
Morphological and Genome-Wide Evidence of Homoploid Hybridisation in Urospermum (Asteraceae)

 , , ,
, , , 
Abstract
1. Introduction
2. Results
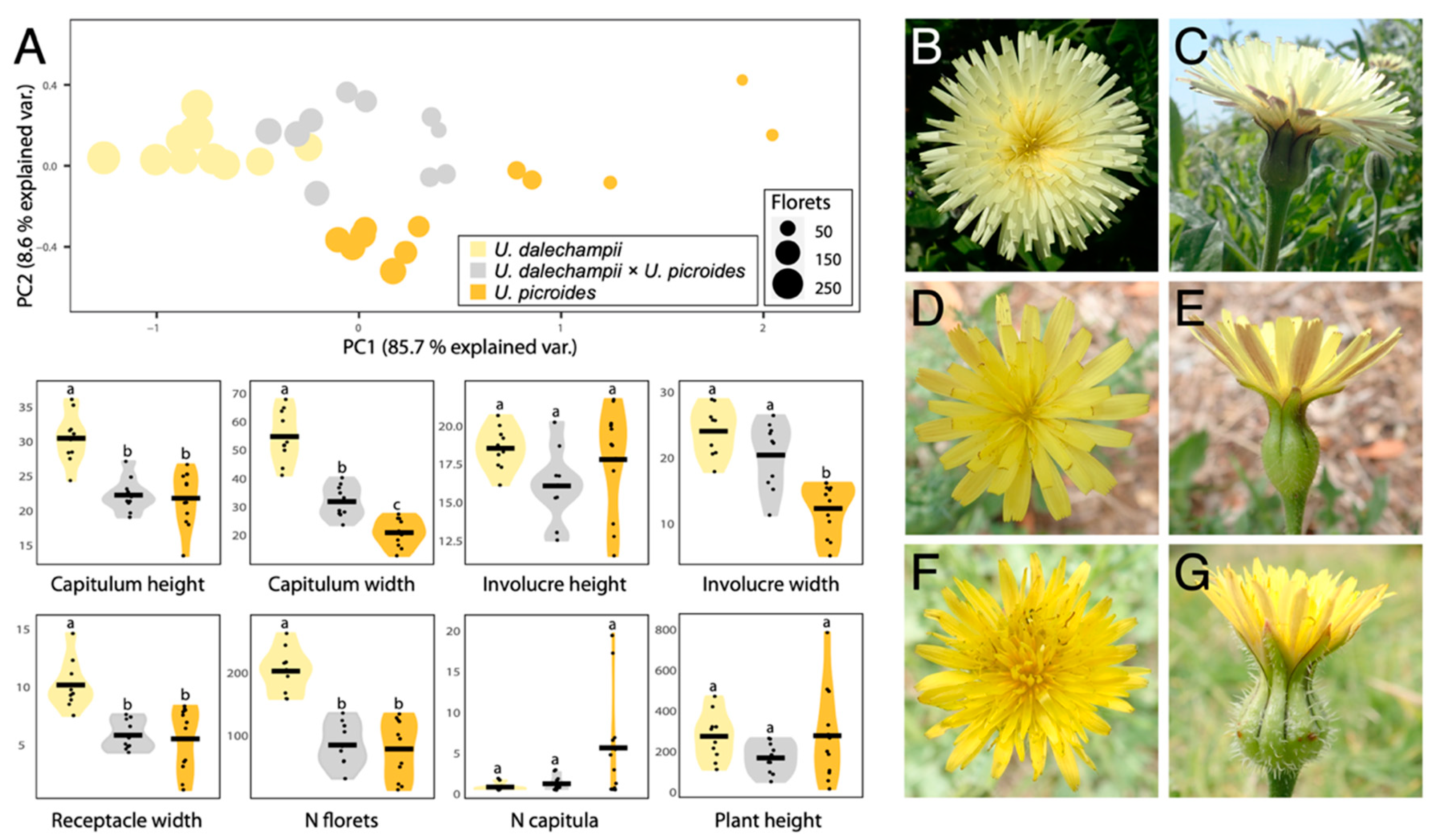
2.1. Morphological Characterisation of the Genus Urospermum
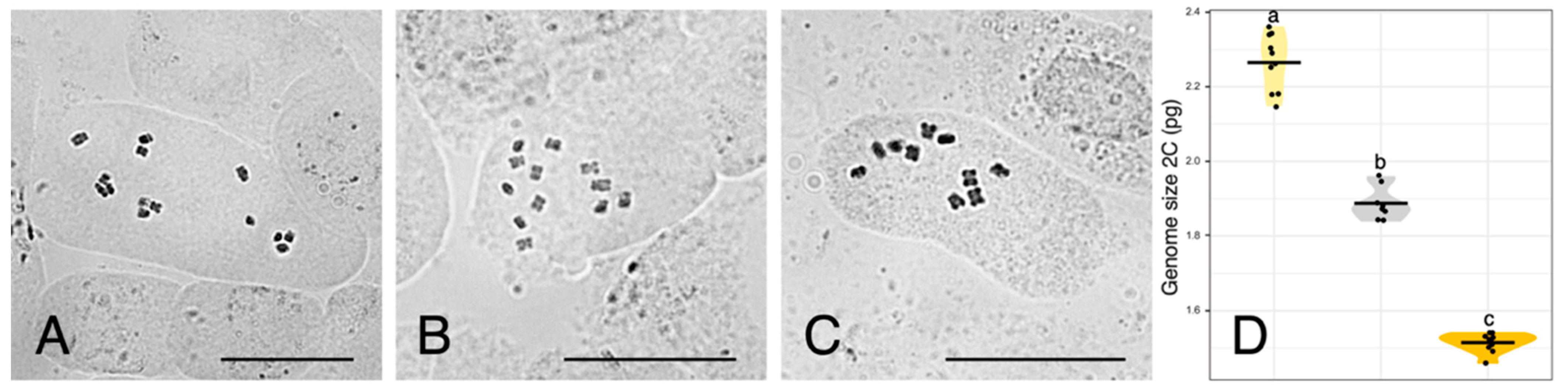
2.2. Chromosome Counts
2.3. Genome Size Measurements
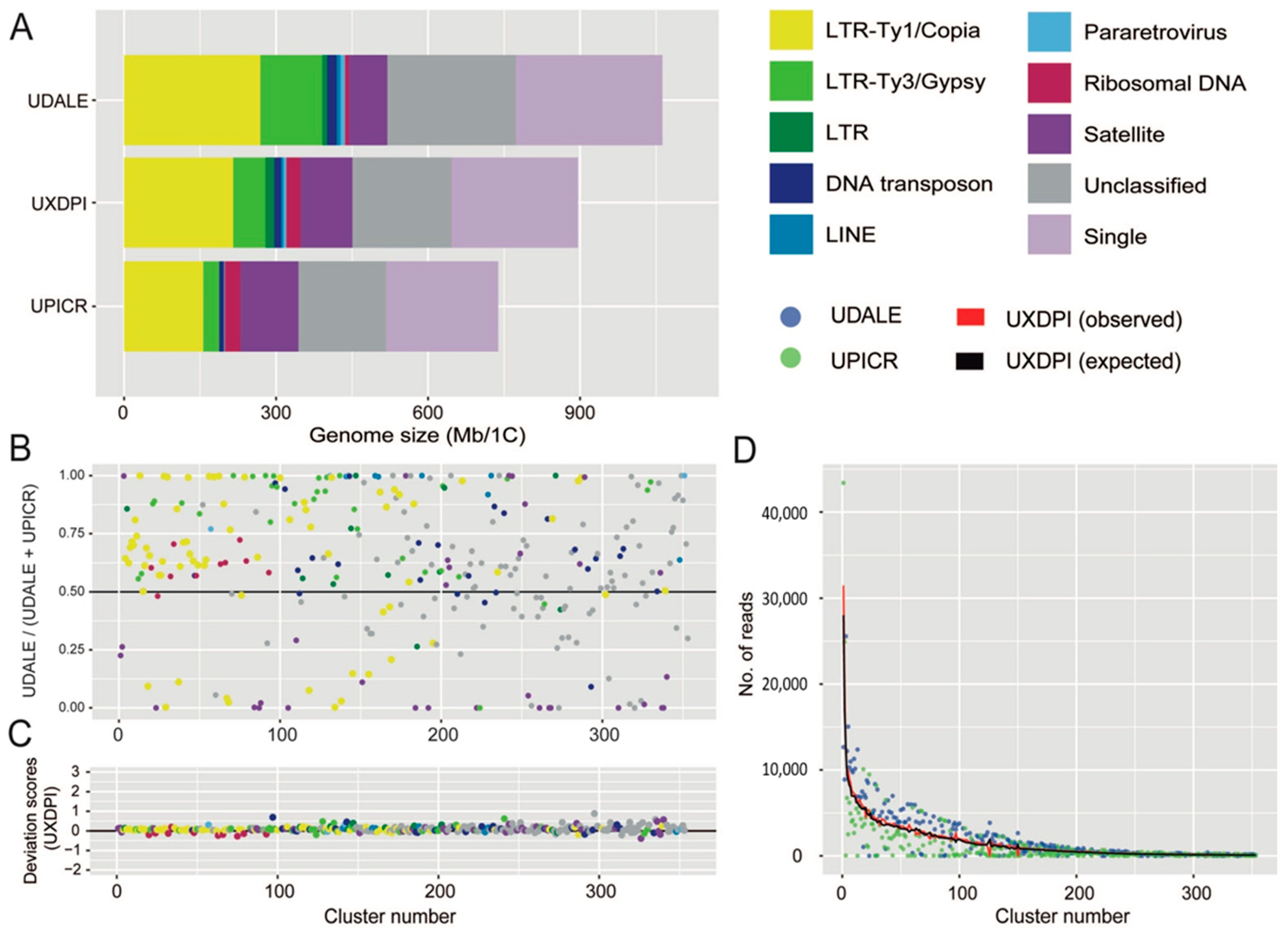
2.4. Individual Repeat Content and TE Annotation
2.5. Comparative Repeat Dynamics in Urospermum and the Putative Hybrid
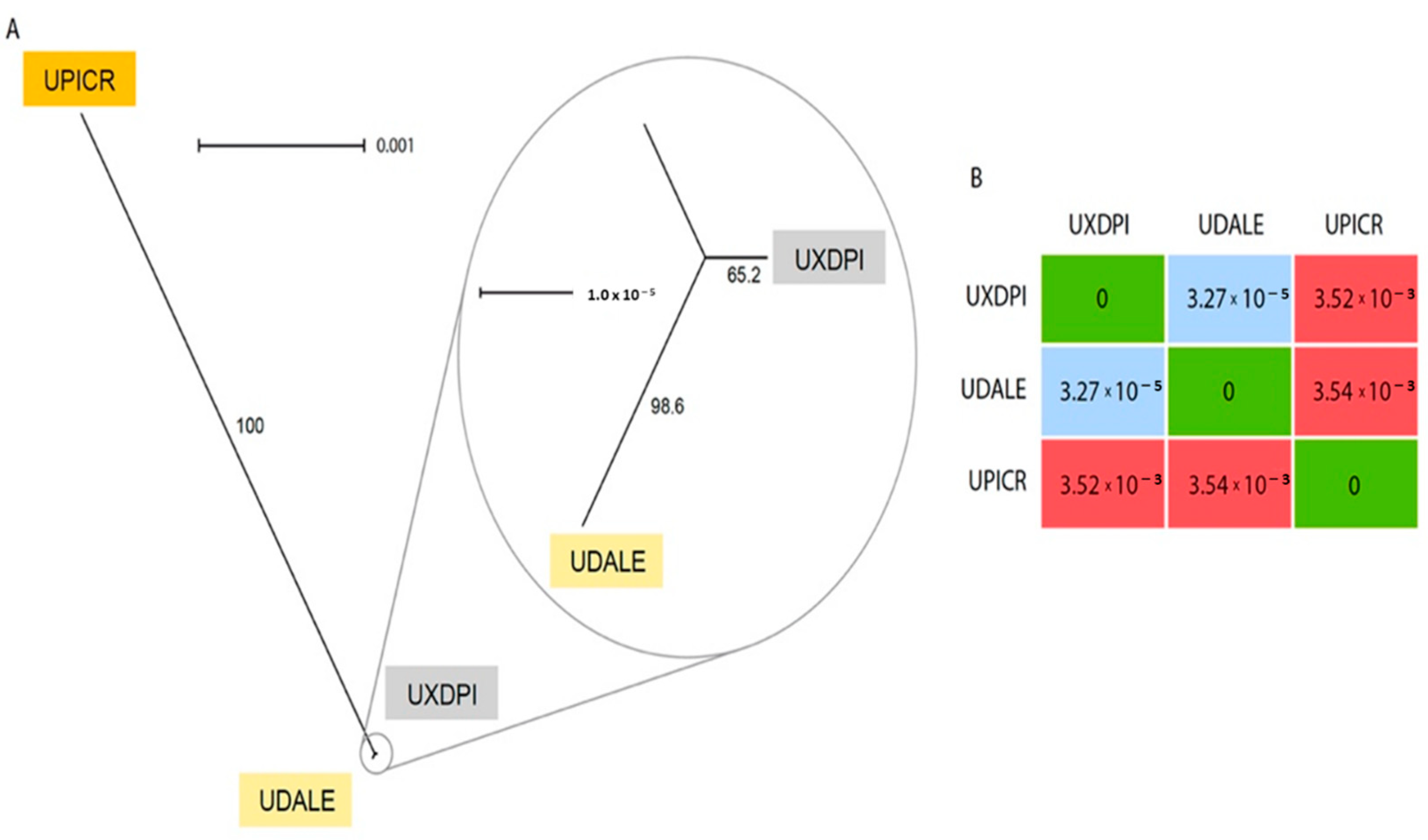
2.6. Chloroplast Reconstruction and Network Analysis
3. Discussion
3.1. A Hybrid with a Very Distinctive Involucral Bract Indumentum
3.2. Genome Size Provides Support for Homoploid Hybridisation in Urospermum
3.3. Chloroplast Analysis Provides Insights into the Parentage Origin of the Hybrid
3.4. Analysis of Repetitive DNA in Urospermum and Consequences of Hybridisation in the Genome Organisation of Hybrid Taxa
4. Materials and Methods
4.1. Plant Sampling
4.2. Chromosome Counts
4.3. Flow Cytometry Measurements
4.4. Statistical Analyses
4.5. DNA Isolation and Next Generation Sequencing
4.6. Graph-Based Clustering in RepeatExplorer 2 and Transposable Element Annotation
4.7. Assembly of Chloroplast Genomes and Network Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Talavera, S.; Talavera, M.; Berjano, R. Urospermum Scop. [nom. cons.]. In Flora iberica. Plantas Vasculares de la Península Ibérica e Islas Baleares; Talavera, S., Buira, A., Quintanar, A., García, M.Á., Talavera, M., Fernández Piedra, P., Aedo, C., Eds.; Real Jardín Botánico, CSIC: Madrid, Spain, 2017; Volume XVI (II), pp. 1063–1068. [Google Scholar]
- Fernández-Mazuecos, M.; Jiménez-Mejías, P.; Martín-Bravo, S.; Buide, M.L.; Álvarez, I.; Vargas, P. Narrow endemics on coastal plains: Miocene divergence of the critically endangered genus Avellara (Compositae). Plant Biol. 2016, 18, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Vallès, J.; Malik, S.; Gomez, M.; Siljak-Yakovlev, S. Contribution to knowledge about nuclear DNA amounts in the family Asteraceae: First assessments in one genus and 12 species, with chromosome counts for three taxa. Bot. Serbica 2017, 41, 213–219. [Google Scholar] [CrossRef]
- Garcia, S.; Hidalgo, O.; Jakovljević, I.; Siljak-Yakovlev, S.; Vigo, J.; Garnatje, T.; Vallès, J. New data on genome size in 128 Asteraceae species and subspecies, with first assessments for 40 genera, 3 tribes and 2 subfamilies. Plant Biosyst. 2013, 147, 1219–1227. [Google Scholar] [CrossRef]
