guo, H.; Jenkins, J.; Jin, D.; Llewellyn, D.; Showmaker, K.C.; Shu, S.; Udall, J.; et al. Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature 2012, 492, 423–427. [Google Scholar] [CrossRef] [Green Version]
- Abdelraheem, A.; Thyssen, G.N.; Fang, D.D.; Jenkins, J.N.; Mccarty, J.C.; Wedegaertner, T.; Zhang, J. GWAS reveals consistent QTL for drought and salt tolerance in a MAGIC population of 550 lines derived from intermating of 11 Upland cotton (Gossypium hirsutum) parents. Mol. Genet. Genom. 2020, 296, 119–129. [Google Scholar] [CrossRef]
- Taghizadeh, N.; Ranjbar, G.A.; Nematzadeh, G.A.; Moghaddam, M.R.R. Salt-related Genes Expression in Salt-Tolerant and Salt-Sensitive Cultivars of Cotton (Gossypium sp. L.) under NaCl Stress. J. Plant Mol. Breed. 2018, 6, 1–15. [Google Scholar]
- Frouin, J.; Languillaume, A.; Mas, J.; Mieulet, D.; Boisnard, A.; Labeyrie, A.; Bettembourg, M.; Bureau, C.; Lorenzini, E.; Portefaix, M.; et al. Tolerance to mild salinity stress in japonica rice: A genome-wide association mapping study highlights calcium signaling and metabolism genes. PLoS ONE 2018, 13, e0190964. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, N.; Khan, M.O.; Islam, E.; Wei, Z.-Y.; McAusland, L.; Lawson, T.; Johnson, G.N.; Nixon, P.J. Contrasting Responses to Stress Displayed by Tobacco Overexpressing an Algal Plastid Terminal Oxidase in the Chloroplast. Front. Plant Sci. 2020, 11, 501. [Google Scholar] [CrossRef] [PubMed]
- Stich, B.; Maurer, H.P.; Melchinger, A.E.; Frisch, M.; Heckenberger, M.; Van Der Voort, J.R.; Peleman, J.; Sørensen, A.P.; Reif, J.C. Comparison of Linkage Disequilibrium in Elite European Maize Inbred Lines using AFLP and SSR Markers. Mol. Breed. 2006, 17, 217–226. [Google Scholar] [CrossRef]
- Yang, X.; Yan, J.; Zheng, Y.; Yu, J.; Li, J. Reviews of association analysis for quantitative traits in plants. Acta Agron. Sin. 2007, 33, 523–530. [Google Scholar]
- Page, J.T.; Liechty, Z.S.; Alexander, R.H.; Clemons, K.; Hulse-Kemp, A.M.; Ashrafi, H.; Van Deynze, A.; Stelly, D.M.; Udall, J.A. DNA Sequence Evolution and Rare Homoeologous Conversion in Tetraploid Cotton. PLoS Genet. 2016, 12, e1006012. [Google Scholar] [CrossRef]
- Wang, M.; Tu, L.; Yuan, D.; Zhu, D.; Shen, C.; Li, J.; Liu, F.; Pei, L.; Wang, P.; Zhao, G.; et al. Reference genome sequences of two cultivated allotetraploid cottons, Gossypium hirsutum and Gossypium barbadense. Nat. Genet. 2018, 51, 224–229. [Google Scholar] [CrossRef] [Green Version]
- Hatzig, S.V.; Frisch, M.; Breuer, F.; Nesi, N.; Ducournau, S.; Wagner, M.-H.; Leckband, G.; Abbadi, A.; Snowdon, R.J. Genome-wide association mapping unravels the genetic control of seed germination and vigor in Brassica napus. Front. Plant Sci. 2015, 6, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Song, Q.; Cregan, P.B.; Nelson, R.L.; Wang, X.; Wu, J.; Jiang, G.-L. Genome-wide association study for flowering time, maturity dates and plant height in early maturing soybean (Glycine max) germplasm. BMC Genom. 2015, 16, 217. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L.; Guan, X.; Chen, J.; Zhang, J.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M.; et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Han, B. Natural variations and genome—wide assoclatton studies crop plants. Annu. Rev. Plant Bio. 2014, 165, 531–551. [Google Scholar] [CrossRef] [PubMed]
