
Strategies for Intraspecific Genotyping of Duckweed: Comparison of Five Orthogonal Methods Applied to the Giant Duckweed Spirodela polyrhiza

 , , , ,
, , , ,  , , and
, , and 
Abstract
:1. Introduction
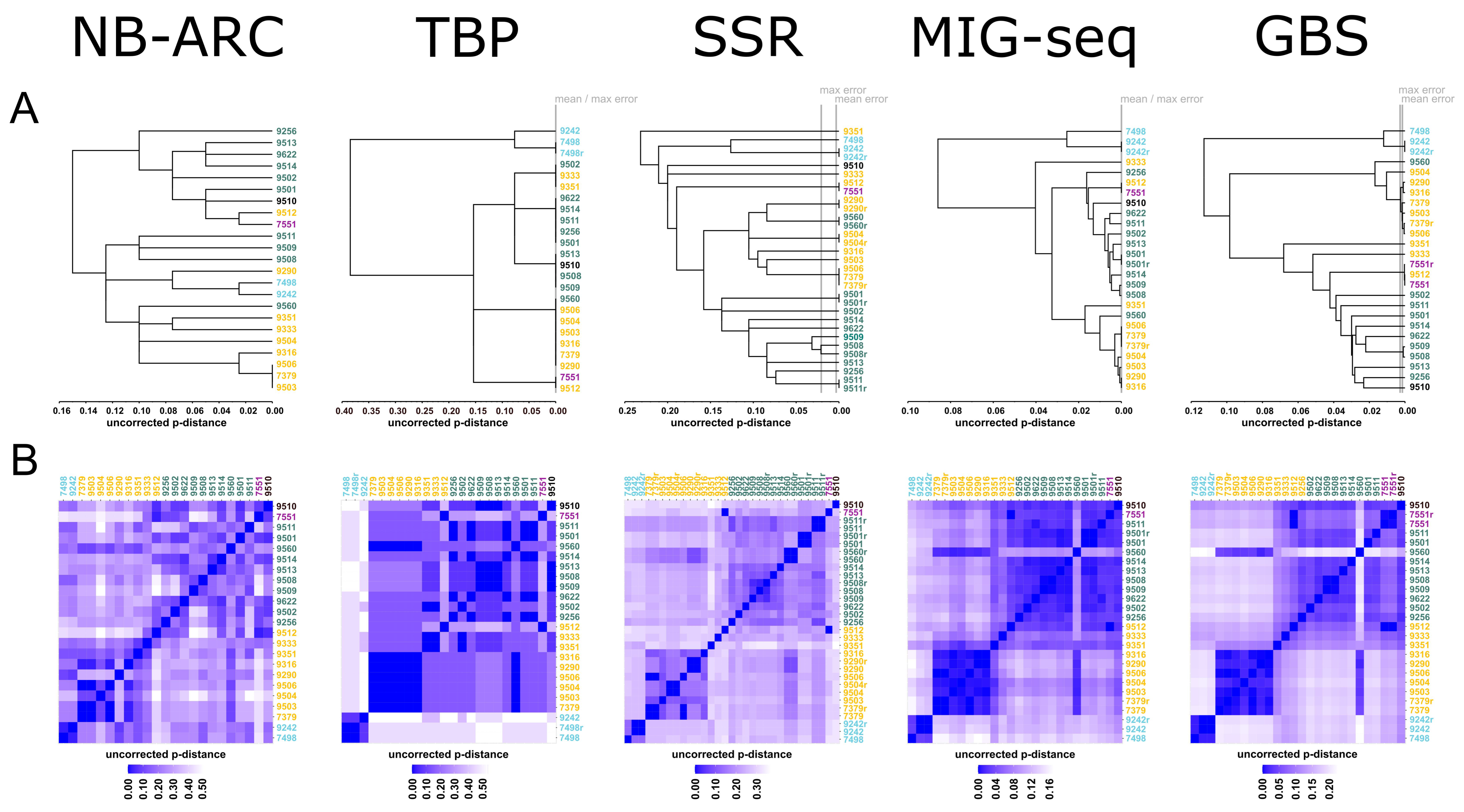
2. Results
2.1. NB-ARC-Related Genes (NB-ARC)
2.2. Tubulin-Gene-Based Polymorphism (TBP)
2.3. Simple Sequence Repeats (SSR)
2.4. Multiplexed ISSR Genotyping by Sequencing (MIG-Seq)
2.5. Genotyping-by-Sequencing (GBS)
3. Discussion
4. Materials and Methods
4.1. Plant Material and Cultivation
4.2. DNA Isolation and Downstream Lab Work
4.3. Data Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Acosta, K.; Appenroth, K.J.; Borisjuk, L.; Edelman, M.; Heinig, U.; Jansen, M.A.K.; Oyama, T.; Pasaribu, B.; Schubert, I.; Sorrels, S.; et al. Return of the Lemnaceae: Duckweed as a model plant system in the genomics and postgenomics era. Plant Cell 2021, 33, 3207–3234. [Google Scholar] [CrossRef] [PubMed]
- Tippery, N.P.; Les, D.H.; Appenroth, K.J.; Sree, K.S.; Crawford, D.J.; Bog, M. Lemnaceae and Orontiaceae are phylogenetically and morphologically distinct from Araceae. Plants 2021, 10, 2639. [Google Scholar] [CrossRef] [PubMed]
- Les, D.H.; Crawford, D.J.; Landolt, E.; Gabel, J.D.; Kimball, R.T. Phylogeny and systematics of Lemnaceae, the duckweed family. Syst. Bot. 2002, 27, 221–240. [Google Scholar] [CrossRef]
- Bog, M.; Appenroth, K.J.; Sree, K.S. Duckweed (Lemnaceae): Its molecular taxonomy. Front. Sustain. Food Syst. 2019, 3, 117. [Google Scholar] [CrossRef]
- Landolt, E. The Family of Lemnaceae—A Monographic Study. Vol. 1. Biosystematic Investigations in the Family of Duckweeds (Lemnaceae); Veröffentlichungen des Geobotanischen Institutes der ETH, Stiftung Ruebel: Zurich, Switzerland, 1986. [Google Scholar]
- Bog, M.; Appenroth, K.J.; Sree, K.S. Key to the determination of taxa of Lemnaceae: An update. Nordic J. Bot. 2020, 8, e02658. [Google Scholar] [CrossRef]
- Bog, M.; Xu, S.; Himmelbach, A.; Brandt, R.; Wagner, F.; Appenroth, K.J.; Sree, K.S. Genotyping-By-Sequencing for species delimitation in the section Uninerves Hegelm. (Genus Lemna L.). In Compendium of Plant Genomes: The Duckweed Genomes, 1st ed.; Cao, X.H., Fourounjian, P., Wang, W., Eds.; Springer Nature: Cham, Switzerland, 2020; pp. 115–123. [Google Scholar] [CrossRef]
- Bog, M.; Sree, K.S.; Fuchs, J.; Hoang, P.T.N.; Schubert, I.; Kuever, J.; Rabenstein, A.; Paolacci, S.; Jansen, M.A.K.; Appenroth, K.J. A taxonomic revision of Lemna sect. Uninerves (Lemnaceae). Taxon 2020, 69, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Vunsh, R.; Heinig, U.; Malitsky, S.; Aharoni, A.; Avidov, A.; Lerner, A.; Edelman, M. Manipulating duckweed through genome duplication. Plant Biol. 2015, 17, 115–119. [Google Scholar] [CrossRef]
