
A Selection of Reliable Reference Genes for Gene Expression Analysis in the Female and Male Flowers of Salix suchowensis


Abstract
:1. Introduction
2. Results
2.1. RNA Quality and Primer Specificity Analysis
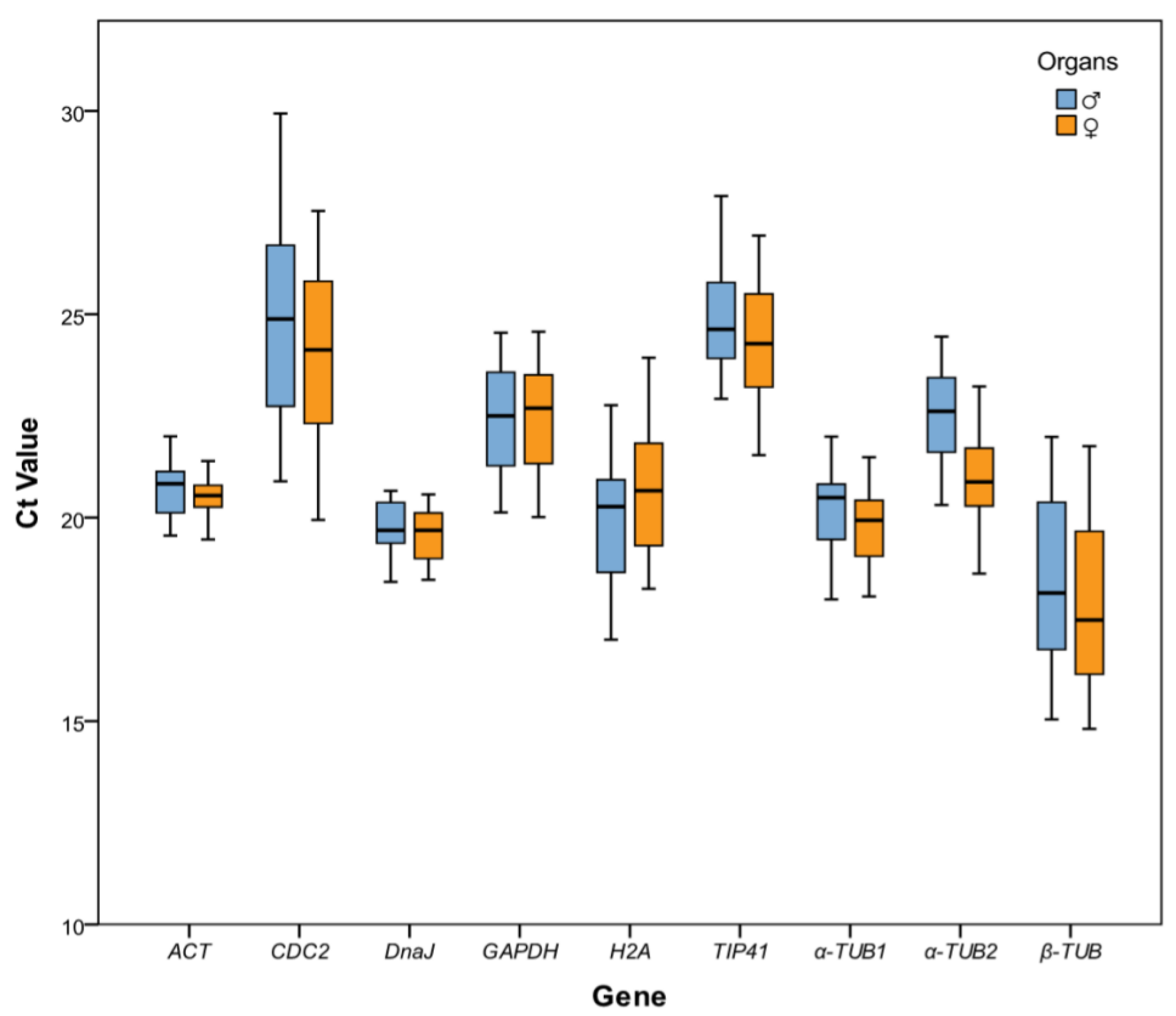
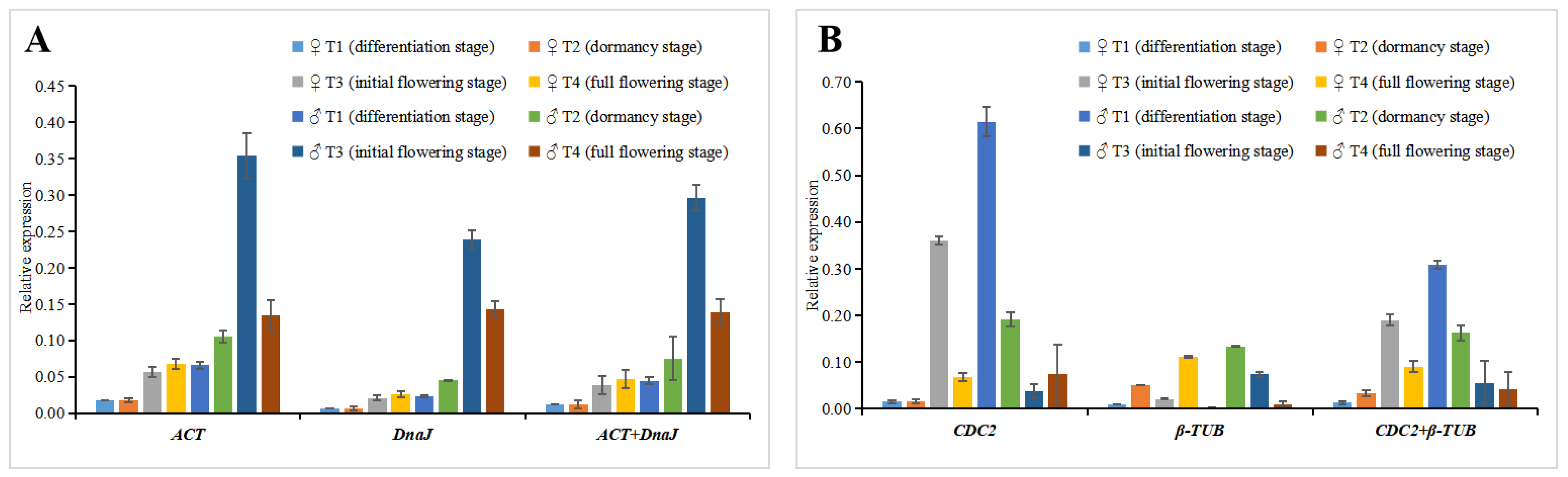
2.2. Reference Gene Expression Levels
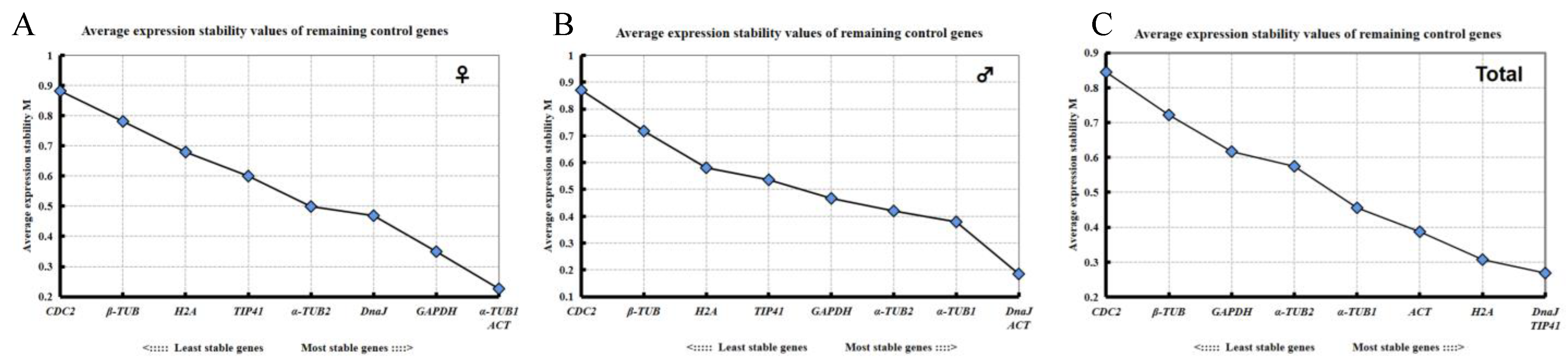
2.3. Expression Stability Analysis of the Candidate Reference Genes
2.3.1. geNorm Analysis
2.3.2. NormFinder Analysis
2.3.3. ΔCt Analysis
2.3.4. BestKeeper Analysis
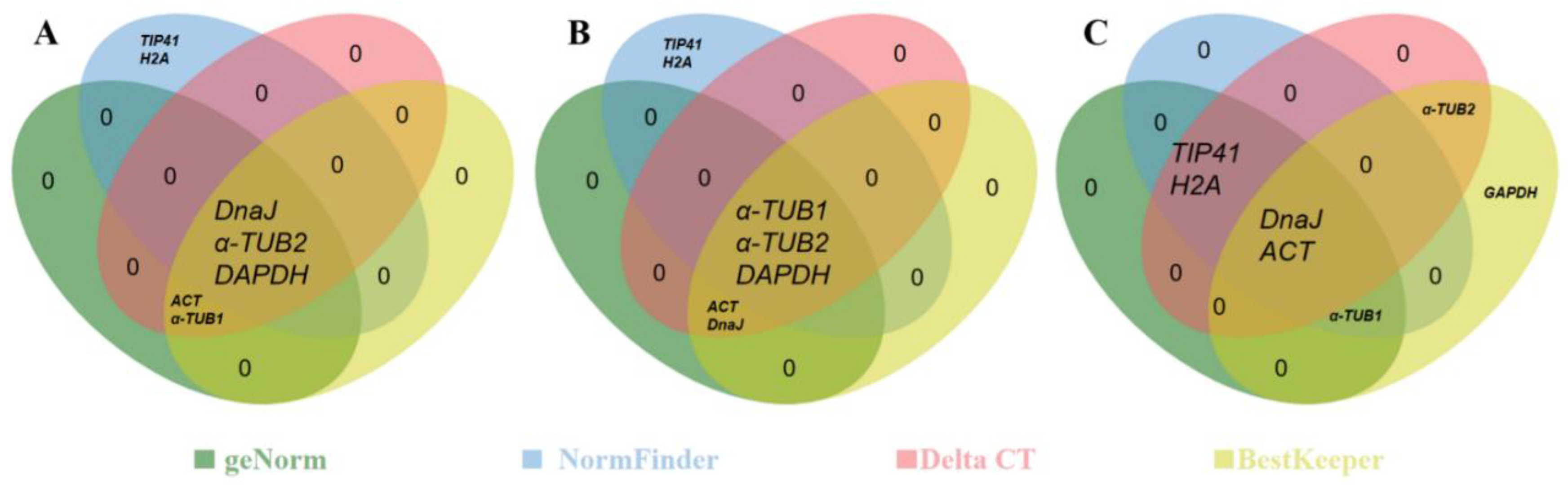
2.3.5. RefFinder Analysis
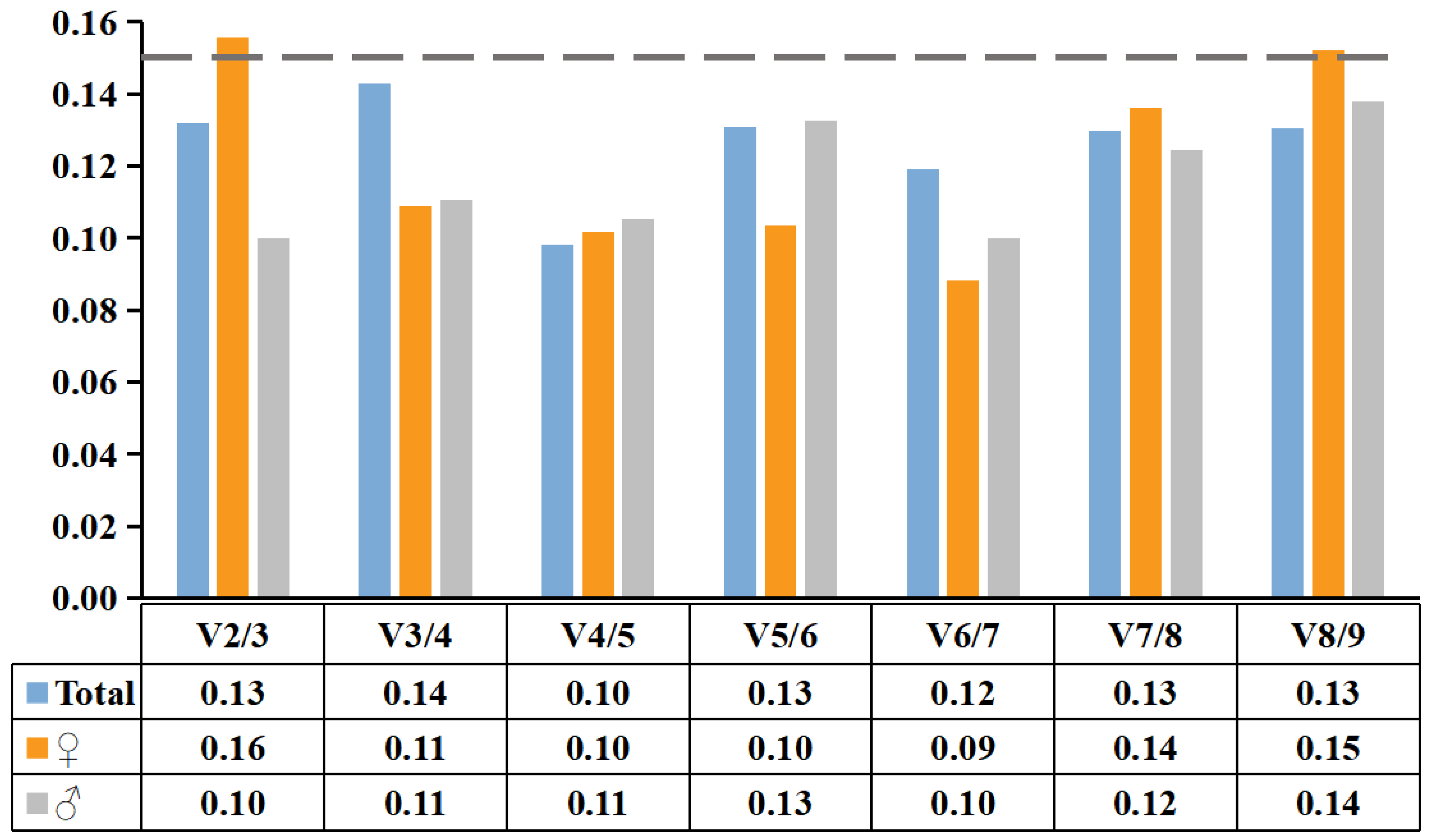
2.4. Reference Gene Stability Verification
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. Extraction of Total RNA from Plant Tissues and cDNA Aynthesis
4.3. Selection of Reference Genes and Primer Design
4.4. qRT-PCR of Candidate Reference Genes
4.5. Establishment of Reference Gene Primer Standard Curve
4.6. Data Processing
4.7. Reference Gene Verification
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Argus, G.W. Infrageneric classification of Salix (Salicaceae) in the new world. Syst. Bot. Monogr. 1997, 52, 1–121. [Google Scholar] [CrossRef]
- Karp, A.; Hanley, S.J.; Trybush, S.O.; Macalpine, W.; Pei, M.; Shield, I. Genetic improvement of willow for bioenergy and biofuels. J. Integr. Plant Biol. 2011, 53, 151–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Yuan, H.; Li, Y.; Chen, Y.; Liu, G.; Ye, M.; Yu, C.; Lian, B.; Zhong, F.; Jiang, Y.; et al. Genome sequencing and phylogenetic analysis of allotetraploid Salix matsudana Koidz. Hortic. Res. 2020, 7, 201. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Wu, H.; Chen, Y.; Li, X.; Hou, J.; Lu, J.; Wei, S.; Dai, X.; Olson, M.S.; Liu, J.; et al. Evidences for a role of two Y-specific genes in sex determination in Populus deltoides. Nat. Commun. 2020, 11, 5893. [Google Scholar] [CrossRef]
- Li, J.; Xia, X.; Xu, S.; Wu, J.; Peng, L.; Zhao, L. Development, structure and evolutionary significance of seed appendages in Salix matsudana (Salicaceae). PLoS ONE 2018, 13, e0203061. [Google Scholar] [CrossRef]