- Platt, A.; Vilhjálmsson, B.J.; Nordborg, M. Conditions under whmh genome-wlde association studies will be positively mlsleadmg. Genetics 2010, 186, 1045–1052. [Google Scholar] [CrossRef] [Green Version]
- Korte, A.; Farlow, A. The advantages and limitations of trait analysis with GWAS: A review. Plant Methods 2013, 9, 29. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Li, H.; Zhang, Y.; Li, Z.; Ke, H.; Wu, L.; Zhang, G.; Wang, X.; Ma, Z. Identification of SNPs and Candidate Genes Associated With Salt Tolerance at the Seedling Stage in Cotton (Gossypium hirsutum L.). Front. Plant Sci. 2018, 9, 1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, L.; Cai, C.; Wu, S.; Zhang, F.; Hou, S.; Guo, W. Evaluation and Exploration of Favorable QTL Alleles for Salt Stress Related Traits in Cotton Cultivars (G. hirsutum L.). PLoS ONE 2016, 11, e0151076. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.-H.; Sun, J.-L.; Wang, X.-W.; Zhou, Z.-L.; Pan, Z.-E.; He, S.-P.; Pang, B.-Y.; Wang, L.-R.; DU, X.-M. Molecular Diversity and Association Analysis of Drought and Salt Tolerance in Gossypium hirsutum L. Germplasm. J. Integr. Agric. 2014, 13, 1845–1853. [Google Scholar] [CrossRef] [Green Version]
- Reddy, U.K.; Nimmakayala, P.; Abburi, V.L.; Reddy, C.V.C.M.; Saminathan, T.; Percy, R.G.; John, Z.Y.; Frelichowski, J.; Udall, J.A.; Page, J.T.; et al. Genome-wlde divergence, haplotype distribution and population demographic hmtones for Gossypmm hwsutum and Gossypmm barbadense as revealed by genome-anchored SNPs. Sci. Rep. 2017, 7, 41285. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.; Wu, J.; Li, Y.; Nan, Z.; Guo, X.; Wang, Y.; Zhang, A.; Wang, Z.; Xia, G.; Tian, Y. Synergistic Effects of GhSOD1 and GhCAT1 Overexpression in Cotton Chloroplasts on Enhancing Tolerance to Methyl Viologen and Salt Stresses. PLoS ONE 2013, 8, e54002. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Yu, Y.; Wang, D.; Wu, C.; Yang, G.; Huang, J.; Zheng, C. GhZFP1, a novel CCCH-type zinc finger protein from cotton, enhances salt stress tolerance and fungal disease resistance in transgenic tobacco by interacting with GZIRD21A and GZIPR5. New Phytol. 2009, 183, 62–75. [Google Scholar] [CrossRef]
- Meng, C.; Cai, C.; Zhang, T.; Guo, W. Characterization of six novel NAC genes and their responses to abiotic stresses in Gossypium hirsutum L. Plant Sci. 2009, 176, 352–359. [Google Scholar] [CrossRef]
- Johnson, K.L.; Jones, B.J.; Bacic, A.; Schultz, C.J. The fasciclin-like arabinogalactan pro- teins of Arabidopsis. A multigene family of putative cell adhesion molecules. Plant Physiol. 2003, 133, 1911–1925. [Google Scholar] [CrossRef] [Green Version]
- Xue, T.; Li, X.; Zhu, W.; Wu, C.; Yang, G.; Zheng, C. Cotton metallothionein GhMT3a, a reactive oxygen species scavenger, increased tolerance against abiotic stress in transgenic tobacco and yeast. J. Exp. Bot. 2008, 60, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X.; Li, Z.; Zhang, G.; Ma, Z. Assessing genetic diversity of cotton cultivars using genomic and newly developed expressed sequence tag-derived microsatellite markers. Genet. Mol. Res. 2011, 10, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.A.; Yang, G.D.; Meng, Q.W.; Zheng, C.C. The cotton GhNHX1 gene encoding a novel putative tonoplast Na+/H+ antiporter plays an important role in salt stress. Plant Cell Physiol. 2004, 45, 600–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelraheem, A.; Thyssen, G.; Jenkins, J.; McCarty, J.; Fang, D.D.; Zhang, J.F. A genome-wide association study of tolerance to biotic and abiotic stresses in a MAGIC population of upland cotton. In Proceedings of the 2018 Beltwild Cotton Conferences, San Antonio, TX, USA, 3–5 January 2018; p. 565. [Google Scholar]
