

Targeted Metabolomics Provide Chemotaxonomic Insights of Medicago ruthenica, with Coupled Transcriptomics Elucidating the Mechanism Underlying Floral Coloration
Abstract
:1. Introduction
2. Results
2.1. Floral Color Phenotypes
2.2. Sample Quality Control and the Discrimination of Samples in the Validation Set
2.3. Composition of Anthocyanins and Flavonoids and Concentrations’ Discrepancy in YFM and PFM
2.4. Identification of 3319 Differentially Expressed Genes (DEGs) Using Transcriptome Sequencing
2.5. Differential ABP Gene Expression and Their Quantitative Real-Time PCR (qRT-PCR) Validation
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Metabolite Extraction and Detection
4.3. RNA Extraction and Transcriptomic Analysis
4.4. qRT-PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zamir, D. Improving Plant Breeding with Exotic Genetic Libraries. Nat. Rev. Genet. 2001, 2, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Ernest, S. Alfalfa and Relatives: Evolution and Classification of Medicago: The Other Species (Indicates a Species of Uncertain Status) 84; NRC Research Press: Ottawa, ON, Canada, 2011; pp. 565–570. [Google Scholar]
- Campbell, T.A.; Bao, G.; Xia, Z.L. Completion of the Agronomic Evaluations of Medicago ruthenica (L.) Ledebour Germplasm Collected in Inner Mongolia. Genet. Resour. Crop Evol. 1999, 46, 477–484. [Google Scholar] [CrossRef]
- Campbell, T.A.; Bao, G.; Xia, Z.L. Agronomic Evaluation of Medicago ruthenica Collected in Inner Mongolia. Crop Sci. 1997, 37, 599–604. [Google Scholar] [CrossRef]
- Li, H.Y.; Li, Z.Y.; Cai, L.Y.; Shi, W.G.; Mi, F.G.; Shi, F.L. Analysis of Genetic Diversity of Ruthenia Medic (Medicago ruthenica (L.) Trautv.) in Inner Mongolia Using ISSR and SSR Markers. Genet. Resour. Crop Evol. 2013, 60, 1687–1694. [Google Scholar] [CrossRef]
- Wang, D.K.; Luo, X.Y.; Li, H. Crossbreeding of Melilotoides ruthenicus and Medicago sativa. Acta Agrestia Sin. 2008, 16, 458–465. (In Chinese) [Google Scholar]
- Fu, H.C. Medicago ruthenica (L.) Trautv. Flora of China; Science Press: Beijing, China, 2010; p. 10. [Google Scholar]
- Xu, C.L. Melilotoides ruthenica var. Tianzhuensis, a New Variety of Melilotoides (Leguminosae) from Gansu, China. Guihaia 2016, 36, 1023–1025. (In Chinese) [Google Scholar]
- Wang, T.; Ren, L.; Li, C.; Zhang, D.; Zhang, X.; Zhou, G.; Gao, D.; Chen, R.; Chen, Y.; Wang, Z.; et al. The Genome of a Wild Medicago species Provides Insights into the Tolerant Mechanisms of Legume Forage to Environmental Stress. BMC Biol. 2021, 19, 96. [Google Scholar] [CrossRef]
