
Diversity and Functional Evolution of Terpene Synthases in Rosaceae

 ,
,
Abstract
:1. Introduction
2. Results
2.1. Identification of TPS Family Members in Rosaceae
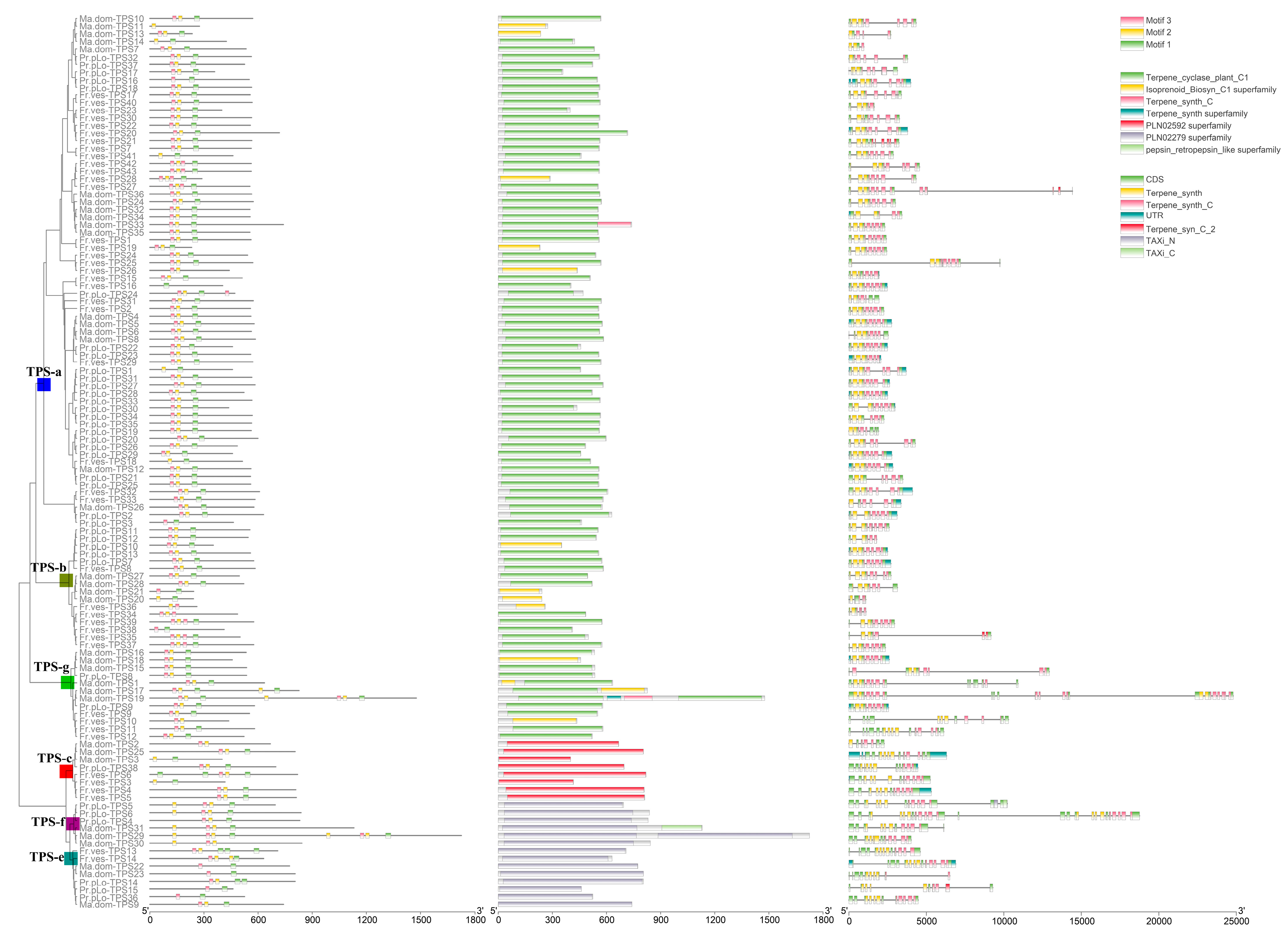