- Abdelraheem, A.; Liu, F.; Song, M.; Zhang, J.F. A meta-analysis of quantitative trait loci for abiotic and biotic stress resistance in tetraploid cotton. Mol. Genet. Genom. 2017, 292, 1221–1235. [Google Scholar] [CrossRef]
- Baroja-Fernández, E.; Muñoz, F.J.; Li, J.; Bahaji, A.; Almagro, G.; Montero, M.; Etxeberria, E.; Hidalgo, M.; Sesma, M.T.; Pozueta-Romero, J. Sucrose synthase activity in the sus1/sus2/sus3/sus4 Arabidopsis mutant is sufficient to support normal cellulose and starch production. Proc. Natl. Acad. Sci. USA 2012, 109, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Li-Na, L.I.; Jian-Qiang, K. Gene Organization, Function and Application of Plant Sucrose Synthase. Chin. J. Biochem. Mol. Biol. 2015, 9, 904–913. [Google Scholar]
- Angeles-Núñez, J.G.; Tiessen, A. Arabidopsis sucrose synthase 2 and 3 modulate metabolic homeostasis and direct carbon towards starch synthesis in developing seeds. Planta 2010, 232, 701–718. [Google Scholar] [CrossRef] [PubMed]
- Angeles-Núñez, J.G.; Tiessen, A. Regulation of AtSUS2 and AtSUS3 by glucose and the transcription factor LEC2 in different tissues and at different stages of Arabidopsis seed development. Plant Mol. Biol. 2012, 78, 377–392. [Google Scholar] [CrossRef]
- Wen, X.; Zhang, W.; Feng, Y.; Yu, X. Cloning and characterization of a sucrose synthase-encoding gene from muskmelon. Mol. Biol. Rep. 2009, 37, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Baud, S.; Vaultier, M.; Rochat, C. Structure and expression profile of the sucrose synthase multigene family in Arabidopsis. J. Exp. Bot. 2004, 55, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Poschet, G.; Hannich, B.; Büttner, M. Identification and Characterization of AtSTP14, a Novel Galactose Transporter from Arabidopsis. Plant Cell Physiol. 2010, 51, 1571–1580. [Google Scholar] [CrossRef] [Green Version]
- Bavnhøj, L.; Paulsen, P.A.; Flores-Canales, J.C.; Schiøtt, B.; Pedersen, B.P. Molecular mechanism of sugar transport in plants unveiled by structures of glucose/H+ symporter STP10. Nat. Plants 2021, 7, 1409–1419. [Google Scholar] [CrossRef]
- Nørholm, M.H.; Nour-Eldin, H.H.; Brodersen, P.; Mundy, J.; Halkier, B.A. Expression of the Arabidopsis high-affinity hexose transporter STP13 correlates with programmed cell death. FEBS Lett. 2006, 580, 2381–2387. [Google Scholar] [CrossRef] [Green Version]
- Rottmann, T.M.; Klebl, F.; Schneider, S.; Kischka, D.; Rüscher, D.; Sauer, N.; Stadler, R. Sugar Transporter STP7 Specificity for l-Arabinose and d-Xylose Contrasts with the Typical Hexose Transporters STP8 and STP12. Plant Physiol. 2018, 176, 2330–2350. [Google Scholar] [CrossRef] [Green Version]
- McBride, Z.; Chen, D.; Reick, C.; Xie, J.; Szymanski, D.B. Global Analysis of Membrane-associated Protein Oligomerization Using Protein Correlation Profiling. Mol. Cell. Proteom. 2017, 16, 1972–1989. [Google Scholar] [CrossRef] [Green Version]
- Bissaro, B.; Monsan, P.; Fauré, R.; O’Donohue, M.J. Glycosynthesis in a waterworld: New insight into the molecular basis of transglycosylation in retaining glycoside hydrolases. Biochem. J. 2015, 467, 17–35. [Google Scholar] [CrossRef]