- Ren, T.; Li, Z.X.; Xie, D.F.; Gui, L.J.; Peng, C.; Wen, J.; He, X.J. Plastomes of Eight Ligusticum species: Characterization, Genome Evolution, and Phylogenetic Relationships. BMC Plant Biol. 2020, 20, 519. [Google Scholar] [CrossRef]
- Eujayl, I.; Sledge, M.K.; Wang, L.; May, G.D.; Chekhovskiy, K.; Zwonitzer, J.C.; Mian, M.A.R. Medicago truncatula EST-SSRs Reveal Cross-Species Genetic Markers for Medicago spp. Theor. Appl. Genet. 2004, 108, 414–422. [Google Scholar] [CrossRef]
- Liu, K.; Abdullah, A.A.; Huang, M.; Nishioka, T.; Altaf-Ul-Amin, M.; Kanaya, S. Novel Approach to Classify Plants Based on Metabolite-Content Similarity. BioMed Res. Int. 2017, 2017, 5296729. [Google Scholar] [CrossRef]
- Bielecka, M.; Pencakowski, B.; Stafiniak, M.; Jakubowski, K.; Rahimmalek, M.; Gharibi, S.; Matkowski, A.; Ślusarczyk, S. Metabolomics and DNA-Based Authentication of Two Traditional Asian Medicinal and Aromatic Species of Salvia subg. Perovskia. Cells. 2021, 10, 112. [Google Scholar] [CrossRef] [PubMed]
- Mongrand, S.; Badoc, A.; Patouille, B.; Lacomblez, C.; Chavent, M.; Bessoule, J.J. Chemotaxonomy of the Rubiaceae Family Based on Leaf Fatty Acid Composition. Phytochemistry 2005, 66, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.X.; Xiong, W.; Zhao, G.D.; Peng, Y.; Zhong, Z.F.; Xu, L.; Duan, R.; Tsim, K.W.K.; Yu, H.; Wang, Y.T. Discrimination of Three Siegesbeckiae Herba Species Using UPLC-QTOF/MS-Based Metabolomics Approach. Food Chem. Toxicol. 2018, 119, 400–406. [Google Scholar] [CrossRef]
- Fayek, N.M.; Mekky, R.H.; Dias, C.N.; Kropf, M.; Heiss, A.G.; Wessjohann, L.A.; Farag, M.A. UPLC-MS Metabolome-Based Seed Classification of 16 Vicia species: A Prospect for Phyto-equivalency and Chemotaxonomy of Different Accessions. J. Agric. Food Chem. 2021, 69, 5252–5266. [Google Scholar] [CrossRef]
- Hegnauer, R. Chemical Characters in Plant Taxonomy: Some Possibilities and Limitations. Pure Appl. Chem. 1967, 14, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, C. Physiological Biochemistry of Floral Color; Forestry Press: Beijing, China, 1989; p. 2. [Google Scholar]
- Tanaka, Y.; Sasaki, N.; Ohmiya, A. Biosynthesis of Plant Pigments: Anthocyanins, Betalains and Carotenoids. Plant J. 2008, 54, 733–749. [Google Scholar] [CrossRef]