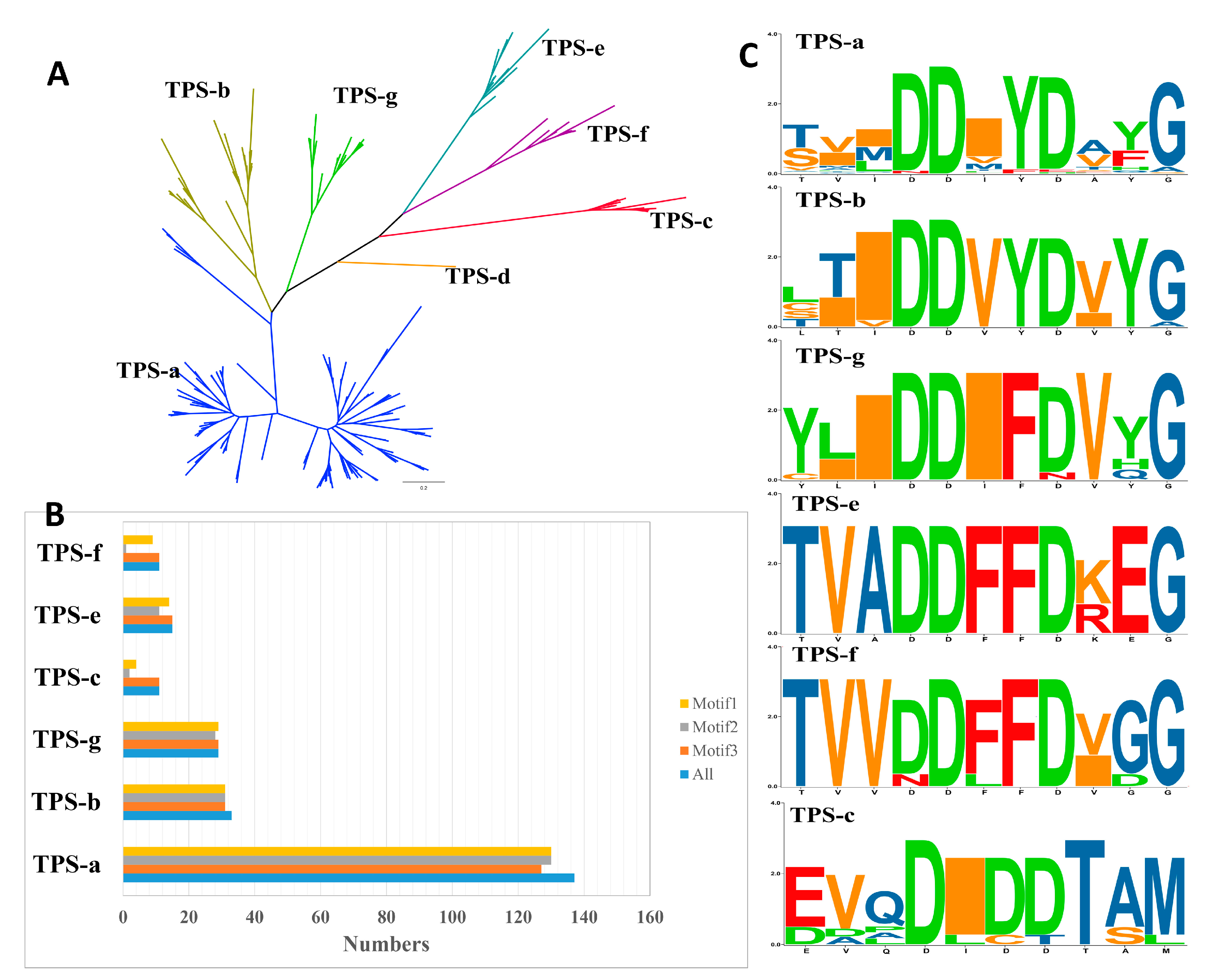
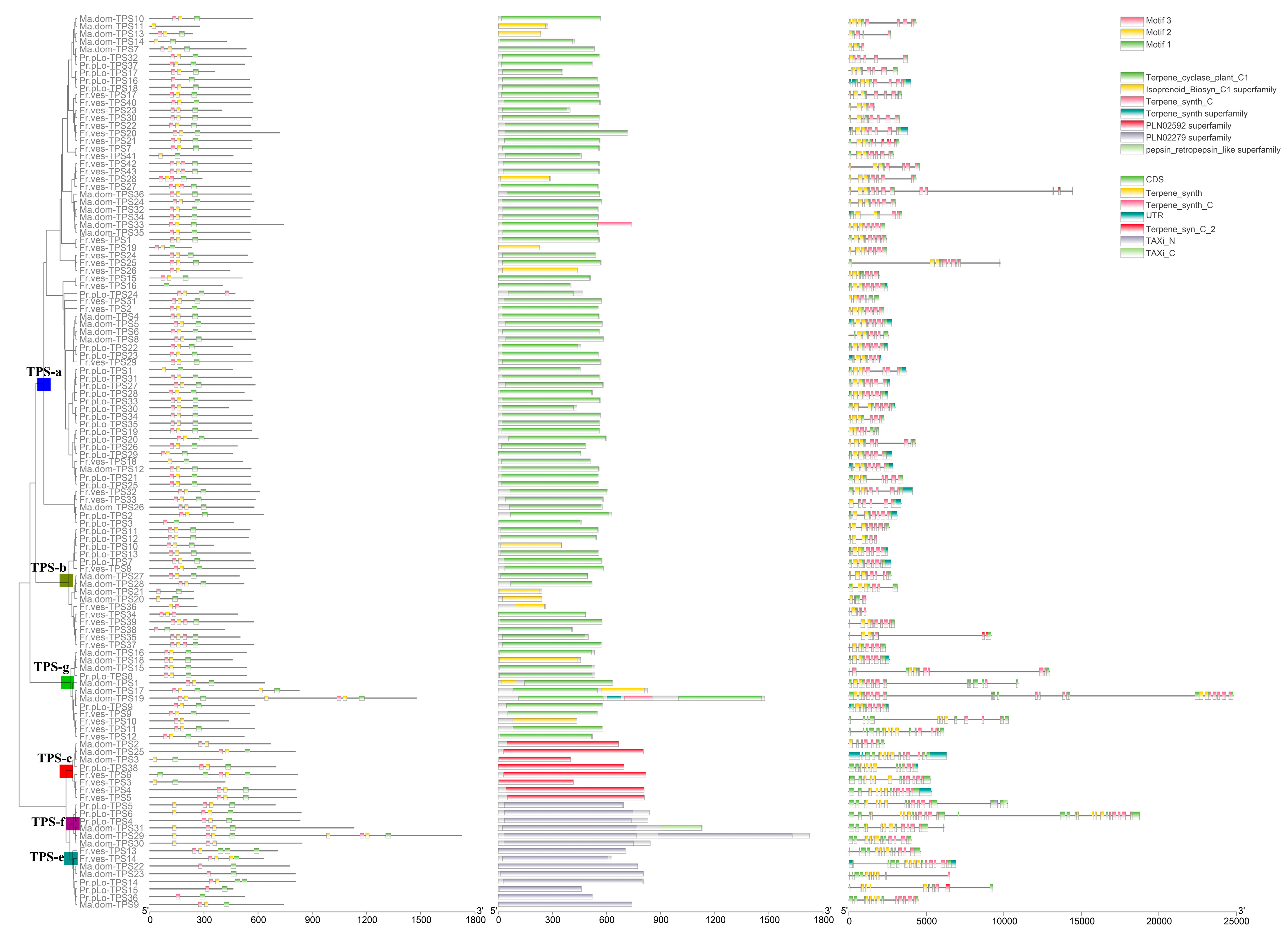
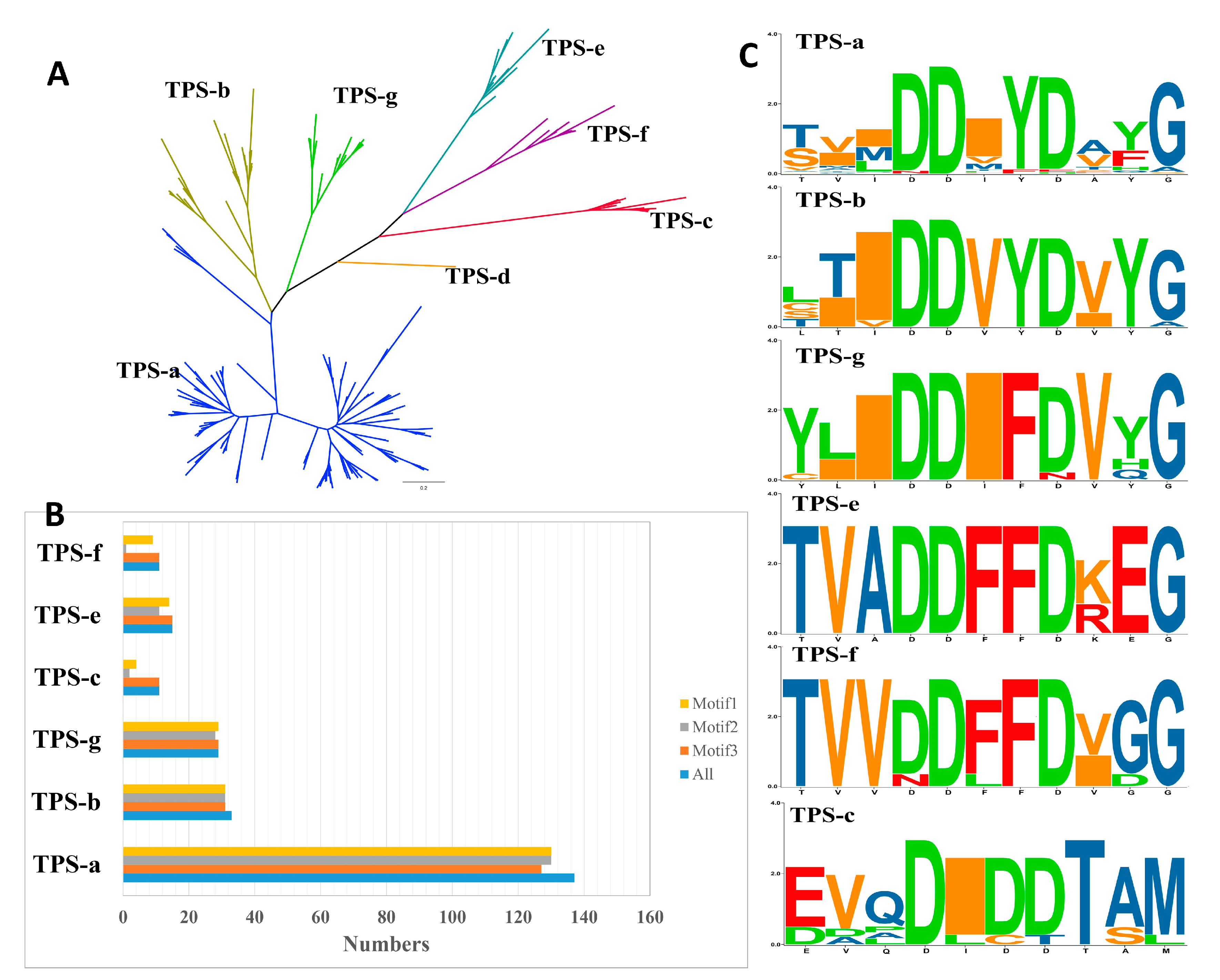