- Honer, J.; Niemeyer, K.M.; Fercher, C.; Tissera, A.L.D.; Jaberolansar, N.; Jafrani, Y.M.; Zhou, C.; Caramelo, J.J.; Shewan, A.M.; Schulz, B.L.; et al. TorsinA folding and N-linked glycosylation are sensitive to redox homeostasis. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 119073. [Google Scholar] [CrossRef]
- Konishi, M.; Donner, T.J.; Scarpella, E.; Yanagisawa, S. MONOPTEROS directly activates the auxin-inducible promoter of the Dof5.8 transcription factor gene in Arabidopsis thaliana leaf provascular cells. J. Exp. Bot. 2014, 66, 283–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Möller, B.K.; Colette, A.; Xiang, D.; Williams, N.; López, L.G.; Yoshida, S.; Smit, M.; Datla, R.; Weijers, D. Auxin response cell-autonomously controls ground tissue initiation in the early Arabidopsis embryo. Proc. Natl. Acad. Sci. USA 2017, 114, E2533–E2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odat, O.; Gardiner, J.; Sawchuk, M.G.; Verna, C.; Donner, T.J.; Scarpella, E. Characterization of an allelic series in the MONOPTEROS gene of Arabidopsis. Genesis 2014, 52, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Ckurshumova, W.; Smirnova, T.; Marcos, D.; Zayed, Y.; Berleth, T. Irrepressible MONOPTEROS/ARF 5 promotes de novo shoot formation. New Phytol. 2014, 204, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Fendrych, M.; Leung, J.; Friml, J. TIR1/AFB-Aux/IAA auxin perception mediates rapid cell wall acidification and growth of Arabidopsis hypocotyls. eLife 2016, 5, e19048. [Google Scholar] [CrossRef] [PubMed]
- Niemeyer, M.; Castillo, E.M.; Ihling, C.H.; Iacobucci, C.; Wilde, V.; Hellmuth, A.; Hoehenwarter, W.; Samodelov, S.L.; Zurbriggen, M.D.; Kastritis, P.L.; et al. Author Correction: Flexibility of intrinsically disordered degrons in AUX/IAA proteins reinforces auxin co-receptor assemblies. Nat. Commun. 2021, 12, 1768. [Google Scholar] [CrossRef] [PubMed]
- Spartz, A.K.; Lee, S.H.; Wenger, J.P.; Gonzalez, N.; Itoh, H.; Inzé, D.; Peer, W.A.; Murphy, A.S.; Overvoorde, P.J.; Gray, W.M. The SAUR19 subfamily of SMALL AUXIN UP RNA genes promote cell expansion. Plant J. 2012, 70, 978–990. [Google Scholar] [CrossRef] [Green Version]
- Spartz, A.K.; Ren, H.; Park, M.Y.; Grandt, K.N.; Lee, S.H.; Murphy, A.S.; Sussman, M.R.; Overvoorde, P.J.; Gray, W.M. SAUR Inhibition of PP2C-D Phosphatases Activates Plasma Membrane H+-ATPases to Promote Cell Expansion in Arabidopsis. Plant Cell 2014, 26, 2129–2142. [Google Scholar] [CrossRef] [Green Version]
- Stortenbeker, N.; Bemer, M. The SAUR gene family: The plant’s toolbox for adaptation of growth and development. J. Exp. Bot. 2019, 70, 17–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.-E.; Kim, Y.-S.; Yoon, H.-K.; Park, C.-M. Functional characterization of a small auxin-up RNA gene in apical hook development in Arabidopsis. Plant Sci. 2007, 172, 150–157. [Google Scholar] [CrossRef]
- Leyser, O. Auxin signaling. Plant Physiol. 2018, 176, 465–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.; Duan, X.; Luo, L.; Dai, S.; Ding, Z.; Xia, G. How Plant Hormones Mediate Salt Stress Responses. Trends Plant Sci. 2020, 25. [Google Scholar] [CrossRef]
- Parida, A.K.; Das, A.B.; Mohanty, P. Investigations on the antioxidative defence responses to NaCl stress in a mangrove, Bruguiera parviflora: Differential regulations of isoforms of some antioxidative enzymes. Plant Growth Regul. 2004, 42, 213–226. [Google Scholar] [CrossRef]