- Pan, Y.Z.; Guan, Y.; Wei, Z.F.; Peng, X.; Li, T.T.; Qi, X.L.; Zu, Y.G.; Fu, Y.J. Flavonoid C-Glycosides from Pigeon Pea Leaves as Color and Anthocyanin Stabilizing Agent in Blueberry Juice. Ind. Crops Prod. 2014, 58, 142–147. [Google Scholar] [CrossRef]
- Borghi, M.; Fernie, A.R. Outstanding Questions in Flower Metabolism. Plant J. 2020, 103, 1275–1288. [Google Scholar] [CrossRef]
- Santos-Buelga, C.; Mateus, N.; De Freitas, V. Anthocyanins. Plant Pigments and Beyond. J. Agric. Food Chem. 2014, 62, 6879–6884. [Google Scholar] [CrossRef]
- Saigo, T.; Wang, T.; Watanabe, M.; Tohge, T. Diversity of Anthocyanin and Proanthocyanin Biosynthesis in Land Plants. Curr. Opin. Plant Biol. 2020, 55, 93–99. [Google Scholar] [CrossRef]
- Ishiga, Y.; Uppalapati, S.R.; Gill, U.S.; Huhman, D.; Tang, Y.; Mysore, K.S. Transcriptomic and Metabolomic Analyses Identify a Role for Chlorophyll Catabolism and Phytoalexin during Medicago Nonhost Resistance against Asian Soybean Rust. Sci. Rep. 2015, 5, 13061. [Google Scholar] [CrossRef] [PubMed]
- Cooper, R.L.; Elliott, F.C. Flower Pigments in Diploid Alfalfa 1. Crop Sci. 1964, 4, 367–371. [Google Scholar] [CrossRef]
- Yu, M.Q. Study on Mechanism of Medicago falcata Adapting to Saline-Alkali Stress Based on Metabolomic and Transcriptomic Analyses; Jilin Agriculture University: Changchun, China, 2021. [Google Scholar] [CrossRef]
- Hongmei, N.; Fugui, M.; Hongyan, L.; Bingzhe, F.; Guozhan, L. Karyotype Analysis of Wild Yellow Type Medicago ruthenica L. Acta Agrestia Sin. 2008, 16, 316–318. (In Chinese) [Google Scholar]
- Jiao, F.; Zhao, L.; Wu, X.; Song, Z.; Li, Y. Metabolome and Transcriptome Analyses of the Molecular Mechanisms of Flower Color Mutation in Tobacco. BMC Genom. 2020, 21, 611. [Google Scholar] [CrossRef]
- Campanella, J.J.; Smalley, J.V.; Dempsey, M.E. A Phylogenetic Examination of the Primary Anthocyanin Production Pathway of the Plantae. Bot. Stud. 2014, 55, 10. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.R.; Wang, L.R.; Cui, G.X.; Zhou, X.H.; Duan, X.R.; Yang, H.S. Identification of the Regulatory Networks and Hub Genes Controlling Alfalfa Floral Pigmentation Variation Using RNA-Sequencing Analysis. BMC Plant Biol. 2020, 20, 110. [Google Scholar] [CrossRef]
- Zhu, H.H.; Yang, J.X.; Xiao, C.H.; Mao, T.Y.; Zhang, J.; Zhang, H.Y. Differences in Flavonoid Pathway Metabolites and Transcripts Affect Yellow Petal Colouration in the Aquatic Plant Nelumbo nucifera. BMC Plant Biol. 2019, 19, 277. [Google Scholar] [CrossRef]