- Futamura, N.; Mori, H.; Kouchi, H.; Shinohara, K. Male flower-specific expression of genes for polygalacturonase, pectin methylesterase and beta-1,3-glucanase in a dioecious willow (Salix gilgiana Seemen). Plant Cell Physiol. 2000, 41, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Costache, A.; Berghi, O.N.; Cergan, R.; Dumitru, M.; Neagos, A.; Popa, L.G.; Giurcaneanu, C.; Vrinceanu, D. Respiratory allergies: Salicaceae sensitization (Review). Exp. Ther. Med. 2021, 21, 609. [Google Scholar] [CrossRef]
- Cai, Z.; Yang, C.; Liao, J.; Song, H.; Zhang, S. Sex-biased genes and metabolites explain morphologically sexual dimorphism and reproductive costs in Salix paraplesia catkins. Hortic. Res. 2021, 8, 125. [Google Scholar] [CrossRef]
- Dai, X.; Hu, Q.; Cai, Q.; Feng, K.; Ye, N.; Tuskan, G.A.; Milne, R.; Chen, Y.; Wan, Z.; Wang, Z.; et al. The willow genome and divergent evolution from poplar after the common genome duplication. Cell Res. 2014, 24, 1274–1277. [Google Scholar] [CrossRef]
- Wei, S.Y.; Yang, Y.H.; Yin, T.M. The chromosome-scale assembly of the willow genome provides insight into Salicaceae genome evolution. Hortic. Res. 2020, 7, 45. [Google Scholar] [CrossRef] [Green Version]
- Gibson, U.E.; Heid, C.A.; Williams, P.M. A novel method for real time quantitative RT-PCR. Genome Res. 1996, 6, 995–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gachon, C.; Mingam, A.; Charrier, B. Real-time PCR: What relevance to plant studies? J. Exp. Bot. 2004, 55, 1445–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, D. Quantifification using real-time PCR technology: Applications and limitations. Trends Mol. Med. 2002, 8, 257–260. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.; et al. The need for transparency and good practices in the qPCR literature. Nat. Methods 2013, 10, 1063–1067. [Google Scholar] [CrossRef]
- Zhang, Y.; Han, X.; Chen, S.; Zheng, L.; He, X.; Liu, M.; Qiao, G.; Wang, Y.; Zhou, R. Selection of suitable reference genes for quantitative real-time PCR gene expression analysis in Salix matsudana under different abiotic stresses. Sci. Rep. 2017, 7, 40290. [Google Scholar] [CrossRef] [Green Version]
- Ambroise, V.; Legay, S.; Guerriero, G.; Hausman, J.F.; Cuypers, A.; Sergeant, K. Selection of appropriate reference genes for gene expression analysis under abiotic stresses in Salix viminalis. Int. J. Mol. Sci. 2019, 20, 4210. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, L.; Mauriat, M.; Guénin, S.; Pelloux, J.; Lefebvre, J.F.; Louvet, R.; Rusterucci, C.; Moritz, T.; Guerineau, F.; Bellini, C.; et al. The lack of a systematic validation of reference genes: A serious pitfall undervalued in reverse transcription-polymerase chain reaction (RT-PCR) analysis in plants. Plant Biotechnol. J. 2008, 6, 609–618. