
Genomic Insights into Cultivated Mexican Vanilla planifolia Reveal High Levels of Heterozygosity Stemming from Hybridization



Abstract
:1. Introduction
2. Results
2.1. Review of Plant Levels of Genomic Heterozygosity Inferred Using GenomeScope
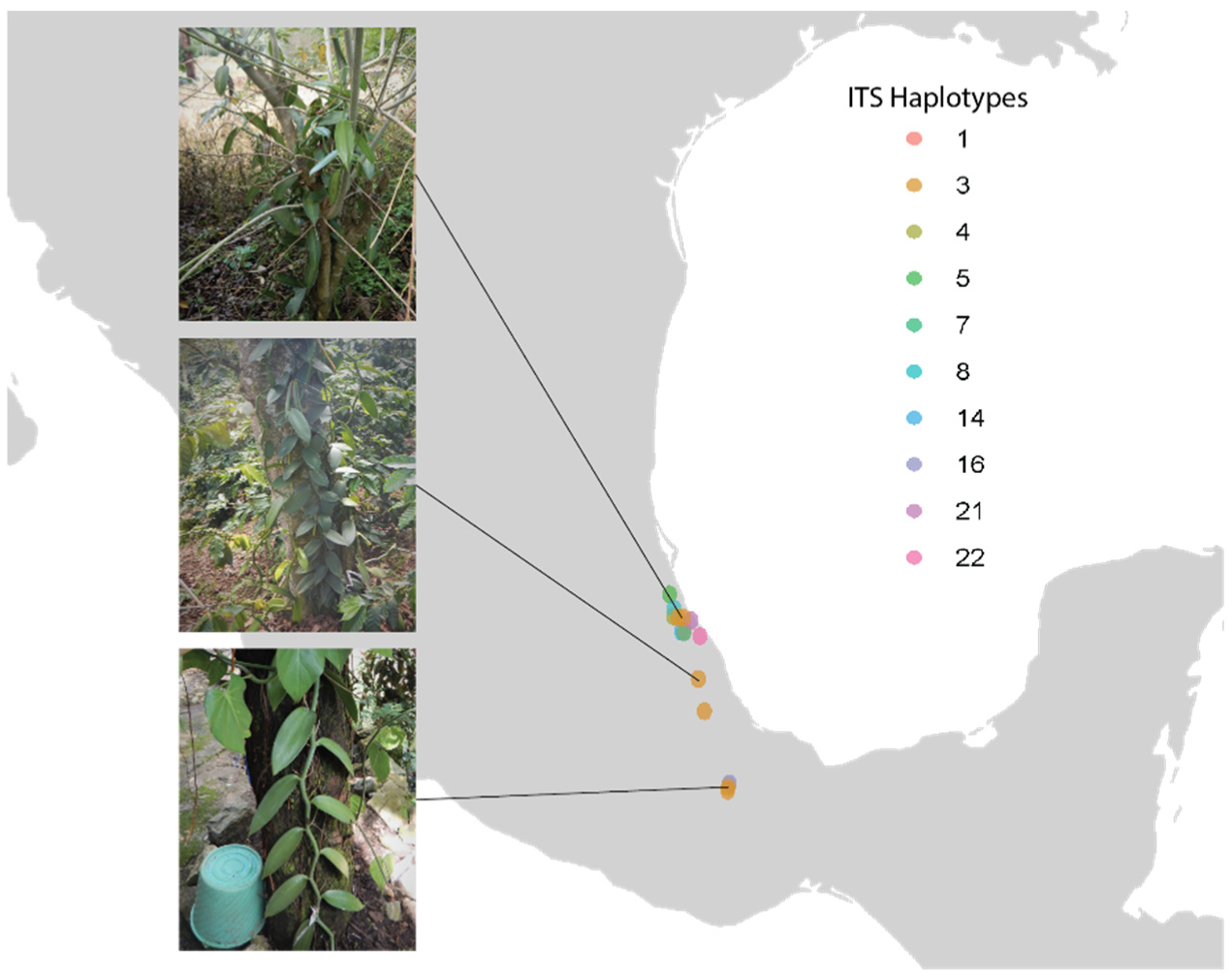
2.2. Sampling, DNA Extraction, and Whole Genome Re-Sequencing
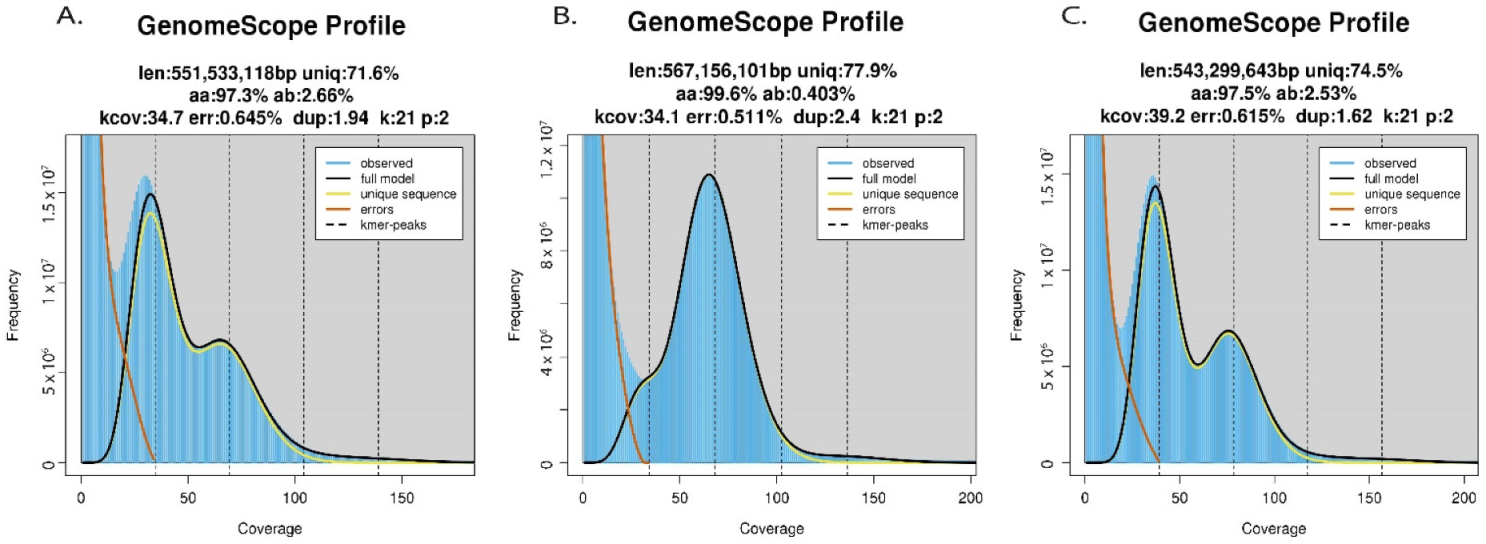
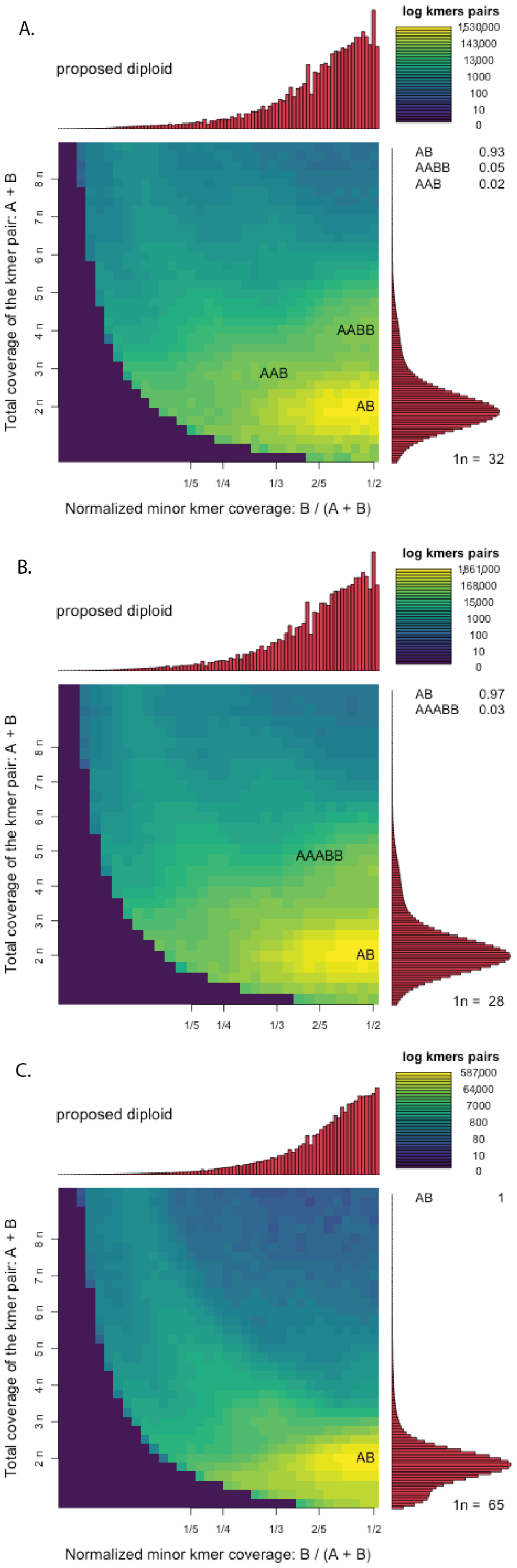
2.3. Genomic Heterozygosity, Ploidy, and Complexity
2.4. Genome Reconstructions to Infer Structural Variation and Synteny
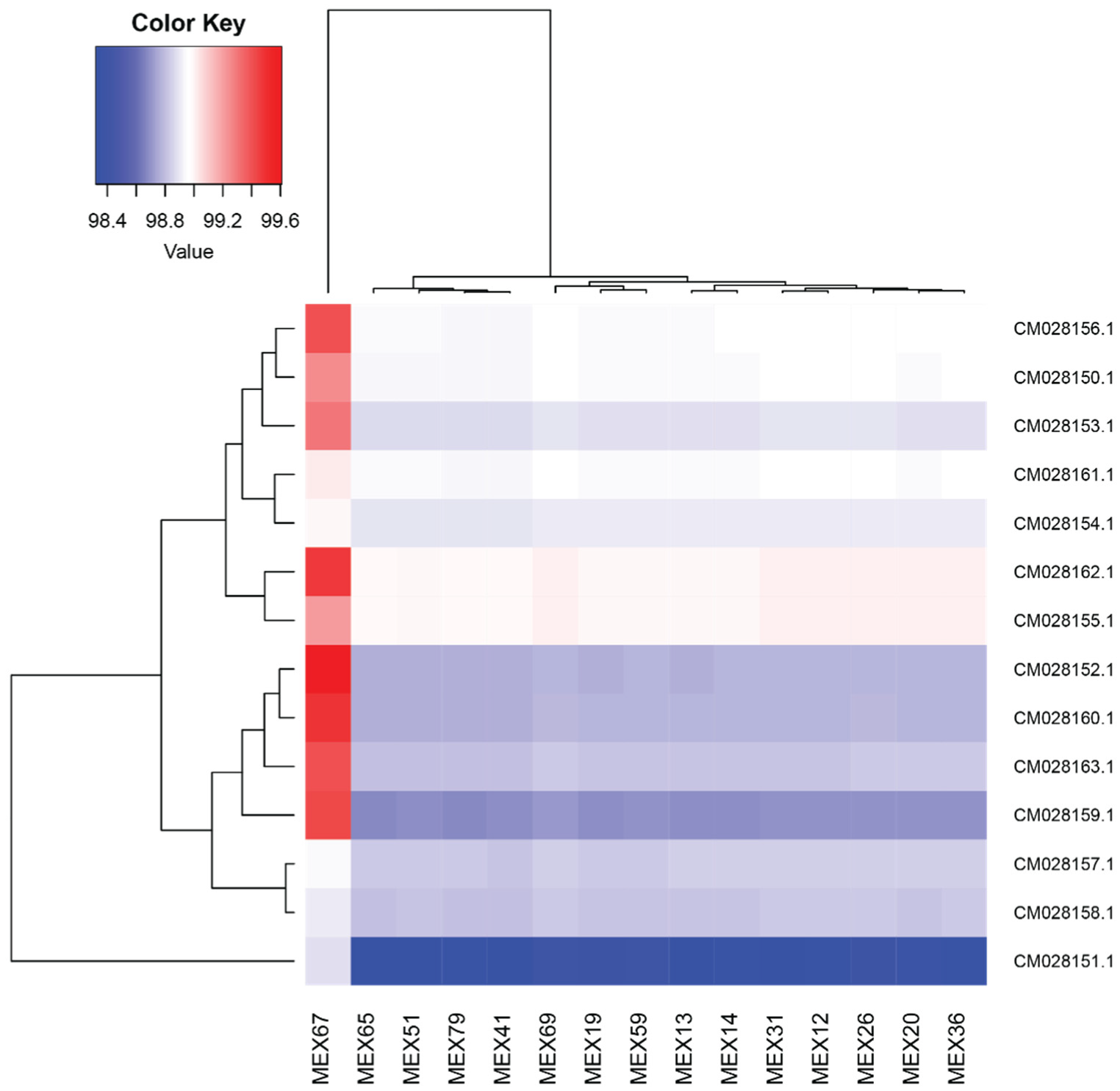
2.5. SNP Calling and Clustering Analyses
3. Discussion
3.1. High Genome-Wide Heterozygosity Stemming from Hybridization
3.2. Comparative Chromosomal Analyses Suggest Multiple Domestication Events in Mexico