- Pang, Y.Z.; Peel, G.J.; Wright, E.; Wang, Z.Y.; Dixon, R.A. Early Steps in Proanthocyanidin Biosynthesis in the Model Legume Medicago truncatula. Plant Physiol. 2007, 145, 601–615. [Google Scholar] [CrossRef]
- Zha, H.H.; Cai, Y.P.; Yin, Y.D.; Wang, Z.Z.; Li, K.; Zhu, Z.J. SWATH to MRM: Development of High-Coverage Targeted Metabolomics Method Using SWATH Technology for Biomarker Discovery. Anal. Chem. 2018, 90, 4062–4070. [Google Scholar] [CrossRef]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing Mass Spectrometry Data for Metabolite Profiling Using Nonlinear Peak Alignment, Matching, and Identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]

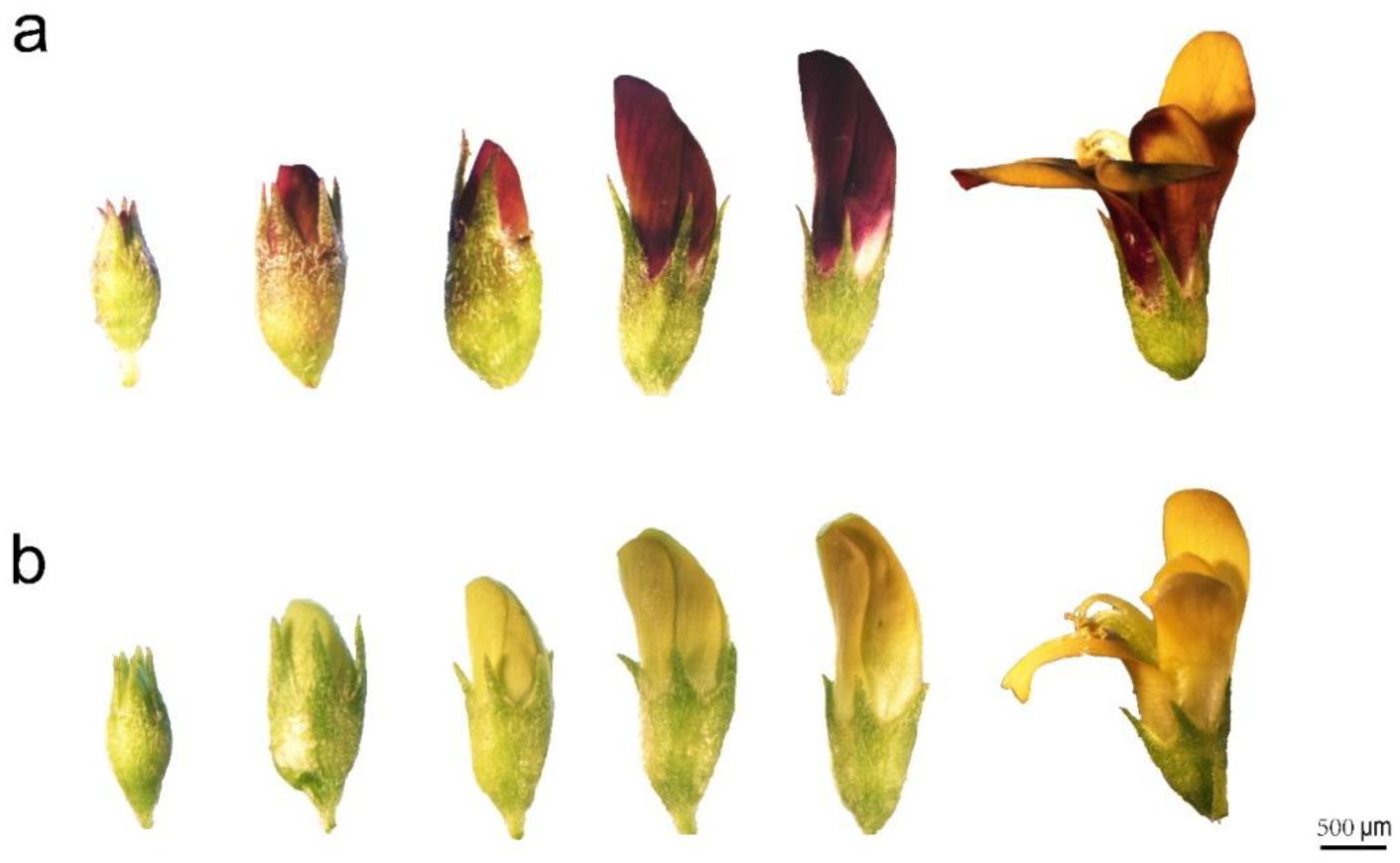







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NO. | Metabolite | Types | Polarity | Rt (min) | Related Contents (μmol/g) | p-Value | FC (YFM/PFM) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| YFM-1 | YFM-2 | YFM-3 | PFM-1 | PFM-2 | PFM-3 | |||||||
| 1 | Cyanidin-3-O-glucoside | anthocyanidins | 1 | 3.36 | 12.96 | 11.15 | 9.77 | 18,153.86 | 19,748.49 | 18,996.23 | 0.001 | 0.0006 |
| 2 | (-)-Epigallocatechin | flavanols | 1 | 3.07 | 0.02 | 0.00 | 0.01 | 0.35 | 0.39 | 0.39 | 0.000 | 0.0225 |
| 3 | Genkwanin | flavonoids | 1 | 13.64 | 1.22 | 0.93 | 0.85 | 38.05 | 41.18 | 40.75 | 0.025 | 0.0250 |
| 4 | Vitexin | flavonoids | 1 | 5.91 | 4.03 | 4.05 | 4.25 | 134.06 | 135.03 | 139.94 | 0.030 | 0.0302 |
| 5 | Homoorientin | flavonoids | 1 | 5.03 | 1.06 | 1.17 | 1.20 | 13.13 | 12.95 | 14.06 | 0.085 | 0.0854 |
| 6 | Cianidanol | flavanols | 1 | 3.64 | 0.03 | 0.01 | 0.00 | 0.11 | 0.13 | 0.10 | 0.105 | 0.1046 |
| 7 | Isorhamnetin | flavonols | 1 | 10.64 | 18.05 | 18.21 | 19.49 | 122.01 | 122.26 | 120.99 | 0.153 | 0.1526 |
| 8 | Orientin | flavonoids | 1 | 5.32 | 0.71 | 0.49 | 0.72 | 3.61 | 4.51 | 3.67 | 0.163 | 0.1633 |
| 9 | L-Epicatechin | flavanols | 1 | 4.5 | 0.04 | 0.02 | 0.03 | 0.11 | 0.19 | 0.14 | 0.193 | 0.1934 |
| 10 | Isosakuranetin | dihydroflavonoids | 1 | 13.52 | 0.93 | 0.95 | 0.94 | 4.77 | 4.87 | 4.93 | 0.194 | 0.1938 |
| 11 | kaempferol | flavonols | 1 | 10.37 | 28.23 | 28.48 | 28.37 | 141.99 | 144.90 | 141.06 | 0.199 | 0.1988 |
| 12 | Tectochrysin | flavonoids | 1 | 16.86 | 0.47 | 0.35 | 0.38 | 1.85 | 2.01 | 1.92 | 0.207 | 0.2068 |
| 13 | Kaempferitrin | flavonols | 1 | 6.1 | 0.01 | 0.03 | 0.04 | 0.11 | 0.09 | 0.09 | 0.281 | 0.2807 |
| 14 | Myricetin | flavonoids | 1 | 7.4 | 2.07 | 2.54 | 2.85 | 8.69 | 9.46 | 6.24 | 0.305 | 0.3054 |