- Sree, K.S.; Sudakaran, S.; Appenroth, K.J. How fast can angiosperms grow? Species and clonal diversity of growth rates in the genus Wolffia (Lemnaceae). Acta Physiol. Plant. 2015, 37, 204. [Google Scholar] [CrossRef]
- Ziegler, P.; Adelmann, K.; Zimmer, S.; Schmidt, C.; Appenroth, K.J. Relative in vitro growth rates of duckweeds (Lemnaceae), the most rapidly growing higher plants. Plant Biol. 2015, 17, 33–41. [Google Scholar] [CrossRef]
- Sree, K.S.; Appenroth, K.J. Starch accumulation in duckweeds (Lemnaceae) induced by nutrient deficiency. Emir. J. Food Agric. 2022, 34, 204–212. [Google Scholar] [CrossRef]
- Sree, K.S.; Adelmann, K.; Garcia, C.; Lam, E.; Appenroth, K.J. Natural variance in salt tolerance and induction of starch accumulation in duckweeds. Planta 2015, 241, 1395–1404. [Google Scholar] [CrossRef]
- Appenroth, K.J.; Ziegler, P.; Sree, K.S. Accumulation of starch in duckweeds (Lemnaceae), potential energy plants. Physiol. Mol. Biol. Plants. 2021, 27, 2621–2633. [Google Scholar] [CrossRef]
- Kaplan, A.; Zelicha, H.; Tsaban, G.; Yaskolka, M.A.; Rinott, E.; Kovsan, J.; Novack, L.; Thiery, J.; Ceglarek, U.; Burkhardt, R.; et al. Protein bioavailability of Wolffia globosa duckweed, a novel aquatic plant—A randomized controlled trial. Clin. Nutr. 2019, 38, 2576–2582. [Google Scholar] [CrossRef] [PubMed]
- Bog, M.; Lautenschlager, U.; Landrock, M.F.; Landolt, E.; Fuchs, J.; Sree, K.S.; Oberprieler, C.; Appenroth, K.J. Genetic characterization and barcoding of taxa in the genera Landoltia and Spirodela (Lemnaceae) by three plastidic markers and amplified fragment length polymorphism (AFLP). Hydrobiologia 2015, 749, 169–182. [Google Scholar] [CrossRef]
- Hoang, P.T.N.; Fuchs, J.; Schubert, V.; Tran, T.B.N.; Schubert, I. Chromosome Numbers and Genome Sizes of All 36 Duckweed Species (Lemnaceae). Plants 2022, 11, 2674. [Google Scholar] [CrossRef] [PubMed]
- Michael, T.P.; Bryant, D.; Gutierrez, R.; Borisjuk, N.; Chu, P.; Zhang, H.; Xia, J.; Zhou, J.; Peng, H.; El Baidouri, M.; et al. Comprehensive definition of genome features in Spirodela polyrhiza by high-depth physical mapping and short-read DNA sequencing strategies. Plant J. 2017, 89, 617–635. [Google Scholar] [CrossRef] [Green Version]
- Gan, X.; Stegle, O.; Behr, J.; Steffen, J.; Drewe, P.; Hildebrand, K.; Lyngsoe, R.; Schultheiss, S.J.; Osborne, E.J.; Sreedharan, K.L.; et al. Multiple reference genomes and transcriptomes for Arabidopsis thaliana. Nature 2011, 477, 419–423. [Google Scholar] [CrossRef] [Green Version]