- Berjano, R.; Talavera, M.; Talavera, S. El género Urospermum en el oeste de la región mediterránea. Acta Bot. Malacit. 2014, 39, 117–128. [Google Scholar] [CrossRef][Green Version]
- Abbott, R.; Albach, D.; Ansell, S.; Arntzen, J.W.; Baird, S.J.E.; Bierne, N.; Boughman, J.; Brelsford, A.; Buerkle, C.A.; Buggs, R.; et al. Hybridization and speciation. J. Evol. Biol. 2013, 26, 229–246. [Google Scholar] [CrossRef]
- Roux, C.; Fraïsse, C.; Romiguier, J.; Anciaux, Y.; Galtier, N.; Bierne, N. Shedding light on the grey zone of speciation along a continuum of genomic divergence. PLoS Biol. 2016, 14, e2000234. [Google Scholar] [CrossRef] [PubMed]
- Nieto Feliner, G.; Álvarez, I.; Fuertes-Aguilar, J.; Heuertz, M.; Marques, I.; Moharrek, F.; Piñeiro, R.; Riina, R.; Rosselló, J.A.; Soltis, P.S.; et al. Is homoploid hybrid speciation that rare? An empiricist’s view. Heredity 2017, 118, 513–516. [Google Scholar] [CrossRef] [PubMed]
- Fjellheim, S.; Jørgensen, M.H.; Kjos, M.; Borgen, L. A molecular study of hybridization and homoploid hybrid speciation in Argyranthemum (Asteraceae) on Tenerife, the Canary Islands. Bot. J. Linn. Soc. 2009, 159, 19–31. [Google Scholar] [CrossRef][Green Version]
- Pellicer, J.; López-Pujol, J.; Aixarch, M.; Garnatje, T.; Vallès, J.; Hidalgo, O. Detecting introgressed populations in the Iberian endemic Centaurea podospermifolia through genome size. Plants 2021, 10, 1492. [Google Scholar] [CrossRef] [PubMed]
- Gross, B.L.; Rieseberg, L.H. The ecological genetics of homoploid hybrid speciation. J. Hered. 2005, 96, 241–252. [Google Scholar] [CrossRef]
- Chester, M.; Gallagher, J.P.; Symonds, V.V.; Cruz da Silva, A.V.; Mavrodiev, E.V.; Leitch, A.R.; Soltis, P.S.; Soltis, D.E. Extensive chromosomal variation in a recently formed natural allopolyploid species, Tragopogon miscellus (Asteraceae). Proc. Natl. Acad. Sci. USA 2012, 109, 1176–1181. [Google Scholar] [CrossRef] [PubMed]
- Pellicer, J.; Clermont, S.; Houston, L.; Rich, T.C.G.; Fay, M.F. Cytotype diversity in the Sorbus complex (Rosaceae) in Britain: Sorting out the puzzle. Ann. Bot. 2012, 110, 1185–1193. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Kelly, L.J.; McAllister, H.A.; Zohren, J.; Buggs, R.J.A. Resolving phylogeny and polyploid parentage using genus-wide genome-wide sequence data from birch trees. Mol. Phylogenet. Evol. 2021, 160, 107126. [Google Scholar] [CrossRef]