- Said, J.I.; Lin, Z.; Zhang, X.; Song, M.; Zhang, J. A comprehen- sive meta QTL analysis for fiber quality, yield, yield related and morphological traits, drought tolerance, and disease resistance in tetraploid cotton. BMC Genom. 2013, 14, 776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Said, J.I.; Song, M.; Wang, H.; Lin, Z.; Zhang, X.; Fang, D.D.; Zhang, J. A comparative Meta- analysis of QTL between intraspe- cific Gossypium hirsutum and interspecific G. hirsutum x G. bar- badense populations. Mol. Genet. Genom. 2015, 290, 1003–1025. [Google Scholar] [CrossRef]
- Said, J.I.; Knapka, J.A.; Song, M.; Zhang, J. Cotton QTLdb: A cotton QTL database for QTL analysis, visualization, and comparison between Gossypium hirsutum and G. hirsutum x G. barbadense populations. Mol. Genet. Genom. 2015, 290, 1615–1625. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [Green Version]
- Hardy, O.J.; Vekemans, X. SPAGeDi: A versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol. Ecol. Notes 2002, 2, 618–620. [Google Scholar] [CrossRef] [Green Version]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higbie, S.M.; Wang, F.; Stewart, J.M.; Sterling, T.M.; Lindemann, W.C.; Hughs, E.; Zhang, J. Physiological Response to Salt (NaCl) Stress in Selected Cultivated Tetraploid Cottons. Int. J. Agron. 2010, 2010, 643475. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, R.S.; Picchioni, G.A.; Steiner, R.L.; Hughs, S.E.; Jones, D.C.; Zhang, J. Genetic variation in salt tolerance during seed germina- tion in a backcross inbred line population and advanced breeding lines derived from Gossypium hirsutum x G. barbadense. Crop Sci. 2013, 53, 1974–1982. [Google Scholar] [CrossRef]
- Tiwari, R.S.; Picchioni, G.A.; Steiner, R.L.; Jones, D.C.; Hughs, S.E.; Zhang, J. Genetic variation in salt tolerance at the seedling stage in an interspecific backcross inbred line population of cotton. Euphytica 2013, 194, 1–11. [Google Scholar] [CrossRef]
- Barrick, B.; Steiner, R.; Picchioni, G.; Ulery, A.; Zhang, J. Salinity responses of selected introgression cotton grown in two soils. J. Cotton Sci. 2015, 19, 268–278. [Google Scholar]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Bi, I.V.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3d Germination Potential Index | 7d Germination Rate Index | 7d Radicle Weight Index | 3d Radicle Length Index | 7d Radicle Length Index | |
|---|---|---|---|---|---|
| 3d Germination potential index | 1 | 0.565096 | 0.330095 | 0.006608 | 0.178595 |
| 7d Germination rate index | 0.565096 | 1 | 0.484635 | 0.000286 | −0.03288 |
| 7d Radicle weight index | 0.330095 | 0.484635 | 1 | 0.475708 | 0.192883 |
| 3d Radicle length index | 0.006608 | 0.000286 | 0.475708 | 1 | 0.230305 |
| 7d Radicle length index | 0.178595 | −0.03288 | 0.192883 | 0.230305 | 1 |
| Traits | Chromosomes | Position | p-Value | −log10(p) | Alleles |
|---|---|---|---|---|---|
| 7d Radicle length | D08 | 64,320,143 | 0.000019 | 4.731944 | T/G |
| D13 | 35,877,478 | 0.000046 | 4.334531 | C/T | |
| scaffold33030 | 93 | 0.000099 | 4.004747 | A/C | |
| 7d Radicle length drop rate | D01 | 16,458,717 | 0.000000 | 6.588126 | T/C |
| A01 | 22,140,802 | 0.000001 | 6.1328 | T/C | |
| A01 | 22,174,537 | 0.000001 | 6.1328 | T/C | |
| A01 | 22,350,123 | 0.000001 | 6.1328 | G/A | |
| A01 | 22,376,230 | 0.000001 | 6.1328 | G/A | |
| A01 | 22,387,777 | 0.000001 | 6.1328 | T/G | |
| D01 | 16,458,204 | 0.000001 | 6.1328 | C/A | |
| D05 | 58,225,028 | 0.000006 | 5.226497 | T/C | |
| D05 | 58,227,014 | 0.000015 | 4.83936 | T/C | |
| D11 | 58,447,888 | 0.000017 | 4.762019 | C/T | |