| 15 | Quercetin | flavonols | −1 | 8.95 | 44.20 | 44.90 | 46.54 | 134.23 | 137.88 | 128.82 | 0.338 | 0.3383 |
| 16 | Baicalein | flavonoids | −1 | 8.15 | 0.68 | 0.82 | 0.78 | 2.06 | 1.86 | 1.96 | 0.388 | 0.3885 |
| 17 | Cynaroside | flavonoids | 1 | 6.21 | 339.96 | 354.97 | 358.97 | 748.84 | 717.89 | 754.48 | 0.474 | 0.4745 |
| 18 | Tiliroside | flavonoids | 1 | 8.83 | 0.67 | 0.57 | 0.69 | 1.41 | 1.340 | 1.26 | 0.481 | 0.4809 |
| 19 | (-)-Epicatechin gallate | flavanols | −1 | 6.1 | 0.30 | 0.35 | 0.29 | 0.59 | 0.55 | 0.64 | 0.528 | 0.5280 |
| 20 | Acacetin | flavonoids | 1 | 13.39 | 0.84 | 0.86 | 0.90 | 1.51 | 1.70 | 1.65 | 0.534 | 0.5340 |
| 21 | Hesperidin | dihydroflavonoids | −1 | 7.24 | 0.32 | 0.41 | 0.38 | 0.65 | 0.70 | 0.60 | 0.567 | 0.5669 |
| 22 | Dihydromyricetin | flavonols | 1 | 4.81 | 3.08 | 3.34 | 3.43 | 5.75 | 5.62 | 5.61 | 0.579 | 0.5792 |
| 23 | Hydroxygenkwanin | flavonoids | 1 | 12.08 | 6.75 | 6.73 | 6.76 | 11.52 | 11.23 | 11.32 | 0.594 | 0.5939 |
| 24 | Luteolin | flavonoids | 1 | 8.9 | 205.21 | 206.01 | 214.63 | 319.76 | 319.67 | 326.94 | 0.648 | 0.6476 |
| 25 | Apigenin-7-glucoside | flavonoids | 1 | 7.05 | 179.48 | 183.14 | 186.88 | 260.15 | 248.46 | 261.00 | 0.708 | 0.7076 |
| 26 | Astragalin | flavonols | 1 | 6.75 | 1835.43 | 1902.50 | 1880.48 | 2573.41 | 2492.87 | 2579.29 | 0.735 | 0.7349 |
| 27 | Icariin | flavonoids | 1 | 9.42 | 0.09 | 0.05 | 0.04 | 0.06 | 0.06 | 0.11 | 0.753 | 0.7526 |
| 28 | Phloretin | chalcone | 1 | 10.27 | 0.35 | 0.52 | 0.30 | 0.56 | 0.47 | 0.45 | 0.789 | 0.7888 |
| 29 | Apigenin | flavonoids | 1 | 10.13 | 363.68 | 357.08 | 379.27 | 448.01 | 456.86 | 482.19 | 0.794 | 0.7931 |
| 30 | Genistein | flavonoids | 1 | 10.13 | 141.81 | 143.86 | 150.57 | 175.34 | 178.58 | 187.64 | 0.806 | 0.8055 |
| 31 | Schaftoside | flavonoids | 1 | 4.97 | 78.80 | 77.17 | 83.68 | 77.88 | 81.38 | 83.61 | 0.987 | 0.9867 |
| 31 | Liquiritin | dihydroflavonoids | 1 | 6.13 | 9.60 | 9.86 | 9.72 | 10.07 | 9.94 | 9.23 | 1.00 | 0.9975 |
| 33 | Narcissoside | flavonoids | −1 | 6.56 | 2.94 | 2.74 | 2.71 | 2.32 | 2.27 | 2.33 | 1.21 | 1.2121 |
| 34 | Taxifolin | flavonols | 1 | 6.35 | 2.17 | 2.32 | 2.28 | 1.73 | 1.94 | 1.82 | 1.24 | 1.2351 |
| 35 | Chrysin | flavonoids | 1 | 13.34 | 1.14 | 0.98 | 0.98 | 0.67 | 0.62 | 0.66 | 1.59 | 1.5868 |
| 36 | Spinosin | flavonoids | −1 | 5.75 | 17.89 | 16.45 | 17.06 | 10.47 | 10.40 | 11.16 | 1.61 | 1.6050 |
| 37 | Eriodictyol | dihydroflavonoids | 1 | 8.85 | 8.68 | 8.95 | 9.23 | 5.49 | 5.39 | 5.48 | 1.64 | 1.6421 |