3.3. Conservation of Mexican Vanilla Landraces and Implications for Production
4. Materials and Methods
4.1. Review of Plant Levels of Genomic Heterozygosity Inferred Using GenomeScope
4.2. Sampling, DNA Extraction, and Whole Genome Resequencing
4.3. Genomic Heterozygosity, Ploidy, and Complexity
4.4. Genome Reconstructions to Infer Structural Variation and Synteny
4.5. SNP Calling and Clustering Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bruman, H. The Culture History of Mexican Vanilla. Hisp. Am. Hist. Rev. 1948, 28, 360. [Google Scholar] [CrossRef]
- Lubinsky, P.; Bory, S.; Hernández, J.H.; Kim, S.-C.; Gómez-Pompa, A. Origins and Dispersal of Cultivated Vanilla (Vanilla Planifolia Jacks. [Orchidaceae]). Econ. Bot. 2008, 62, 127–138. [Google Scholar] [CrossRef]
- Food and Agriculture Organization of the United Nations (FAO) FAOSTAT. Available online: http://www.fao.org/faostat/en/#rankings/countries_by_commodity (accessed on 28 April 2020).
- Bory, S.; Grisoni, M.; Duval, M.-F.; Besse, P. Biodiversity and preservation of vanilla: Present state of knowledge. Genet. Resour. Crop Evol. 2007, 55, 551–571. [Google Scholar] [CrossRef]
- Cameron, K. Vanilla Orchids: Natural History and Cultivation; Timber Press: Portland, OR, USA, 2011; ISBN 0881929891. [Google Scholar]
- Hu, Y.; Resende, M.F.R., Jr.; Bombarely, A.; Brym, M.; Bassil, E.; Chambers, A.H. Genomics-based diversity analysis of Vanilla species using a Vanilla planifolia draft genome and Genotyping-By-Sequencing. Sci. Rep. 2019, 9, 3416. [Google Scholar] [CrossRef]
- Chambers, A.; Cibrián-Jaramillo, A.; Karremans, A.P.; Martinez, D.M.; Hernandez-Hernandez, J.; Brym, M.; Resende, M.F.; Moloney, R.; Sierra, S.N.; Hasing, T.; et al. Genotyping-By-Sequencing diversity analysis of international Vanilla collections uncovers hidden diversity and enables plant improvement. Plant Sci. 2021, 311, 111019. [Google Scholar] [CrossRef]
- Soto Arenas, M.A.; Dressler, R.L. A revision of the Mexican and Central American species of Vanilla plumier ex miller with a characterization of their its region of the nuclear ribosomal DNA. Lankesteriana Int. J. Orchid. 2013, 9, 285–354. [Google Scholar] [CrossRef]
- Varela, M.; Guerrero, H.; de Miguel, T. La Sequía Que Abrasa México, Una Tragedia Predecible y Devastadora. Available online: https://elpais.com/mexico/2021-04-24/la-sequia-que-abrasa-mexico-una-tragedia-predecible-y-devastadora.html?prm=enviar_email (accessed on 9 May 2021).