| 7d Germination rate | D05 | 12,154,352 | 0.000028 | 4.560614 | C/A |
| D05 | 12,155,655 | 0.000030 | 4.516439 | C/T | |
| A05 | 12,181,224 | 0.000078 | 4.109102 | G/A | |
| 7d Germination weight | D08 | 2,191,589 | 0.000054 | 4.264409 | G/A |
| D05 | 12,154,352 | 0.000056 | 4.255444 | C/A | |
| D05 | 12,155,655 | 0.000092 | 4.035935 | C/T | |
| Relative germination rate | D08 | 43,495,093 | 0.000022 | 4.65032 | A/G |
| D08 | 43,541,365 | 0.000027 | 4.57704 | A/G | |
| D08 | 43,557,843 | 0.000029 | 4.5365 | T/C | |
| D08 | 43,483,056 | 0.000033 | 4.48071 | A/G | |
| D08 | 43,501,088 | 0.000034 | 4.473257 | C/T | |
| D08 | 43,479,511 | 0.000035 | 4.458785 | C/T | |
| D01 | 54,316,248 | 0.000051 | 4.28993 | T/C | |
| D01 | 54,289,162 | 0.000078 | 4.106987 | C/A |
| Traits | Chromosomes | Position | p-Value | −log10(p) | Alleles |
|---|---|---|---|---|---|
| 7d Radicle length index | A11 | 85,527,572 | 0.000058 | 4.236572 | T/C |
| 7d Germination rate index | A10 | 79,275,413 | 0.000229 | 3.640165 | C/T |
| D08 | 43,479,511 | 0.000485 | 3.314258 | C/T | |
| D08 | 43,483,056 | 0.000485 | 3.314258 | A/G | |
| D08 | 43,495,093 | 0.00034 | 3.468521 | A/G | |
| D08 | 43,501,088 | 0.000485 | 3.314258 | C/T | |
| D08 | 43,541,365 | 0.000384 | 3.415669 | A/G | |
| D08 | 43,557,843 | 0.000495 | 3.305395 | T/C | |
| 3d Germination potential index | A05 | 12,472,578 | 0.000299 | 3.524329 | A/G |
| A05 | 12,473,346 | 0.000675 | 3.170696 | A/C | |
| D02 | 56,360,435 | 0.000872 | 3.059484 | A/G | |
| D05 | 3,861,440 | 0.000815 | 3.088842 | T/C | |
| D05 | 3,870,101 | 0.000887 | 3.052076 | A/G | |
| D05 | 3,873,693 | 0.000301 | 3.521434 | G/A | |
| 3d Radicle length index | D01 | 883,613 | 0.000872 | 3.059484 | C/T |
| Chromosomes | Gene | Position |
|---|---|---|
| D01 | Gh_D01G0943 | 15,976,403–15,982,586 |
| Gh_D01G0945 | 16,034,346–16,041,810 | |
| A01 | Gh_A01G0906 | 21,801,560–21,801,880 |
| Gh_A01G0908 | 22,204,069–22,208,876 | |
| D08 | Gh_D08G1308 | 43,063,260–43,063,640 |
| Gh_D08G1309 | 43,067,294–43,072,264 |
| Chromosomes | Gene | Arabidopsis Homology Gene | Homology Index |
|---|---|---|---|
| D01 | Gh_D01G0943 | AT1G75680 | 76% |
| Gh_D01G0945 | AT1G77210 | 75% | |
| A01 | Gh_A01G0906 | AT2G21220 | 78% |
| Gh_A01G0908 | AT1G19850 | 83% | |
| D08 | Gh_D08G1308 | AT5G18010 | 83% |
| Gh_D08G1309 | AT4G02280 | 75% |
| Number | Breed Name | Origin | Number | Breed Name | Origin | Number | Breed Name | Origin |
|---|---|---|---|---|---|---|---|---|
| 1 | Xinluzao No. 1 | Inland Northwest | 51 | Xinluzhong No. 2 | Inland Northwest | 101 | Coker310 | Central Asia |
| 2 | Xinluzao No. 2 | Inland Northwest | 52 | Xinluzhong No. 6 | Inland Northwest | 102 | Dunhuang 77-116 | Inland Northwest |
| 3 | Xinluzao No. 3 | Inland Northwest | 53 | Xinluzhong No. 7 | Inland Northwest | 103 | Shan 63-1 | Yellow River Basin |
| 4 | Xinluzao No. 4 | Inland Northwest | 54 | Xinluzhong No. 8 | Inland Northwest | 104 | Shanmian No. 9 | Yellow River Basin |
| 5 | Xinluzao No. 5 | Inland Northwest | 55 | Xinluzhong No. 9 | Inland Northwest | 105 | G164 2 20 | Yellow River Basin |
| 6 | Xinluzao No. 7 | Inland Northwest | 56 | Xinluzhong No. 10 | Inland Northwest | 106 | Jimian No. 11 | Yellow River Basin |
| 7 | Xinluzao No. 8 | Inland Northwest | 57 | Xinluzhong No. 11 | Inland Northwest | 107 | Lumian No. 17 | Yellow River Basin |
| 8 | Xinluzao No. 9 | Inland Northwest | 58 | Xinluzhong No. 12 | Inland Northwest | 108 | Lumian No. 28 | Yellow River Basin |