- Lu, P.; Han, X.; Qi, J.; Yang, J.; Wijeratne, A.J.; Li, T.; Ma, H. Analysis of Arabidopsis genome-wide variations before and after meiosis and meiotic recombination by resequencing Landsberg erecta and all four products of a single meiosis. Genome Res. 2012, 22, 508–518. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Stapley, J.; Gablenz, S.; Boyer, J.; Appenroth, K.J.; Sree, K.S.; Gershenzon, J.; Widmer, A.; Huber, M. Low genetic variation is associated with low mutation rate in the giant duckweed. Nat. Commun. 2019, 10, 1243. [Google Scholar] [CrossRef]
- Ho, E.K.H.; Bartkowska, M.; Wright, S.I.; Agrawal, A.F. Population genomics of the facultatively asexual duckweed Spirodela polyrhiza. New Phytol. 2019, 224, 1361–1371. [Google Scholar] [CrossRef]
- Chu, P.; Wilson, G.M.; Michael, T.P.; Vaiciunas, J.; Honig, J.; Lam, E. Sequence-guided approach to genotyping plant clones and species using polymorphic NB-ARC-related genes. Plant Mol. Biol. 2018, 98, 219–231. [Google Scholar] [CrossRef]
- Clark, R.M.; Schweikert, G.; Toomajian, C.; Ossowski, S.; Zeller, G.; Shinn, P.; Warthmann, N.; Gu, T.T.; Fu, G.; Hinds, D.A.; et al. Common sequence polymorphisms shaping genetic diversity in Arabidopsis thaliana. Science 2007, 317, 338–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuehdorf, K.; Jetschke, G.; Ballani, L.; Appenroth, K.J. The clonal dependence of turion formation in the duckweed Spirodela polyrhiza—An ecogeographical approach. Physiol. Plant. 2014, 150, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Braglia, L.; Gavazzi, F.; Morello, L.; Gianì, S.; Nick, P.; Breviario, D. On the applicability of the Tubulin-Based Polymorphism (TBP) genotyping method: A comprehensive guide illustrated through the application on different genetic resources in the legume family. Plant Methods 2020, 16, 86. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Hu, F.; Zhang, W.; Wang, M.; Zhu, M.; Ke, J. Characterization of 19 polymorphic SSR markers in Spirodela polyrhiza (Lemnaceae) and cross amplification in Lemna perpusilla. Appl. Plant Sci. 2018, 6, 1–5. [Google Scholar] [CrossRef]
- Suyama, Y.; Matsuki, Y. MIG-seq: An effective PCR-based method for genome-wide single-nucleotide polymorphism genotyping using the next-generation sequencing platform. Sci. Rep. 2015, 5, 16963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braglia, L.; Lauria, M.; Appenroth, K.J.; Bog, M.; Breviario, D.; Grasso, A.; Gavazzi, F.; Morello, L. Duckweed species genotyping and interspecific hybrid discovery by tubulin-based polymorphism fingerprinting. Front. Plant Sci. 2021, 12, 625670. [Google Scholar] [CrossRef]