- Pinaria, A.G.; Liew, E.C.Y.; Burgess, L.W. Fusarium Species Associated with Vanilla Stem Rot in Indonesia. Australas. Plant Pathol. 2010, 39, 176–183. [Google Scholar] [CrossRef]
- Hinsley, A.; De Boer, H.J.; Fay, M.F.; Gale, S.W.; Gardiner, L.M.; Gunasekara, R.S.; Kumar, P.; Masters, S.; Metusala, D.; Roberts, D.; et al. A review of the trade in orchids and its implications for conservation. Bot. J. Linn. Soc. 2017, 186, 435–455. [Google Scholar] [CrossRef]
- Rain, P.; Lubinsky, P. Vanilla Use in Colonial Mexico and Traditional Totonac. In Vanilla. Medicinal and Aromatic Plants—Industrial Profiles; Odoux, G., Ed.; CRC Press: Boca Raton, FL, USA; Taylor & Francis Group: New York, NY, USA, 2011; pp. 251–259. [Google Scholar]
- Rain, P. Vanilla: The Cultural History of the World’s Most Popular Flavor and Fragrance; Tarcher, J.P., Ed.; Tarcher/Penguin: New York, NY, USA, 2004; ISBN 1585423637. [Google Scholar]
- Ellestad, P.; Forest, F.; Serpe, M.; Novak, S.J.; Buerki, S. Harnessing large-scale biodiversity data to infer the current distribution of Vanilla planifolia (Orchidaceae). Bot. J. Linn. Soc. 2021, 196, 407–422. [Google Scholar] [CrossRef]
- Minoo, D.; Jayakumar, V.N.; Veena, S.S.; Vimala, J.; Basha, A.; Saji, K.V.; Babu, K.N.; Peter, K.V. Genetic variations and interrelationships in Vanilla planifolia and few related species as expressed by RAPD polymorphism. Genet. Resour. Crop Evol. 2007, 55, 459–470. [Google Scholar] [CrossRef]
- Schlüter, P.M.; Arenas, M.A.S.; Harris, S.A.; Harris, S.A.; Schluter, P.M.; Arenas, M.A.S. Genetic Variation in Vanilla planifolia (Orchidaceae). Econ. Bot. 2007, 61, 328–336. [Google Scholar] [CrossRef]
- Villanueva-Viramontes, S.; Hernández-Apolinar, M.; Carnevali Fernández-Concha, G.; Dorantes-Euán, A.; Dzib, G.R.; Martínez-Castillo, J. Wild Vanilla planifolia and its relatives in the Mexican Yucatan Peninsula: Systematic analyses with ISSR and ITS. Bot. Sci. 2017, 95, 169. [Google Scholar] [CrossRef]
- Ramos-Castellá, A.L.; Iglesias-Andreu, L.G.; Martínez-Castillo, J.; Ortíz-García, M.; Andueza-Noh, R.H.; Octavio-Aguilar, P.; Luna-Rodríguez, M. Evaluation of Molecular Variability in Germplasm of Vanilla (Vanilla Planifolia G. Jackson in Andrews) in Southeast Mexico: Implications for Genetic Improvement and Conservation. Plant Genet. Resour. Characterisation Util. 2016, 15, 310–320. [Google Scholar] [CrossRef]
- Ellestad, P.; Farrera, M.A.P.; Forest, F.; Buerki, S. Uncovering haplotype diversity in cultivated Mexican vanilla species. Am. J. Bot. 2022, 109, 1120–1138. [Google Scholar] [CrossRef]
- Hasing, T.; Tang, H.; Brym, M.; Khazi, F.; Huang, T.; Chambers, A.H. A phased Vanilla planifolia genome enables genetic improvement of flavour and production. Nat. Food 2020, 1, 811–819. [Google Scholar] [CrossRef]