- Brandrud, M.K.; Baar, J.; Lorenzo, M.T.; Athanasiadis, A.; Bateman, R.M.; Chase, M.W.; Hedrén, M.; Paun, O. Phylogenomic relationships of diploids and the origins of allotetraploids in Dactylorhiza (Orchidaceae). Syst. Biol. 2020, 69, 91–109. [Google Scholar] [CrossRef] [PubMed]
- Piñeiro, R.; Karrman-Bailey, F.; Cowan, R.S.; Fay, M.F. Isolation and characterization of microsatellite loci in Sorbus porrigentiformis and cross-amplification in S. aria and S. rupicola (Rosaceae). Appl. Plant Sci. 2017, 5, 1600150. [Google Scholar] [CrossRef]
- Bennetzen, J.L.; Wang, H. The contributions of transposable elements to the structure, function, and evolution of plant genomes. Annu. Rev. Plant Biol. 2014, 65, 505–530. [Google Scholar] [CrossRef] [PubMed]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef]
- Sahebi, M.; Hanafi, M.M.; van Wijnen, A.J.; Rice, D.; Rafii, M.Y.; Azizi, P.; Osman, M.; Taheri, S.; Bakar, M.F.A.; Isa, M.N.M.; et al. Contribution of transposable elements in the plant’s genome. Gene 2018, 665, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Heyduk, K.; McAssey, E.V.; Grimwood, J.; Shu, S.; Schmutz, J.; McKain, M.R.; Leebens-Mack, J. Hybridization history and repetitive element content in the genome of a homoploid hybrid, Yucca gloriosa (Asparagaceae). Front. Plant Sci. 2021, 11, 2220. [Google Scholar] [CrossRef]
- Zagorski, D.; Hartmann, M.; Bertrand, Y.J.K.; Paštová, L.; Slavíková, R.; Josefiová, J.; Fehrer, J. Characterization and dynamics of repeatomes in closely related species of Hieracium (Asteraceae) and their synthetic and apomictic hybrids. Front. Plant Sci. 2020, 11, 1689. [Google Scholar] [CrossRef]
- Pellicer, J.; Fernández, P.; Fay, M.F.; Michálková, E.; Leitch, I.J. Genome size doubling arises from the differential repetitive DNA dynamics in the genus Heloniopsis (Melanthiaceae). Front. Genet. 2021, 12, 1685. [Google Scholar]
- McCann, J.; Jang, T.-S.; Macas, J.; Schneeweiss, G.M.; Matzke, N.J.; Novák, P.; Stuessy, T.F.; Villaseñor, J.L.; Weiss-Schneeweiss, H. Dating the species network: Allopolyploidy and repetitive DNA evolution in American Daisies (Melampodium sect. Melampodium, Asteraceae). Syst. Biol. 2018, 67, 1010–1024. [Google Scholar] [CrossRef]