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Jia, H.; Han, X.; Zhang, J.; Sun, P.; Lu, M.; Hu, J. Selection of reliable reference genes for gene expression analysis under abiotic stresses in the desert biomass willow, Salix psammophila. Front Plant Sci. 2016, 7, 1505. [Google Scholar] [CrossRef] [Green Version]
- Saha, P.; Blumwald, E. Assessing reference genes for accurate transcript normalization using quantitative real-time PCR in Pearl millet [Pennisetum glaucum (L.) R. Br.]. PLoS ONE 2014, 9, e106308. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Mao, Y.; Huang, S.; Ni, J.; Lu, W.; Hou, J.; Wang, Y.; Zhao, W.; Li, M.; Wang, Q.; et al. Selection of suitable reference genes for quantitative real-time PCR in Sapium sebiferum. Front. Plant Sci. 2017, 8, 637. [Google Scholar] [CrossRef] [Green Version]
- Shin, B.; Choi, G.; Yi, H.; Yang, S.; Cho, I.; Kim, J.; Lee, S.; Paek, N.C.; Kim, J.H.; Song, P.S.; et al. AtMYB21, a gene encoding a flower-specific transcription factor, is regulated by COP1. Plant J. 2002, 30, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Fernández, M.; Lukaszewicz, G.; Lamattina, L.; Cassia, R. Selection and optimization of reference genes for RT-qPCR normalization: A case study in Solanum lycopersicum exposed to UV-B. Plant Physiol. Biochem. 2021, 160, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Andersen, C.L.; Jensen, J.L.; Ørntoft, T.F. Normalization of real-time quantitative reverse transcription-PCR data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Li, N.; Fang, H.; Qi, X.; Zhou, Y. Selection and Validation of Reference Genes for Normalisation of Gene Expression in Glehnia littoralis. Sci Rep. 2020, 10, 7374. [Google Scholar] [CrossRef]
- Pfaffl, M.; Tichopad, A.; Prgomet, C.; Neuvians, T.P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper-Excel-based tool using pair-wise correlations. Biotechnol. Lett. 2004, 26, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Xiao, P.; Chen, D.; Xu, L.; Zhang, B. miRDeepFinder: A miRNA analysis tool for deep sequencing of plant small RNAs. Plant Mol. Biol. 2012, 80, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Dan, N.; Zhang, X.; Lin, S.; Bao, M.; Fu, X. Identification, characterization and functional analysis of C-class genes associated with double flower trait in Carnation (Dianthus caryphyllus L.). Plants 2020, 9, 87. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Zhang, S.; Lei, Y.B.; Xu, G.; Zhang, D. Alternative growth and defensive strategies reveal potential and gender specific trade-offs in dioecious plants Salix paraplesia to nutrient availability. Front. Plant Sci. 2016, 7, 1064. [Google Scholar] [CrossRef] [Green Version]
- Jia, H.; Zhang, J.; Li, J.; Sun, P.; Zhang, Y.; Xin, X.; Lu, M.; Hu, J. Genome-wide transcriptomic analysis of a desert willow, Salix psammophila, reveals the function of hub genes SpMDP1 and SpWRKY33 in drought tolerance. BMC Plant Biol. 2019, 19, 356. [Google Scholar] [CrossRef] [Green Version]
- Pucholt, P.; Wright, A.E.; Conze, L.; Mank, J.; Berlin, S. Recent sex chromosome divergence despite ancient dioecy in the willow Salix viminalis. Mol. Biol. Evol. 2017, 34, 1991–2001. [Google Scholar] [CrossRef]
- Zhou, R.; Macaya-Sanz, D.; Carlson, C.H.; Schmutz, J.; Jenkins, J.W.; Kudrna, D.; Sharma, A.; Sandor, L.; Shu, S.; Barry, K.; et al. A willow sex chromosome reveals convergent evolution of complex palindromic repeats. Genome Biol. 2020, 21, 38. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Jia, K.H.; Zhang, R.G.; Wang, Y.; Shi, T.L.; Li, Z.C.; Zeng, S.W.; Cai, X.J.; Wagner, N.D.; Hörandl, E.; et al. Chromosome-scale assembly of the genome of Salix dunnii reveals a male-heterogametic sex determination system on chromosome 7. Mol. Ecol. Resour. 2021, 21, 1966–1982. [Google Scholar] [CrossRef] [PubMed]
- Manechini, J.R.; da Silva Santos, P.H.; Romanel, E.; dos Santos Brito, M.; Scarpari, M.S.; Jackson, S.; Pinto, L.R.; Vicentini, R. Transcriptomic analysis of changes in gene expression during flowering induction in sugarcane under controlled photoperiodic conditions. Front. Plant Sci. 2021, 12, 635784. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.; Horgan, G.; Dempfle, L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002, 30, e36. [Google Scholar] [CrossRef] [PubMed]
- Hellemans, J.; Mortier, G.; De Paepe, A.; Speleman, F.; Vandesompele, J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007, 8, R19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parveen, S.; Rahman, A. Actin isovariant ACT7 modulates root thermomorphogenesis by altering intracellular auxin homeostasis. Int. J. Mol. Sci. 2021, 22, 7749. [Google Scholar] [CrossRef] [PubMed]