- Favre, F.; Jourda, C.; Grisoni, M.; Piet, Q.; Rivallan, R.; Dijoux, J.-B.; Hascoat, J.; Lepers-Andrzejewski, S.; Besse, P.; Charron, C. A genome-wide assessment of the genetic diversity, evolution and relationships with allied species of the clonally propagated crop Vanilla planifolia Jacks. ex Andrews. Genet. Resour. Crop Evol. 2022, 69, 2125–2139. [Google Scholar] [CrossRef]
- Ranallo-Benavidez, T.R.; Jaron, K.S.; Schatz, M. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 2020, 11, 1432. [Google Scholar] [CrossRef]
- Vurture, G.W.; Sedlazeck, F.J.; Nattestad, M.; Underwood, C.J.; Fang, H.; Gurtowski, J.; Schatz, M.C. GenomeScope: Fast reference-free genome profiling from short reads. Bioinformatics 2017, 33, 2202–2204. [Google Scholar] [CrossRef]
- McKey, D.; Elias, M.; Pujol, M.E.; Duputié, A. The evolutionary ecology of clonally propagated domesticated plants. New Phytol. 2010, 186, 318–332. [Google Scholar] [CrossRef]
- Nair, R.R.; Ravindran, P.N. Somatic Association of Chromosomes and Other Mitotic Abnormalities in Vanilla Planifolia (Andrews). Caryologia 1994, 47, 65–73. [Google Scholar] [CrossRef]
- Ravindran, P.N. Nuclear behaviour in the sterile pollen of Vanilla planifolia (Andrews). Cytologia 1979, 44, 391–396. [Google Scholar] [CrossRef]
- Bory, S.; Catrice, O.; Brown, S.; Leitch, I.J.; Gigant, R.; Chiroleu, F.; Grisoni, M.; Duval, M.-F.; Besse, P. Natural Polyploidy in Vanilla Planifolia (Orchidaceae). Genome 2008, 51, 816–826. [Google Scholar] [CrossRef] [PubMed]
- GitHub. Available online: https://github.com/svenbuerki/VanillaGenomicsCode (accessed on 4 July 2022).
- Vanilla Genomic Project. Available online: https://svenbuerki.github.io/VanillaGenomicsCode/index.html (accessed on 4 July 2022).
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Melton, A.E.; Child, A.W.; Beard, R.S.; Dave, C.; Dumaguit, C.; Forbey, J.S.; Germino, M.; de Graaff, M.-A.; Kliskey, A.; Leitch, I.J.; et al. A haploid pseudo-chromosome genome assembly for a keystone sagebrush species of western North American rangelands. G3 Genes Genomes Genet. 2022, 12, jkac122. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Feng, J.; Xiang, X.; Wang, J.; Salojärvi, J.; Liu, C.; Wu, Z.; Zhang, J.; Liang, X.; Jiang, Z.; et al. Two divergent haplotypes from a highly heterozygous lychee genome suggest independent domestication events for early and late-maturing cultivars. Nat. Genet. 2022, 54, 73–83. [Google Scholar] [CrossRef]
- Bellon, M.R.; Gotor, E.; Caracciolo, F. Conserving landraces and improving livelihoods: How to assess the success of on-farm conservation projects? Int. J. Agric. Sustain. 2014, 13, 167–182. [Google Scholar] [CrossRef]
- Lubinsky, P.; Cameron, K.M.; Molina, M.C.; Wong, M.; Lepers-Andrzejewski, S.; Gómez-Pompa, A.; Kim, S.-C. Neotropical roots of a Polynesian spice: The hybrid origin of Tahitian vanilla, Vanilla tahitensis (Orchidaceae). Am. J. Bot. 2008, 95, 1040–1047. [Google Scholar] [CrossRef]
- CBI Exporting Vanilla to Europe. Available online: https://www.cbi.eu/market-information/spices-herbs/vanilla#product-description (accessed on 11 May 2022).