- Michael, T.P.; Ernst, E.; Hartwick, N.; Chu, P.; Bryant, D.; Gilbert, S.; Ortleb, S.; Baggs, E.L.; Sree, K.S.; Appenroth, K.J.; et al. Genome and time-of-day transcriptome of Wolffia australiana link morphological minimization with gene loss and less growth control. Genome Res 2021, 31, 225–238. [Google Scholar] [CrossRef]
- Wang, X.; Wang, L. GMATA: An Integrated Software Package for Genome-Scale SSR Mining, Marker Development and Viewing. Front. Plant Sci. 2016, 7, 1350. [Google Scholar] [CrossRef]
- Braglia, L.; Breviario, D.; Gianì, S.; Gavazzi, F.; De Gregori, J.; Morello, L. New Insights into Interspecific Hybridization in Lemna L. Sect. Lemna (Lemnaceae Martinov). Plants 2021, 10, 2767. [Google Scholar] [CrossRef] [PubMed]
- Gavazzi, F.; Braglia, L.; Mastromauro, F.; Gianì, S.; Morello, L.; Breviario, D. The Tubulin-Based-Polymorphism Method Provides a Simple and Effective Alternative to the Genomic Profiling of Grape. PLoS ONE 2016, 11, e0163335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braglia, L.; Manca, A.; Gianì, S.; Hatzopoulos, P.; Breviario, D. A Simplified Approach for Olive (Olea europaea L.) Genotyping and Cultivars Traceability. Am. J. Plant Sci. 2017, 8, 3475–3489. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Appenroth, K.J.; Landesman, L.; Salmean, A.A.; Lam, E. Duckweed rising at Chengdu: Summary of the 1st International Conference on Duckweed Application and Research. Plant Mol. Biol. 2012, 78, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Hoang, P.N.T.; Michael, T.P.; Gilbert, S.; Chu, P.; Motley, S.T.; Appenroth, K.J.; Schubert, I.; Lam, E. Generating a high-confidence reference genome map of the greater duckweed by integration of cytogenomic, optical mapping, and Oxford Nanopore technologies. Plant J. 2018, 96, 670–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appenroth, K.J.; Teller, S.; Horn, M. Photophysiology of turion formation and germination in Spirodela polyrhiza. Biol. Plant. 1996, 38, 95–106. [Google Scholar] [CrossRef]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Valledor, L.; Escandón, M.; Meijón, M.; Nukarinen, E.; Canal, M.J.; Weckwerth, W. A universal protocol for the combined isolation of metabolites, DNA, long RNAs, small RNAs, and proteins from plants and microorganisms. Plant J. 2014, 79, 173–180. [Google Scholar] [CrossRef]
- National Center for Biotechnolgy Information (NCBI). Available online: https://www.ncbi.nlm.nih.gov/ (accessed on 10 October 2022).