- Fratini, M.; Krishnamoorthy, P.; Stenzel, I.; Riechmann, M.; Matzner, M.; Bacia, K.; Heilmann, M.; Heilmann, I. Plasma membrane nano-organization specifies phosphoinositide effects on Rho-GTPases and actin dynamics in tobacco pollen tubes. Plant Cell. 2021, 33, 642–670. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.F.; Fatema, U.; Peng, X.; Hacker, S.W.; Maruyama, D.; Sun, M.X.; Kawashima, T. ARP2/3-independent WAVE/SCAR pathway and class XI myosin control sperm nuclear migration in flowering plants. Proc. Natl. Acad. Sci. USA 2020, 117, 32757–32763. [Google Scholar] [CrossRef]
- Zhang, K.; Fan, W.; Chen, D.; Jiang, L.; Li, Y.; Yao, Z.; Yang, Y.; Qiu, D. Selection and validation of reference genes for quantitative gene expression normalization in Taxus spp. Sci. Rep. 2020, 10, 22205. [Google Scholar] [CrossRef]
- Czechowski, T.; Stitt, M.; Altmann, T.; Udvardi, M.K.; Scheible, W.R. Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol. 2005, 139, 5–17. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Hou, S.; Cui, G.; Chen, S.; Wei, J.; Huang, L. Characterization of reference genes for quantitative real-time PCR analysis in various tissues of Salvia miltiorrhiza. Mol. Biol. Rep. 2010, 37, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Yan, H.; Jiang, X.; Zhang, Y.; Zhang, X.; Ji, Y.; Zeng, B.; Xu, B.; Yin, G.; Lee, S.; et al. Reference gene selection for quantitative real-time reverse-transcriptase PCR in orchardgrass subjected to various abiotic stresses. Gene 2014, 553, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuka, T.; Saito, M.; Yamada, E.; Fujita, K.; Yamagishi, N.; Yoshikawa, N.; Nishihara, M. Isolation and characterization of the C-class MADS-box gene involved in the formation of double flowers in Japanese gentian. BMC Plant Biol. 2015, 15, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yim, A.K.; Wong, J.W.; Ku, Y.S.; Qin, H.; Chen, T.F.; Lam, H.M. Using RNA-Seq data to evaluate reference genes suitable for gene expression studies in soybean. PLoS ONE 2015, 10, e0136343. [Google Scholar]
- Li, G.; Xu, G.; Lin, Z.; Li, H.; Liu, Z.; Xu, Y.; Zhang, H.; Ji, R.; Luo, W.; Qiu, Y.; et al. Selection of suitable reference genes for RT-qPCR normalisation in sweet potato (Ipomoea batatas L.) under different stresses. J. Hortic. Sci. Biotechnol. 2021, 2, 209–219. [Google Scholar]