- U.S FDA CFR—Code of Federal Regulations Title 21. Available online: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=169.3 (accessed on 11 May 2022).
- Fantini, D. easyPubMed 2019. Available online: https://www.data-pulse.com/dev_site/easypubmed/ (accessed on 29 March 2022).
- Winter, D.J. Rentrez: An R Package for the NCBI EUtils API. R J. 2017, 9, 520–526. [Google Scholar] [CrossRef]
- Diazgranados, M.; Allkin, B.; Black, N.; Cámara-Leret, R.; Canteiro, C.; Carretero, J.; Eastwood, R.; Hargreaves, S.; Hudson, A.; Milliken, W.; et al. World Checklist of Useful Plant Species. Knowl. Netw. Biocomplex. 2020. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Kokot, M.; Długosz, M.; Deorowicz, S. KMC 3: Counting and manipulating k-mer statistics. Bioinformatics 2017, 33, 2759–2761. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing 2020. R Foundation for Statistical Computing, Vienna, Austria. Available online: https://www.R-project.org/ (accessed on 5 May 2022).
- Winter, D. Pafr: Reading, Manipulating and Plotting Genome Alignments in the PAF Format 2020. Available online: https://dwinter.github.io/pafr/ (accessed on 5 April 2022).
- Warnes, G.R.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Huber, W.; Liaw, A.; Lumley, T.; Maechler, M.; Magnusson, A.; Moeller, S.; et al. Gplots: Various R Programming Tools for Plotting Data2022. Available online: https://github.com/talgalili/gplots (accessed on 7 May 2022).
- Zheng, X.; Levine, D.; Shen, J.; Gogarten, S.M.; Laurie, C.; Weir, B.S. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 2012, 28, 3326–3328. [Google Scholar] [CrossRef]
- Charif, D.; Lobry, J.R. SeqinR 1.0-2: A Contributed Package to the R project for Statistical Computing Devoted to Biological Sequences and Analysis. In Structural Approaches to Sequence Evolution: Molecules, Networks, Populations; Bastolla, U., Porto, M., Roman, H.E., Vendruscolo, M., Eds.; Biological and Medical Physics, Biomedical Engineering; Springer: New York, NY, USA, 2007; pp. 207–232. [Google Scholar]
- Zhang, H.; Meltzer, P.; Davis, S. RCircos: An R package for Circos 2D track plots. BMC Bioinform. 2013, 14, 244. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample_ID | BioSample | SRA | Number of Reads | Yield (Mbases) | Sequencing Coverage (X) |
|---|---|---|---|---|---|
| MEX12 | SAMN28632720 | SRR19374418 | 187,296,525 | 5618.896 | 76.27 |
| MEX13 | SAMN28632721 | SRR19374411 | 191,490,389 | 5744.712 | 77.97 |
| MEX14 | SAMN28632722 | SRR19374410 | 186,065,732 | 5581.972 | 75.76 |
| MEX19 | SAMN28632723 | SRR19374409 | 169,276,636 | 5078.299 | 68.93 |
| MEX20 | SAMN28632724 | SRR19374408 | 185,396,535 | 5561.896 | 75.49 |
| MEX26 | SAMN28632725 | SRR19374407 | 181,142,904 | 5434.287 | 73.76 |
| MEX31 | SAMN28632726 | SRR19374406 | 187,560,091 | 5626.803 | 76.37 |