- Novák, P.; Neumann, P.; Pech, J.; Steinhaisl, J.; Macas, J. RepeatExplorer: A Galaxy-based web server for genome-wide characterization of eukaryotic repetitive elements from next-generation sequence reads. Bioinformatics 2013, 29, 792–793. [Google Scholar] [CrossRef] [PubMed]
- Macas, J.; Novák, P.; Pellicer, J.; Čížková, J.; Koblížková, A.; Neumann, P.; Fuková, I.; Doležel, J.; Kelly, L.J.; Leitch, I.J. In depth characterization of repetitive DNA in 23 plant genomes reveals sources of genome size variation in the legume tribe Fabeae. PLoS ONE 2015, 10, e0143424. [Google Scholar] [CrossRef]
- McCann, J.; Macas, J.; Novák, P.; Stuessy, T.F.; Villaseñor, J.L.; Weiss-Schneeweiss, H. Differential genome size and repetitive DNA evolution in diploid species of Melampodium sect. Melampodium (Asteraceae). Front. Plant Sci. 2020, 11, 362. [Google Scholar] [CrossRef] [PubMed]
- Vitales, D.; Álvarez, I.; Garcia, S.; Hidalgo, O.; Nieto Feliner, G.; Pellicer, J.; Vallès, J.; Garnatje, T. Genome size variation at constant chromosome number is not correlated with repetitive DNA dynamism in Anacyclus (Asteraceae). Ann. Bot. 2020, 125, 611–623. [Google Scholar] [CrossRef]
- Doležel, J.; Bartoš, J.; Voglmayr, H.; Greilhuber, J. Nuclear DNA content and genome size of trout and human. Cytom. Part A 2003, 51, 127–128. [Google Scholar] [CrossRef]
- Pellicer, J.; Kelly, L.J.; Magdalena, C.; Leitch, I.J. Insights into the dynamics of genome size and chromosome evolution in the early diverging angiosperm lineage Nymphaeales (water lilies). Genome 2013, 56, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Baack, E.J.; Whitney, K.D.; Rieseberg, L.H. Hybridization and genome size evolution: Timing and magnitude of nuclear DNA content increases in Helianthus homoploid hybrid species. New Phytol. 2005, 167, 623–630. [Google Scholar] [CrossRef]
- Twyford, A.D.; Ness, R.W. Strategies for complete plastid genome sequencing. Mol. Ecol. Resour. 2017, 17, 858–868. [Google Scholar] [CrossRef]
- Pickup, M.; Brandvain, Y.; Fraïsse, C.; Yakimowski, S.; Barton, N.H.; Dixit, T.; Lexer, C.; Cereghetti, E.; Field, D.L. Mating system variation in hybrid zones: Facilitation, barriers and asymmetries to gene flow. New Phytol. 2019, 224, 1035–1047. [Google Scholar] [CrossRef]
- Novák, P.; Guignard, M.S.; Neumann, P.; Kelly, L.J.; Mlinarec, J.; Koblížková, A.; Dodsworth, S.; Kovařík, A.; Pellicer, J.; Wang, W.; et al. Repeat-sequence turnover shifts fundamentally in species with large genomes. Nat. Plants 2020. [Google Scholar] [CrossRef]