- Rüdiger, S.; Schneider-Mergener, J.; Bukau, B. Its substrate specificity characterizes the DnaJ co-chaperone as a scanning factor for the DnaK chaperone. EMBO J. 2001, 20, 1042–1050. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.K.; Mangalam, A.K.; Dwivedi, S.; Naik, S. Primer premier: Program for design of degenerate primers from a protein sequence. Biotechniques 1998, 24, 318–319. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Lu, M.; Chen, Y.; Zhan, Z.; Cui, Q.; Wang, Y. Selection of reliable reference genes for gene expression studies using real-time PCR in tung tree during seed development. PLoS ONE 2012, 7, e43084. [Google Scholar] [CrossRef]
- Silver, N.; Best, S.; Jiang, J.; Thein, S.L. Selection of housekeeping genes for gene expression studies in human reticulocytes using real-time PCR. BMC Mol. Biol. 2006, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Plackett, A.R.; Thomas, S.G.; Wilson, Z.A.; Hedden, P. Gibberellin control of stamen development: A fertile field. Trends Plant Sci. 2011, 16, 568–578. [Google Scholar] [CrossRef]
- Zhou, F.; Chen, Y.; Wu, H.; Yin, T. Genome-Wide Comparative Analysis of R2R3 MYB Gene Family in Populus and Salix and Identification of Male Flower Bud Development-Related Genes. Front. Plant Sci. 2021, 12, 721558. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Gene Description | Locus Name | Primer Sequence F/R(5′-3′) | Product Size (bp) | Efficiency (%) | R2 |
|---|---|---|---|---|---|---|
| α-TUB1 | alpha-tubulin1 | EVM0011215.1 | CTCTTGGAGCGTTTGTCAGT | 175 | 100.25 | 0.968 |
| TCGTAGATGGCTTCATTGTC | ||||||
| α-TUB2 | alpha-tubulin2 | EVM0036214.1 | TTTCAAGTGCGGTATCAACT | 250 | 96.45 | 0.981 |
| CTTCCTCGTAGTCTTTCTCAA | ||||||
| β-TUB | β-Tubulin | EVM0022575.1 | AATGCGGCAACCAAATAGGA | 164 | 86.66 | 0.996 |
| TAGAACAGCACGAGGGACAA | ||||||
| DnaJ | chaperone protein DnaJ 49 | EVM0005127.1 | GAGCCTATTTATGCCCTTTC | 162 | 95.45 | 0.990 |
| GACGTAGTCCTTATCCACCTT | ||||||
| CDC2 | Cyclin Dependent Kinase-putative | EVM0002694.1 | GATGTATGGTCAGTGGGATG | 158 | 106.62 | 0.983 |
| TTGAAGTCAGGCAAAGAAGT | ||||||
| ACT | actin | EVM0032804.1 | CCAAGGAGGCTGCTGGTAAT | 189 | 98.90 | 0.993 |
| CCAAGGCCACCATGTCTGTC | ||||||