| 38 | Isoquercitrin | flavonols | 1 | 6.08 | 784.94 | 706.57 | 695.53 | 341.99 | 490.93 | 419.78 | 1.75 | 1.7459 |
| 39 | Naringenin | dihydroflavonoids | 1 | 10.18 | 100.29 | 99.78 | 102.12 | 55.23 | 56.57 | 58.11 | 1.78 | 1.7786 |
| 40 | Naringenin chalcone | chalcone | 1 | 10.17 | 64.49 | 65.83 | 65.93 | 34.79 | 35.45 | 37.02 | 1.83 | 1.8299 |
| 41 | Hyperoside | flavonols | 1 | 6.07 | 1286.82 | 1385.39 | 1189.61 | 652.77 | 740.89 | 685.34 | 1.86 | 1.8575 |
| 42 | Glycitein | isoflavones | 1 | 8.77 | 20.60 | 21.39 | 21.02 | 10.92 | 11.21 | 11.09 | 1.90 | 1.8968 |
| 43 | Biochanin A | isoflavones | 1 | 13.93 | 20.79 | 20.01 | 20.95 | 9.29 | 9.58 | 9.71 | 2.16 | 2.1608 |
| 44 | Rutin | flavonols | 1 | 5.79 | 112.67 | 122.59 | 107.68 | 43.37 | 44.62 | 55.40 | 2.39 | 2.3916 |
| 45 | Myricitrin | flavonols | −1 | 6.07 | 0.58 | 0.63 | 0.64 | 0.25 | 0.23 | 0.25 | 2.53 | 2.5284 |
| 46 | Ononin | flavonoids | 1 | 8.3 | 123.76 | 124.57 | 124.68 | 51.60 | 47.81 | 45.40 | 2.58 | 2.5759 |
| 47 | Pinocembrin | dihydroflavonoids | 1 | 13.68 | 1.34 | 1.19 | 1.31 | 0.46 | 0.45 | 0.46 | 2.81 | 2.8057 |
| 48 | Isoliquiritigenin | chalcone | 1 | 11.34 | 789.70 | 788.05 | 797.03 | 196.61 | 197.93 | 203.57 | 3.97 | 3.9705 |
| Gene | Gene ID | FPKM | YFM-FPKM (Mean) | FPKM | PFM-FPKM (Mean) | log2FC | Regulation | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| YFM-1 | YFM-2 | YFM-3 | PFM-1 | PFM-2 | PFM-3 | ||||||
| CHS | MruG02985 | 45.409 | 41.605 | 42.382 | 43.132 | 96.761 | 95.369 | 94.306 | 95.479 | −1.0933 | down |
| CHS | MruG02563 | 8.449 | 8.315 | 9.041 | 8.602 | 22.411 | 20.558 | 21.163 | 21.377 | −1.2513 | down |
| CHS | MruG02992 | 0.907 | 0.749 | 0.582 | 0.746 | 6.559 | 4.906 | 2.970 | 4.812 | −2.3438 | down |
| CHS | MruG02987 | 11.239 | 7.972 | 8.767 | 9.326 | 262.238 | 272.324 | 267.033 | 267.198 | −4.7440 | down |
| CHI | MruG008873 | 1.740 | 1.866 | 0.923 | 1.510 | 3.450 | 3.373 | 3.136 | 3.320 | −1.0093 | down |
| CHI | MruG006531 | 134.095 | 146.950 | 156.917 | 145.988 | 311.336 | 298.964 | 306.483 | 305.595 | −1.0119 | down |
| F3H | MruG015074 | 0.000 | 0.000 | 0.000 | 0.000 | 2.918 | 2.166 | 1.886 | 2.323 | −4.3940 | down |
| F3H | MruG016463 | 2.274 | 2.341 | 2.052 | 2.222 | 13.713 | 13.612 | 14.737 | 14.021 | −2.5314 | down |
| F3H | MruG018452 | 5.761 | 4.335 | 7.982 | 6.026 | 23.731 | 25.329 | 25.807 | 24.956 | −1.9320 | down |