| MEX36 | SAMN28632727 | SRR19374405 | 169,478,017 | 5084.341 | 69.01 |
| MEX41 | SAMN28632728 | SRR19374404 | 220,584,559 | 6617.537 | 89.82 |
| MEX51 | SAMN28632729 | SRR19374417 | 215,541,915 | 6466.257 | 87.77 |
| MEX59 | SAMN28632730 | SRR19374416 | 206,393,194 | 6191.796 | 84.04 |
| MEX65 | SAMN28632731 | SRR19374415 | 214,461,750 | 6433.852 | 87.33 |
| MEX67 | SAMN28632732 | SRR19374414 | 194,615,015 | 5838.450 | 79.25 |
| MEX69 | SAMN28632733 | SRR19374413 | 207,909,587 | 6237.288 | 84.66 |
| MEX79 | SAMN28632734 | SRR19374412 | 213,402,841 | 6402.085 | 86.90 |
| Sample ID | Ploidy | Genome-Wide HetErozygosity (ab%) | K-mer Coverage (kcov) |
|---|---|---|---|
| ‘Daphna’ | 2x | 2.48 | 99 |
| MEX12 | 2x | 2.74 | 32.3 |
| MEX13 | 2x | 2.67 | 32.6 |
| MEX14 | 2x | 2.66 | 34.7 |
| MEX19 | 2x | 2.52 | 32.4 |
| MEX20 | 2x | 2.56 | 33.7 |
| MEX26 | 2x | 2.57 | 31.7 |
| MEX31 | 2x | 2.61 | 32.5 |
| MEX36 | 2x | 2.52 | 28.2 |
| MEX41 | 2x | 2.77 | 40.9 |
| MEX51 | 2x | 2.62 | 39.3 |
| MEX59 | 2x | 2.79 | 38.5 |
| MEX65 | 2x | 2.57 | 16 |
| MEX67 | 2x | 0.403 | 34.1 |
| MEX69 | 2x | 2.85 | 38.9 |
| MEX79 | 2x | 2.53 | 39.2 |
| Chromosome Number | Chromosome ID | SNP Quantity (LD Threshold = 0.2) | SNP Quantity (LD Threshold = 0.8) |
|---|---|---|---|
| 1 | CM028150.1 | 1248 | 45,148 |
| 2 | CM028151.1 | 1315 | 129,408 |
| 3 | CM028152.1 | 710 | 27,900 |
| 4 | CM028153.1 | 680 | 23,651 |
| 5 | CM028154.1 | 723 | 24,225 |
| 6 | CM028155.1 | 678 | 23,166 |
| 7 | CM028156.1 | 570 | 19,088 |
| 8 | CM028157.1 | 525 | 19,218 |
| 9 | CM028158.1 | 524 | 17,213 |
| 10 | CM028159.1 | 487 | 23,185 |
| 11 | CM028160.1 | 508 | 21,826 |
| 12 | CM028161.1 | 564 | 17,157 |
| 13 | CM028162.1 | 418 | 14,437 |
| 14 | CM028163.1 | 347 | 14,263 |
| Total | 9297 | 419,885 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ellestad, P.; Pérez-Farrera, M.A.; Buerki, S. Genomic Insights into Cultivated Mexican Vanilla planifolia Reveal High Levels of Heterozygosity Stemming from Hybridization. Plants 2022, 11, 2090. https://doi.org/10.3390/plants11162090
Ellestad P, Pérez-Farrera MA, Buerki S. Genomic Insights into Cultivated Mexican Vanilla planifolia Reveal High Levels of Heterozygosity Stemming from Hybridization. Plants. 2022; 11(16):2090. https://doi.org/10.3390/plants11162090
Chicago/Turabian StyleEllestad, Paige, Miguel Angel Pérez-Farrera, and Sven Buerki. 2022. "Genomic Insights into Cultivated Mexican Vanilla planifolia Reveal High Levels of Heterozygosity Stemming from Hybridization" Plants 11, no. 16: 2090. https://doi.org/10.3390/plants11162090
APA StyleEllestad, P., Pérez-Farrera, M. A., & Buerki, S. (2022). Genomic Insights into Cultivated Mexican Vanilla planifolia Reveal High Levels of Heterozygosity Stemming from Hybridization. Plants, 11(16), 2090. https://doi.org/10.3390/plants11162090