- Wendler, N.; Mascher, M.; Nöh, C.; Himmelbach, A.; Scholz, U.; Ruge-Wehling, B.; Stein, N. Unlocking the secondary genepool of barley with next-generation sequencing. Plant Biotechnol. J. 2014, 12, 1122–1131. [Google Scholar] [CrossRef]
- Matschiner, M.; Salzburger, W. TANDEM: Integrating automated allele binning into genetics and genomics workflows. Bioinformatics 2009, 25, 1982–1983. [Google Scholar] [CrossRef] [Green Version]
- Eaton, D.A.R.; Overcast, I. ipyrad: Interactive assembly and analysis of RADseq datasets. Bioinformatics 2020, 36, 2592–2594. [Google Scholar] [CrossRef]
- McCartney-Melstad, E.; Gidis, M.; Shaffer, H.B. An empirical pipeline for choosing the optimal clustering threshold in RADseq studies. Mol. Ecol. Resour. 2019, 19, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Spirodela polyrhiza Genome on Joint Genome Institute (JGI). Available online: https://data.jgi.doe.gov/refine-download/phytozome?organism=Spolyrhiza&expanded=290 (accessed on 13 October 2022).
- R Core Team R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. 2021. Available online: https://www.R-project.org/ (accessed on 6 October 2022).
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef] [PubMed]
- van der Loo, M.P.J. The stringdist package for approximate string matching. R J. 2014, 6, 111–122. [Google Scholar]
- Schliep, K.P. phangorn: Phylogenetic analysis in R. Bioinformatics 2011, 27, 592–593. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. Reshaping Data with the reshape Package. J. Stat. Soft 2007, 21, 1–20. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- FigTree—A Graphical Viewer of Phylogenetic Trees and as a Program for Producing Publication-Ready Figures. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 6 October 2022).



{kind=link}
{kind=link}
| NB-ARC | TBP | SSR | MIG-Seq | GBS | |
|---|---|---|---|---|---|
| Marker Type 1 | FLP + SNP | FLP | FLP | SNP | SNP |
| Number of Characters | 40 (17 + 23) | 13 | 95 | 1292 | 6170 |
| Percentage of Ambiguous Characters | 26 | 0 | 0 | 29 | 14 |
| Average Number of Ambiguous Characters per Sample ± Standard Deviation | 11 ± 5 | 0 ± 0 | 0 ± 0 | 380 ± 69 | 894 ± 286 |
| Error Rate % (Number of Replicates) 2 | n.d. (0) | 0.00 (1) | 0.26 (8) | 0.00 (3) | 0.17 (3) |
| NB-ARC | TBP | SSR | MIG-Seq | GBS | |
|---|---|---|---|---|---|
| DNA requirements | degraded works as well | degraded works as well | degraded works as well | degraded works as well | high quality necessary |
| Establishment | elaborate, genome sequence information needed | easy | elaborate, sequence information needed | easy | easy |
| Universality * | no, primers can be species-specific | yes | (no), low cross-amplification | yes | yes |
| Resolution capacity | high | low | high | high | high |
| Costs per sample | low to moderate, depends on whether genome information is available | low | low | moderate | high |
| Clone ID | Country | Area/State | Specific Turion Yield 1 | Specific Growth Rate (h−1) 2 | Genome Size (Mbp/1C) 3 | Genome Sequencing 4 |
|---|---|---|---|---|---|---|