| F3′H | MruG012738 | 3.721 | 3.505 | 3.884 | 3.704 | 11.608 | 14.313 | 8.023 | 11.315 | −1.4392 | down |
| F3′H | MruG012740 | 4.021 | 2.984 | 2.926 | 3.310 | 11.552 | 9.667 | 9.268 | 10.162 | −1.4914 | down |
| DFR | MruG000195 | 0.000 | 0.000 | 0.000 | 0.000 | 188.822 | 185.284 | 185.727 | 186.611 | −10.2740 | down |
| DFR | MruG000196 | 0.318 | 0.524 | 0.489 | 0.444 | 145.809 | 143.038 | 142.730 | 143.859 | −7.9463 | down |
| DFR | MruG007181 | 25.204 | 25.597 | 27.086 | 25.962 | 52.308 | 57.399 | 54.999 | 54.902 | −1.0256 | down |
| DFR | MruG036456 | 2.354 | 1.770 | 2.822 | 2.316 | 5.361 | 5.806 | 5.354 | 5.507 | −1.1270 | down |
| ANS | MruG0255511 | 0.820 | 0.930 | 0.960 | 0.903 | 1.880 | 1.850 | 1.910 | 1.880 | −1.9634 | down |
| ANS | MruG026083 | 0.685 | 0.169 | 0.242 | 0.365 | 434.075 | 442.808 | 436.607 | 437.830 | −7.8978 | down |
| UFGT | MruG0252594 | 1.880 | 2.400 | 2.320 | 2.200 | 3.620 | 2.910 | 2.990 | 3.173 | −2.0093 | down |
| UFGT | MruG008629 | 1.442 | 1.562 | 1.856 | 1.620 | 4.622 | 4.555 | 4.506 | 4.561 | −1.3855 | down |
| UFGT | MruG042817 | 11.856 | 10.709 | 10.530 | 11.032 | 0.225 | 0.190 | 0.240 | 0.218 | 5.1867 | up |
| UFGT | MruG030584 | 1.283 | 1.288 | 1.260 | 1.277 | 0.832 | 0.446 | 0.440 | 0.573 | 1.0296 | up |
| UFGT | MruG008634 | 6.416 | 5.818 | 6.750 | 6.328 | 1.782 | 1.839 | 1.705 | 1.775 | 1.8024 | up |
| UFGT | MruG019752 | 2.039 | 2.417 | 3.234 | 2.563 | 1.293 | 0.313 | 0.794 | 0.800 | 1.4574 | up |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, L.; Li, H.; Wu, Z.; Li, Z.; Guo, M.; Ning, B.; Liu, W.; Liu, Q.; Liu, L.; Wang, Z.; et al. Targeted Metabolomics Provide Chemotaxonomic Insights of Medicago ruthenica, with Coupled Transcriptomics Elucidating the Mechanism Underlying Floral Coloration. Plants 2022, 11, 2408. https://doi.org/10.3390/plants11182408
Zhu L, Li H, Wu Z, Li Z, Guo M, Ning B, Liu W, Liu Q, Liu L, Wang Z, et al. Targeted Metabolomics Provide Chemotaxonomic Insights of Medicago ruthenica, with Coupled Transcriptomics Elucidating the Mechanism Underlying Floral Coloration. Plants. 2022; 11(18):2408. https://doi.org/10.3390/plants11182408
Chicago/Turabian StyleZhu, Lin, Hongyan Li, Zinian Wu, Zhiyong Li, Maowei Guo, Bu Ning, Wanpeng Liu, Qian Liu, Lei Liu, Zhiyong Wang, and et al. 2022. "Targeted Metabolomics Provide Chemotaxonomic Insights of Medicago ruthenica, with Coupled Transcriptomics Elucidating the Mechanism Underlying Floral Coloration" Plants 11, no. 18: 2408. https://doi.org/10.3390/plants11182408