| 9 | Xinluzao No. 10 | Inland Northwest | 59 | Xinluzhong No. 14 | Inland Northwest | 109 | Lu 24 | Yellow River Basin |
| 10 | Xinluzao No. 11 | Inland Northwest | 60 | Xinluzhong No. 15 | Inland Northwest | 110 | Lu 25 | Yellow River Basin |
| 11 | Xinluzao No. 12 | Inland Northwest | 61 | Xinluzhong No. 16 | Inland Northwest | 111 | Lu 34 | Yellow River Basin |
| 12 | Xinluzao No. 13 | Inland Northwest | 62 | Xinluzhong No. 17 | Inland Northwest | 112 | Yumian No. 17 | Yangtze River Basin |
| 13 | Xinluzao No. 15 | Inland Northwest | 63 | Xinluzhong No. 19 | Inland Northwest | 113 | Baimian No. 1 | Yellow River Basin |
| 14 | Xinluzao No. 16 | Inland Northwest | 64 | Xinluzhong No. 21 | Inland Northwest | 114 | Zhong 93001 | Yellow River Basin |
| 15 | Xinluzao No. 17 | Inland Northwest | 65 | Xinluzhong No. 22 | Inland Northwest | 115 | CCRI No. 12 | Yellow River Basin |
| 16 | Xinluzao No. 18 | Inland Northwest | 66 | Xinluzhong No. 26 | Inland Northwest | 116 | CCRI No. 16 | Yellow River Basin |
| 17 | Xinluzao No. 19 | Inland Northwest | 67 | Xinluzhong No. 27 | Inland Northwest | 117 | Zhongmian 41 | Yellow River Basin |
| 18 | Xinluzao No. 20 | Inland Northwest | 68 | Xinluzhong No. 28 | Inland Northwest | 118 | CCRI No. 43 | Yellow River Basin |
| 19 | Xinluzao No. 21 | Inland Northwest | 69 | Xinluzhong No. 30 | Inland Northwest | 119 | Emian No. 10 | Yangtze River Basin |
| 20 | Xinluzao No. 23 | Inland Northwest | 70 | Xinluzhong No. 32 | Inland Northwest | 120 | Emian No. 12 | Yangtze River Basin |
| 21 | Xinluzao No. 24 | Inland Northwest | 71 | Xinluzhong No. 34 | Inland Northwest | 121 | Emian No. 21 | Yangtze River Basin |
| 22 | Xinluzao No. 25 | Inland Northwest | 72 | Xinluzhong No. 35 | Inland Northwest | 122 | Wanmian 8407 | Yangtze River Basin |
| 23 | Xinluzao No. 26 | Inland Northwest | 73 | Xinluzhong No. 36 | Inland Northwest | 123 | Sumian No. 8 | Yangtze River Basin |
| 24 | Xinluzao No. 27 | Inland Northwest | 74 | Xinluzhong No. 38 | Inland Northwest | 124 | Sumian No. 12 | Yangtze River Basin |
| 25 | Xinluzao No. 29 | Inland Northwest | 75 | Xinluzhong No. 39 | Inland Northwest | 125 | Suyuan 04-129 | Yangtze River Basin |
| 26 | Xinluzao No. 30 | Inland Northwest | 76 | Xinluzhong No. 40 | Inland Northwest | 126 | Ganmian No. 10 | Yangtze River Basin |
| 27 | Xinluzao No. 31 | Inland Northwest | 77 | Xinluzhong No. 41 | Inland Northwest | 127 | Ganmian No. 17 | Yangtze River Basin |
| 28 | Xinluzao No. 32 | Inland Northwest | 78 | Xinluzhong No. 42 | Inland Northwest | 128 | Chuan 7327 20 | Yangtze River Basin |
| 29 | Xinluzao No. 33 | Inland Northwest | 79 | Xinluzhong No. 44 | Inland Northwest | 129 | Yumian No. 1 | Yangtze River Basin |
| 30 | Xinluzao No. 34 | Inland Northwest | 80 | Xinluzhong No. 45 | Inland Northwest | 130 | Liaomian No. 9 | Special precocious cotton area |
| 31 | Xinluzao No. 35 | Inland Northwest | 81 | Xinluzhong No. 46 | Inland Northwest | 131 | Liaomian No. 16 | Special precocious cotton area |
| 32 | Xinluzao No. 36 | Inland Northwest | 82 | Xinluzhong No. 50 | Inland Northwest | 132 | Dai-80 | America |
| 33 | Xinluzao No. 37 | Inland Northwest | 83 | Xinluzhong No. 54 | Inland Northwest | 133 | Montenegro cotton No. 1 | Special precocious cotton area |
| 34 | Xinluzao No. 38 | Inland Northwest | 84 | Xinluzhong No. 56 | Inland Northwest | 134 | Pidcotton | Yellow River Basin |