- Neumann, P.; Oliveira, L.; Čížková, J.; Jang, T.-S.; Klemme, S.; Novák, P.; Stelmach, K.; Koblížková, A.; Doležel, J.; Macas, J. Impact of parasitic lifestyle and different types of centromere organization on chromosome and genome evolution in the plant genus Cuscuta. New Phytol. 2021, 229, 2365–2377. [Google Scholar] [CrossRef]
- Emadzade, K.; Jang, T.-S.; Macas, J.; Kovařík, A.; Novák, P.; Parker, J.; Weiss-Schneeweiss, H. Differential amplification of satellite PaB6 in chromosomally hypervariable Prospero autumnale complex (Hyacinthaceae). Ann. Bot. 2014, 114, 1597–1608. [Google Scholar] [CrossRef]
- Sun, Y.; Lu, Z.; Zhu, X.; Ma, H. Genomic basis of homoploid hybrid speciation within chestnut trees. Nat. Commun. 2020, 11, 3375. [Google Scholar] [CrossRef]
- Ungerer, M.C.; Strakosh, S.C.; Zhen, Y. Genome expansion in three hybrid sunflower species is associated with retrotransposon proliferation. Curr. Biol. 2006, 16, R872–R873. [Google Scholar] [CrossRef]
- Garcia, S.; Wendel, J.F.; Borowska-Zuchowska, N.; Aïnouche, M.; Kuderova, A.; Kovarik, A. The utility of graph clustering of 5S ribosomal DNA homoeologs in plant allopolyploids, homoploid hybrids, and cryptic introgressants. Front. Plant Sci. 2020, 11, 41. [Google Scholar] [CrossRef]
- Zozomová-Lihová, J.; Mandáková, T.; Kovaříková, A.; Mühlhausen, A.; Mummenhoff, K.; Lysak, M.A.; Kovařík, A. When fathers are instant losers: Homogenization of rDNA loci in recently formed Cardamine × schulzii trigenomic allopolyploid. New Phytol. 2014, 203, 1096–1108. [Google Scholar] [CrossRef] [PubMed]
- Kovarik, A.; Pires, J.C.; Leitch, A.R.; Lim, K.Y.; Sherwood, A.M.; Matyasek, R.; Rocca, J.; Soltis, D.E.; Soltis, P.S. Rapid concerted evolution of nuclear ribosomal DNA in two Tragopogon allopolyploids of recent and recurrent origin. Genetics 2005, 169, 931–944. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.; Sahara, K.; Yoshido, A.; Marec, F. Evolutionary dynamics of rDNA clusters on chromosomes of moths and butterflies (Lepidoptera). Genetica 2010, 138, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-F.; Su, T.; Cheng, G.-Q.; Wang, B.-X.; Li, X.; Deng, C.-L.; Gao, W.-J. Chromosome evolution in connection with repetitive sequences and epigenetics in plants. Genes 2017, 8, 290. [Google Scholar] [CrossRef] [PubMed]