| TIP41 | Type 2A phosphatase activator | EVM0041976.1 | GCTAATGAGATTAAGGGACA | 142 | 101.06 | 0.985 |
| TGAAGCAGAGTCAGCAGGAT | ||||||
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase | EVM0035312.1 | CGAAGAAGGGCATTACAGCA | 121 | 96.08 | 0.970 |
| GAGCATCGGAAGTCAACAGA | ||||||
| H2A | histone H2A | EVM0033846.1 | GCAAGTATGCTGAGCGTGTT | 123 | 97.91 | 0.985 |
| TACGGGTCTTCTTGTTGTCTC | ||||||
| Target gene | ||||||
| CFS | cop/fus-specific | EVM0013916.1 | AAGTTGGGAAACAGGTGGTC | 194 | 97.57 | 0.993 |
| GTCTCCATTTGGTCGGTTGT | ||||||
| Rank | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
|---|---|---|---|---|---|---|---|---|---|---|
| ♀ | Gene | DnaJ | α-TUB2 | TIP41 | GAPDH | H2A | α-TUB1 | β-TUB | ACT | CDC2 |
| Stability | 0.022 | 0.023 | 0.257 | 0.315 | 0.377 | 0.573 | 0.612 | 0.624 | 0.823 | |
| ♂ | Gene | ACT | α-TUB2 | α-TUB1 | TIP41 | H2A | DnaJ | GAPDH | β-TUB | CDC2 |
| Stability | 0.038 | 0.074 | 0.213 | 0.215 | 0.303 | 0.536 | 0.563 | 0.631 | 0.951 | |
| Total | Gene | ACT | DnaJ | TIP41 | H2A | α-TUB1 | β-TUB | α-TUB2 | GAPDH | CDC2 |
| Stability | 0.068 | 0.093 | 0.174 | 0.259 | 0.283 | 0.575 | 0.585 | 0.616 | 0.861 | |
| Rank | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
|---|---|---|---|---|---|---|---|---|---|---|
| ♀ | Gene | ACT | α-TUB1 | TIP41 | GAPDH | H2A | DnaJ | β-TUB | α-TUB2 | CDC2 |
| STEDV | 0.68 | 0.68 | 0.75 | 0.76 | 0.83 | 0.96 | 1.01 | 1.03 | 1.23 | |
| ♂ | Gene | ACT | DnaJ | α-TUB1 | TIP41 | H2A | α-TUB2 | GAPDH | β-TUB | CDC2 |
| STEDV | 0.63 | 0.66 | 0.68 | 0.77 | 0.79 | 0.90 | 0.94 | 1.06 | 1.40 | |
| Total | Gene | ACT | DnaJ | TIP41 | α-TUB2 | H2A | α-TUB1 | GAPDH | β-TUB | CDC2 |
| STEDV | 0.64 | 0.65 | 0.68 | 0.72 | 0.73 | 0.94 | 0.98 | 0.98 | 1.27 | |
| Rank | ♀ | ♂ | Total | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Gene | SD | CV | Gene | SD | CV | Gene | SD | CV | |
| 1 | ACT | 0.42 | 2.05 | ACT | 0.55 | 2.68 | ACT | 0.50 | 2.42 |
| 2 | DnaJ | 0.56 | 2.84 | DnaJ | 0.56 | 2.86 | DnaJ | 0.56 | 2.85 |
| 3 | α-TUB1 | 0.77 | 3.88 | α-TUB1 | 0.89 | 4.40 | α-TUB1 | 0.84 | 4.21 |
| 4 | α-TUB2 | 1.01 | 4.84 | α-TUB2 | 1.05 | 4.67 | GAPDH | 1.11 | 4.94 |
| 5 | GAPDH | 1.05 | 4.64 | GAPDH | 1.17 | 5.21 | α-TUB2 | 1.22 | 5.63 |
| 6 | TIP41 | 1.32 | 5.44 | H2A | 1.19 | 5.94 | TIP41 | 1.30 | 5.28 |
| 7 | H2A | 1.47 | 7.07 | TIP41 | 1.24 | 4.95 | H2A | 1.34 | 6.57 |
| 8 | β-TUB | 1.85 | 10.28 | β-TUB | 1.88 | 10.16 | β-TUB | 1.88 | 10.28 |