| 35 | Xinluzao No. 39 | Inland Northwest | 85 | Xinluzhong No. 58 | Inland Northwest | 135 | MacNair 210 | America |
| 36 | Xinluzao No. 40 | Inland Northwest | 86 | Xinluzhong No. 59 | Inland Northwest | 136 | Huazhong 106 | Yangtze River Basin |
| 37 | Xinluzao No. 41 | Inland Northwest | 87 | Xinluzhong No. 60 | Inland Northwest | 137 | Keyuan No. 1 | Yellow River Basin |
| 38 | Xinluzao No. 42 | Inland Northwest | 88 | Xinluzhong No. 61 | Inland Northwest | 138 | Bu 3363 | America |
| 39 | Xinluzao No. 45 | Inland Northwest | 89 | Xinluzhong No. 62 | Inland Northwest | 139 | Bamian No. 1 | Yangtze River Basin |
| 40 | Xinluzao No. 46 | Inland Northwest | 90 | Xinluzhong No. 63 | Inland Northwest | 140 | Chad No. 3 | Africa |
| 41 | Xinluzao No. 47 | Inland Northwest | 91 | Xinluzhong No. 64 | Inland Northwest | 141 | Turkmen upland cotton | Central Asia |
| 42 | Xinluzao No. 48 | Inland Northwest | 92 | Xinluzhong No. 65 | Inland Northwest | 142 | NO Phenphenol phenol No. 1 | Yangtze River Basin |
| 43 | Xinluzao No. 49 | Inland Northwest | 93 | Xinluzhong No. 68 | Inland Northwest | 143 | Miscot7803-52 | America |
| 44 | Xinluzao No. 50 | Inland Northwest | 94 | Xinluzhong No. 69 | Inland Northwest | 144 | Si-6524 | Central Asia |
| 45 | Xinluzao No. 51 | Inland Northwest | 95 | Xinlu 201 | Inland Northwest | 145 | Sparculent H10 | America |
| 46 | Xinluzao No. 52 | Inland Northwest | 96 | Xinlu 202 | Inland Northwest | 146 | Yinmian No. 1 | Yellow River Basin |
| 47 | Xinluzao No. 53 | Inland Northwest | 97 | Nongken No. 5 | Inland Northwest | 147 | America 28114-313 | America |
| 48 | Xinluzao No. 60 | Inland Northwest | 98 | Shache Soil cotton | Inland Northwest | 148 | Columbia | South America |
| 49 | Xinluzao No. 61 | Inland Northwest | 99 | Kuche T94-4 | Inland Northwest | 149 | Miscot 8711ne | America |
| 50 | Xinluzhong No. 1 | Inland Northwest | 100 | Bazhou 6510 | Inland Northwest |
| Salt Tolerance Traits | Salt Tolerance Index |
|---|---|
| 7d Germination rate | 3d Germination potential index |
| 7d Germination weight | 7d Germination rate index |
| 3d Radicle length | 7d Radicle weight index |
| 7d Radicle length | 3d Radicle length index |
| Relative germination potential | 7d Radicle length index |
| Relative germination rate | |
| 7d Radicle weight drop rate | |
| 3d Germination potential | |
| 7d Radicle length drop rate | |
| 3d Radicle length drop rate |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, J.; Zhang, Z.; Gong, Z.; Liang, Y.; Sang, Z.; Xu, Y.; Li, X.; Wang, J. Genome-Wide Association Analysis of Salt-Tolerant Traits in Terrestrial Cotton at Seedling Stage. Plants 2022, 11, 97. https://doi.org/10.3390/plants11010097
Zheng J, Zhang Z, Gong Z, Liang Y, Sang Z, Xu Y, Li X, Wang J. Genome-Wide Association Analysis of Salt-Tolerant Traits in Terrestrial Cotton at Seedling Stage. Plants. 2022; 11(1):97. https://doi.org/10.3390/plants11010097
Chicago/Turabian StyleZheng, Juyun, Zeliang Zhang, Zhaolong Gong, Yajun Liang, Zhiwei Sang, Yanchao Xu, Xueyuan Li, and Junduo Wang. 2022. "Genome-Wide Association Analysis of Salt-Tolerant Traits in Terrestrial Cotton at Seedling Stage" Plants 11, no. 1: 97. https://doi.org/10.3390/plants11010097
APA StyleZheng, J., Zhang, Z., Gong, Z., Liang, Y., Sang, Z., Xu, Y., Li, X., & Wang, J. (2022). Genome-Wide Association Analysis of Salt-Tolerant Traits in Terrestrial Cotton at Seedling Stage. Plants, 11(1), 97. https://doi.org/10.3390/plants11010097