- Wreath, S.; Bartholmes, C.; Hidalgo, O.; Scholz, A.; Gleissberg, S. Silencing of EcFLO, a FLORICAULA/LEAFY gene of the California poppy (Eschscholzia californica, affects flower specification in a perigynous flower context. Int. J. Plant Sci. 2013, 174, 139–153. [Google Scholar] [CrossRef]
- Doležel, J.; Greilhuber, J.; Suda, J. Estimation of nuclear DNA content in plants using flow cytometry. Nat. Protoc. 2007, 2, 2233–2244. [Google Scholar] [CrossRef]
- Clark, J.; Hidalgo, O.; Pellicer, J.; Liu, H.; Marquardt, J.; Robert, Y.; Christenhusz, M.; Zhang, S.; Gibby, M.; Leitch, I.J.; et al. Genome evolution of ferns: Evidence for relative stasis of genome size across the fern phylogeny. New Phytol. 2016, 210, 1072–1082. [Google Scholar] [CrossRef]
- Obermayer, R.; Leitch, I.J.; HansonN, L.; Bennett, M.D. Nuclear DNA C-values in 30 species double the familial representation in pteridophytes. Ann. Bot. 2002, 90, 209–217. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2 Elegant Graphics for Data Analysis (Use R!); Springer: Cham, Switzerland, 2016. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Novák, P.; Neumann, P.; Macas, J. Global analysis of repetitive DNA from unassembled sequence reads using RepeatExplorer2. Nat. Protoc. 2020, 15, 3745–3776. [Google Scholar] [CrossRef]
- Neumann, P.; Novák, P.; Hoštáková, N.; Macas, J. Systematic survey of plant LTR-retrotransposons elucidates phylogenetic relationships of their polyprotein domains and provides a reference for element classification. Mob. DNA 2019, 10, 1. [Google Scholar] [CrossRef]
- Novák, P.; Ávila Robledillo, L.; Koblížková, A.; Vrbová, I.; Neumann, P.; Macas, J. TAREAN: A computational tool for identification and characterization of satellite DNA from unassembled short reads. Nucleic Acids Res. 2017, 45, e111. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Dierckxsens, N.; Mardulyn, P.; Smits, G. NOVOPlasty: De novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017, 45, e18. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Bryant, D.; Moulton, V. Neighbor-Net: An agglomerative method for the construction of phylogenetic networks. Mol. Biol. Evol. 2004, 21, 255–265. [Google Scholar] [CrossRef]
- Vallejo-Marín, M.; Hiscock, S.J. Hybridization and hybrid speciation under global change. New Phytol. 2016, 211, 1170–1187. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Traits | U. dalechampii | U. dalechampii × U. picroides | U. picroides | |||
|---|---|---|---|---|---|---|
| Mean (SD 1) | Min–Max | Mean (SD 1) | Min–Max | Mean (SD 1) | Min–Max | |