| 7379 | India | Tamil Nadu | 1.86 ± 0.26 | 0.315 ± 0.008 | 158 ± 3 | n.d. |
| 7498 | USA | North Carolina | 2.37 ± 0.27 | 0.401 ± 0.016 | 157 ± 2 | + |
| 7551 | Australia | Northern Territory | n.d. | 0.376 ± 0.009 | n.d. | + |
| 9242 | Ecuador | Guayas | 1.37 ± 0.12 | 0.456 ± 0.003 | n.d. | + |
| 9256 | Finland | Uusimaa | 3.73 ± 0.25 | 0.296 ± 0.011 | 160 ± 6 | + |
| 9290 | India | Delhi | 1.40 ± 0.13 | 0.299 ± 0.010 | n.d. | + |
| 9316 | India | Rajasthan | 1.22 ± 0.13 | 0.281 ± 0.009 | 159 ± 2 | + |
| 9333 | China | Hubei | n.d. | 0.311 ± 0.033 | n.d. | + |
| 9351 | Vietnam | Hanoi | n.d. | 0.367 ± 0.024 | n.d. | n.d. |
| 9501 | Albania | Fieri | 5.93 ± 0.03 | 0.357 ± 0.005 | 164 ± 4 | + |
| 9502 | Ireland | Leinster | 1.64 ± 0.14 | 0.337 ± 0.008 | 164 ± 5 | + |
| 9503 | India | Rajasthan | 0.51 ± 0.03 | 0.312 ± 0.007 | 170 ± 5 | + |
| 9504 | India | Rajasthan | 0.34 ± 0.03 | 0.284 ± 0.012 | 168 ± 6 | + |
| 9506 | India | Telangana | 0.97 ± 0.07 | 0.313 ± 0.005 | 161 ± 7 | + |
| 9508 | Poland | Cracow | 0.66 ± 0.04 | 0.360 ± 0.007 | 160 ± 4 | + |
| 9509 | Germany | Thuringia | 0.51 ± 0.07 | 0.323 ± 0.006 | 157 ± 2 | + |
| 9510 | Mozambique | Maputo | 0.98 ± 0.10 | 0.289 ± 0.008 | n.d. | + |
| 9511 | Russia | Moscow | 2.25 ± 0.11 | 0.381 ± 0.009 | 161 ± 5 | + |
| 9512 | Russia | Irkutsk | 2.92 ± 0.49 | 0.386 ± 0.005 | 156 ± 3 | + |
| 9513 | Czech | Jindřichův Hradec | 1.16 ± 0.04 | 0.324 ± 0.006 | 161 ± 5 | + |
| 9514 | Austria | Viena | 1.17 ± 0.16 | 0.333 ± 0.012 | 159 ± 5 | + |
| 9560 | Hungary | Bekes | n.d. | 0.367 ± 0.008 | 160 ± 6 | + |
| 9622 | Germany | Baden-Württemberg | n.d. | 0.310 ± 0.011 | 159 ± 5 | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bog, M.; Braglia, L.; Morello, L.; Noboa Melo, K.I.; Schubert, I.; Shchepin, O.N.; Sree, K.S.; Xu, S.; Lam, E.; Appenroth, K.J. Strategies for Intraspecific Genotyping of Duckweed: Comparison of Five Orthogonal Methods Applied to the Giant Duckweed Spirodela polyrhiza. Plants 2022, 11, 3033. https://doi.org/10.3390/plants11223033
Bog M, Braglia L, Morello L, Noboa Melo KI, Schubert I, Shchepin ON, Sree KS, Xu S, Lam E, Appenroth KJ. Strategies for Intraspecific Genotyping of Duckweed: Comparison of Five Orthogonal Methods Applied to the Giant Duckweed Spirodela polyrhiza. Plants. 2022; 11(22):3033. https://doi.org/10.3390/plants11223033
Chicago/Turabian StyleBog, Manuela, Luca Braglia, Laura Morello, Karen I. Noboa Melo, Ingo Schubert, Oleg N. Shchepin, K. Sowjanya Sree, Shuqing Xu, Eric Lam, and Klaus J. Appenroth. 2022. "Strategies for Intraspecific Genotyping of Duckweed: Comparison of Five Orthogonal Methods Applied to the Giant Duckweed Spirodela polyrhiza" Plants 11, no. 22: 3033. https://doi.org/10.3390/plants11223033
APA StyleBog, M., Braglia, L., Morello, L., Noboa Melo, K. I., Schubert, I., Shchepin, O. N., Sree, K. S., Xu, S., Lam, E., & Appenroth, K. J. (2022). Strategies for Intraspecific Genotyping of Duckweed: Comparison of Five Orthogonal Methods Applied to the Giant Duckweed Spirodela polyrhiza. Plants, 11(22), 3033. https://doi.org/10.3390/plants11223033