| 9 | CDC2 | 2.08 | 8.66 | CDC2 | 2.16 | 8.64 | CDC2 | 2.15 | 8.8 |
| Method | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|---|---|
| Ranking order under female flower bud (Better-Good-Average) | |||||||||
| geNorm | ACT/α-TUB1 | GAPDH | DnaJ | α-TUB2 | TIP41 | H2A | β-TUB | CDC2 | |
| NormFinder | DnaJ | α-TUB2 | TIP41 | GAPDH | H2A | α-TUB1 | β-TUB | ACT | CDC2 |
| Delta CT | ACT | α-TUB1 | DnaJ | GAPDH | α-TUB2 | TIP41 | H2A | β-TUB | CDC2 |
| BestKeeper | ACT | DnaJ | α-TUB1 | α-TUB2 | GAPDH | TIP41 | H2A | β-TUB | CDC2 |
| Comprehensive | ACT | DnaJ | α-TUB1 | GAPDH | α-TUB2 | TIP41 | H2A | β-TUB | CDC2 |
| Ranking order under male flower bud (Better-Good-Average) | |||||||||
| geNorm | ACT/DnaJ | α-TUB1 | α-TUB2 | GAPDH | TIP41 | H2A | β-TUB | CDC2 | |
| NormFinder | GAPDH | α-TUB2 | α-TUB1 | TIP41 | H2A | DnaJ | GAPDH | β-TUB | CDC2 |
| Delta CT | ACT | DnaJ | α-TUB1 | α-TUB2 | GAPDH | TIP41 | H2A | β-TUB | CDC2 |
| BestKeeper | ACT | DnaJ | α-TUB1 | α-TUB2 | GAPDH | H2A | TIP41 | β-TUB | CDC2 |
| Comprehensive | ACT | DnaJ | α-TUB1 | α-TUB2 | GAPDH | TIP41 | H2A | β-TUB | CDC2 |
| Ranking order under total samples (Better-Good-Average) | |||||||||
| geNorm | DnaJ/TIP41 | H2A | ACT | α-TUB1 | α-TUB2 | GAPDH | β-TUB | CDC2 | |
| NormFinder | ACT | DnaJ | TIP41 | H2A | α-TUB1 | β-TUB | α-TUB2 | GAPDH | CDC2 |
| Delta CT | ACT | DnaJ | TIP41 | α-TUB2 | H2A | α-TUB1 | GAPDH | β-TUB | CDC2 |
| BestKeeper | ACT | DnaJ | α-TUB1 | GAPDH | α-TUB2 | TIP41 | H2A | β-TUB | CDC2 |
| Comprehensive | ACT | DnaJ | TIP41 | H2A | α-TUB1 | α-TUB2 | GAPDH | β-TUB | CDC2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, F.; Chen, Y.; Wu, H.; Yin, T. A Selection of Reliable Reference Genes for Gene Expression Analysis in the Female and Male Flowers of Salix suchowensis. Plants 2022, 11, 647. https://doi.org/10.3390/plants11050647
Zhou F, Chen Y, Wu H, Yin T. A Selection of Reliable Reference Genes for Gene Expression Analysis in the Female and Male Flowers of Salix suchowensis. Plants. 2022; 11(5):647. https://doi.org/10.3390/plants11050647
Chicago/Turabian StyleZhou, Fangwei, Yingnan Chen, Huaitong Wu, and Tongming Yin. 2022. "A Selection of Reliable Reference Genes for Gene Expression Analysis in the Female and Male Flowers of Salix suchowensis" Plants 11, no. 5: 647. https://doi.org/10.3390/plants11050647