| Plant height 2,3 | 288.5 (116.60) | 125–485 | 182.10 (74.80) | 65–278 | 291.42 (224.74) | 30–800 |
| N capitula 4 | 1.2 (0.42) | 1–2 | 1.6 (0.84) | 1–3 | 6 (6.52) | 1–20 |
| Receptacle width 2 | 10.43 (2.06) | 7.78–14.89 | 6.08 (1.26) | 4.59–7.87 | 5.78 (2.73) | 1.37–8.62 |
| N Florets 5 | 207 (34.66) | 163–267 | 89.2 (33.9) | 36–140 | 83.08 (45.53) | 18–139 |
| Capitulum width 2 | 55.76 (9.11) | 42.09–68.90 | 32.81 (5.54) | 24.57–41.22 | 21.84 (4.61) | 13.68–28.35 |
| Capitulum height 2 | 30.89 (3.54) | 24.76–36.51 | 22.65 (2.39) | 19.47–27.54 | 22.21 (3.90) | 13.88–27.09 |
| Involucre width 2,4 | 24.46 (3.65) | 18.33–29.28 | 20.82 (5.02) | 11.73–26.85 | 12.74 (3.92) | 5.57–16.6 |
| Involucre height 2 | 18.72 (1.37) | 16.31–2086 | 16.26 (2.31) | 12.72–20.42 | 17.99 (3.43) | 11.7–21.91 |
| Urospermum Species | 2C (pg) ± SD 1 | N 2 | CVplt 3 | CVstd 4 | 1C (pg) | 1C (Mbp) 5 |
|---|---|---|---|---|---|---|
| U. dalechampii | 2.26 ± 0.01 | 10 | 3.99 | 2.59 | 1.13 | 1105.14 |
| U. dalechampii × U. picroides | 1.89 ± 0.01 | 8 | 3.88 | 3.00 | 0.95 | 924.21 |
| U. picroides | 1.51 ± 0.01 | 10 | 3.67 | 2.71 | 0.76 | 738.39 |
| Urospermum Species | 1C/Mbp | No. of PE Reads after QC | No. of Reads Analysed | No. of Reads Analysed * | Coverage |
|---|---|---|---|---|---|
| U. dalechampii | 1066.06 | 19,848,388 | 2,240,000 | 2,049,890 | 0.19× |
| U. picroides | 733.5 | 16,219,976 | 1,585,714 | 1,439,699 | 0.19× |
| U. dalechampii × U. picroides | 899.76 | 19,837,944 | 1,916,000 | 1,799,837 | 0.20× |
| GENOME PROPORTION (GP) | |||||||
|---|---|---|---|---|---|---|---|
| U. dalechampii | U. picroides | Hybrid | |||||
| Repeat Type | Lineage | [%] | [Mb] | [%] | [Mb] | [%] | [Mb] |
| Ty1/Copia | 24.007 | 255.911 | 20.681 | 151.698 | 22.483 | 202.292 | |
| SIRE | 18.026 | 192.154 | 17.064 | 125.168 | 17.690 | 159.163 | |
| Angela | 2.990 | 31.873 | 2.903 | 21.291 | 2.974 | 26.758 | |
| TAR | 0.308 | 3.280 | 0.357 | 2.622 | 0.430 | 3.873 | |
| Bianca | 0.517 | 5.514 | 0.241 | 1.764 | 0.377 | 3.394 | |
| Ale | 0.209 | 2.226 | 0.116 | 0.853 | 0.016 | 0.147 | |
| Ivana | 0.309 | 3.294 | 0.000 | 0.000 | 0.028 | 0.250 | |
| Tork | 1.632 | 17.397 | 0.000 | 0.000 | 0.968 | 8.706 | |
| Ikeros | 0.016 | 0.174 | 0.000 | 0.000 | 0.000 | 0.000 | |
| Ty3/Gypsy | 10.938 | 115.443 | 4.255 | 31.207 | 6.693 | 60.219 | |
| Tekay | 9.718 | 103.591 | 3.745 | 27.467 | 5.632 | 50.677 | |
| Athila | 0.981 | 9.337 | 0.373 | 2.734 | 0.871 | 7.833 | |
| CRM | 0.102 | 1.085 | 0.137 | 1.006 | 0.160 | 1.443 | |
| Retand | 0.118 | 1.256 | 0.000 | 0.000 | 0.030 | 0.266 | |
| Reina | 0.020 | 0.174 | 0.000 | 0.000 | 0.000 | 0.000 | |
| LTR | 0.865 | 9.222 | 0.074 | 0.545 | 1.849 | 16.639 | |
| Other repeats | |||||||
| Pararetrovirus | 0.874 | 9.316 | 0.188 | 1.378 | 0.645 | 5.803 | |
| LINE | 0.720 | 7.678 | 0.012 | 0.087 | 0.406 | 3.650 | |
| DNA transposons | 3.016 | 32.999 | 1.168 | 8.571 | 1.337 | 12.029 | |
| TIR/Enspm-CACTA | 0.541 | 5.765 | 0.000 | 0.000 | 0.190 | 1.713 | |
| TIR/MuDR-Mutator | 0.975 | 11.439 | 0.766 | 5.619 | 0.416 | 3.742 | |
| TIR/haT | 0.349 | 3.721 | 0.000 | 0.000 | 0.153 | 1.380 | |
| TIR/PIF-Harbinger | 0.082 | 0.666 | 0.050 | 0.366 | 0.083 | 0.748 | |
| TIR/Mariner | 0.658 | 7.018 | 0.000 | 0.000 | 0.000 | 0.000 | |
| Helitron | 0.412 | 4.390 | 0.353 | 2.586 | 0.494 | 4.445 | |
| Tandem repeats | |||||||
| rDNA | 4.173 | 44.489 | 4.141 | 30.374 | 2.954 | 26.583 | |
| Satellite | 6.942 | 74.004 | 15.638 | 114.703 | 11.657 | 104.884 | |
| Unclassified | 4.486 | 48.151 | 4.822 | 35.368 | 4.834 | 43.498 | |
| GP < 0.01% | 18.142 | 193.390 | 18.765 | 137.645 | 17.934 | 161.367 | |
| Total repeats | 74.164 | 790.602 | 69.744 | 511.575 | 70.793 | 636.964 | |
| Single copy | 25.836 | 275.415 | 30.256 | 221.926 | 29.207 | 262.796 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pellicer, J.; Balant, M.; Fernández, P.; Rodríguez González, R.; Hidalgo, O. Morphological and Genome-Wide Evidence of Homoploid Hybridisation in Urospermum (Asteraceae). Plants 2022, 11, 182. https://doi.org/10.3390/plants11020182
Pellicer J, Balant M, Fernández P, Rodríguez González R, Hidalgo O. Morphological and Genome-Wide Evidence of Homoploid Hybridisation in Urospermum (Asteraceae). Plants. 2022; 11(2):182. https://doi.org/10.3390/plants11020182
Chicago/Turabian StylePellicer, Jaume, Manica Balant, Pol Fernández, Roi Rodríguez González, and Oriane Hidalgo. 2022. "Morphological and Genome-Wide Evidence of Homoploid Hybridisation in Urospermum (Asteraceae)" Plants 11, no. 2: 182. https://doi.org/10.3390/plants11020182
APA StylePellicer, J., Balant, M., Fernández, P., Rodríguez González, R., & Hidalgo, O. (2022). Morphological and Genome-Wide Evidence of Homoploid Hybridisation in Urospermum (Asteraceae). Plants, 11(2), 182. https://doi.org/10.3390/plants11020182