2.2. TPS Classification and Motif/Domain Annotation
2.3. Varied Family Number of TPSs among Different Clades in Rosaceae
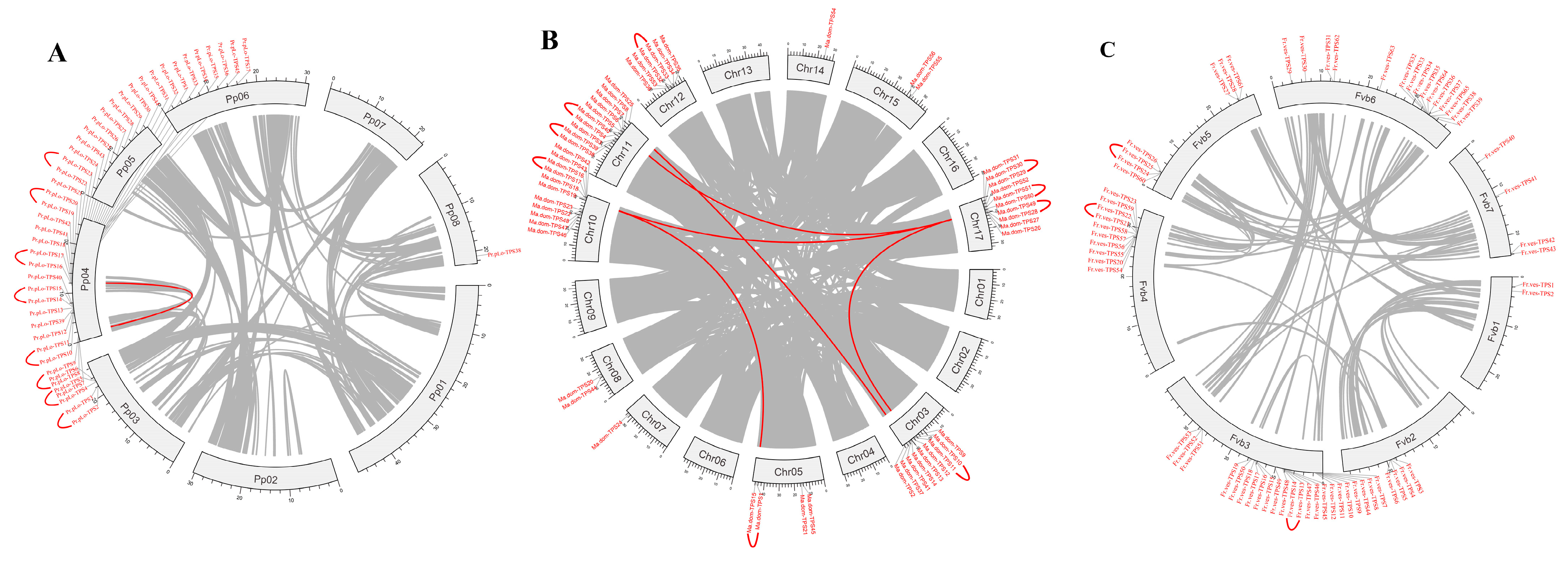
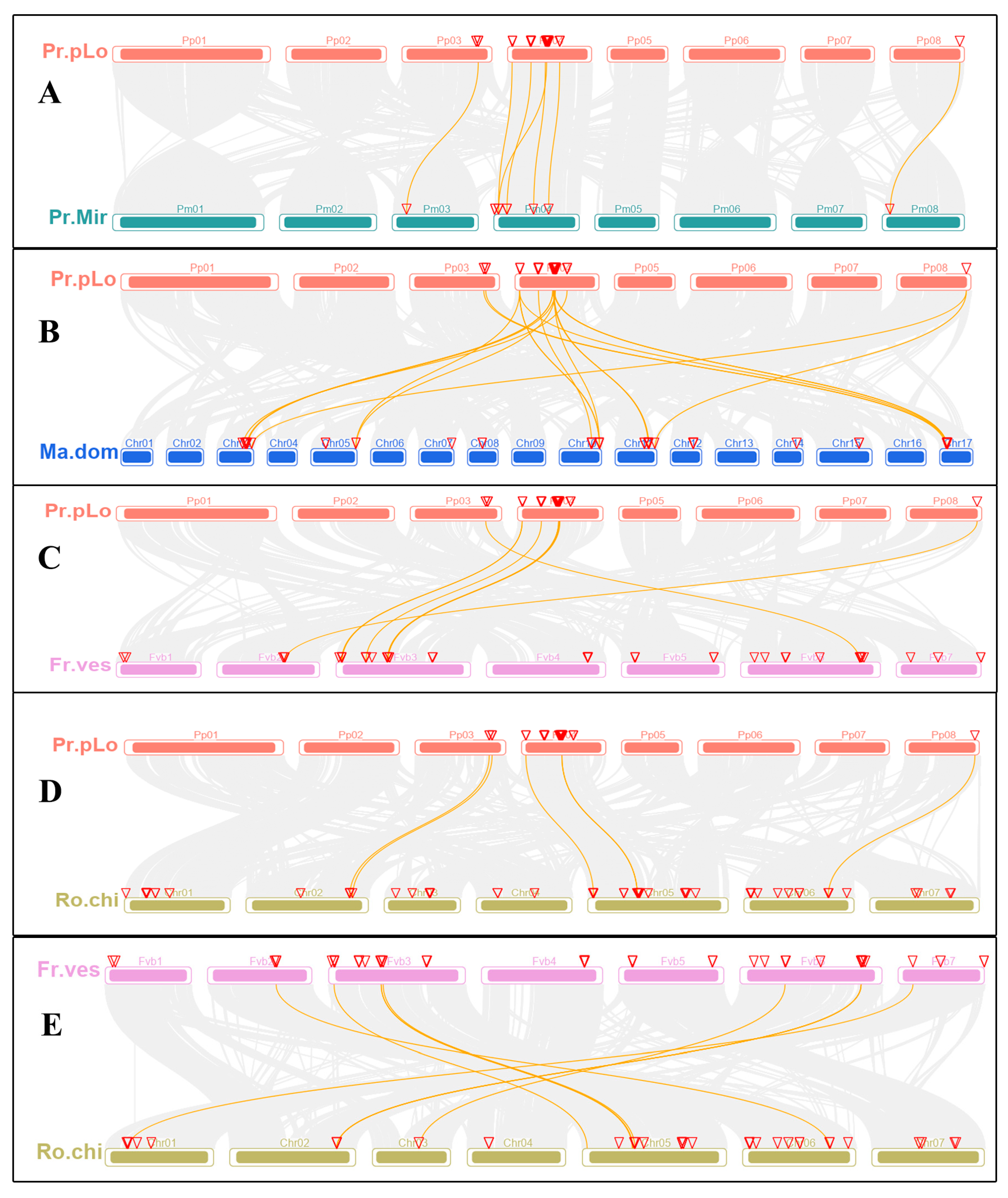
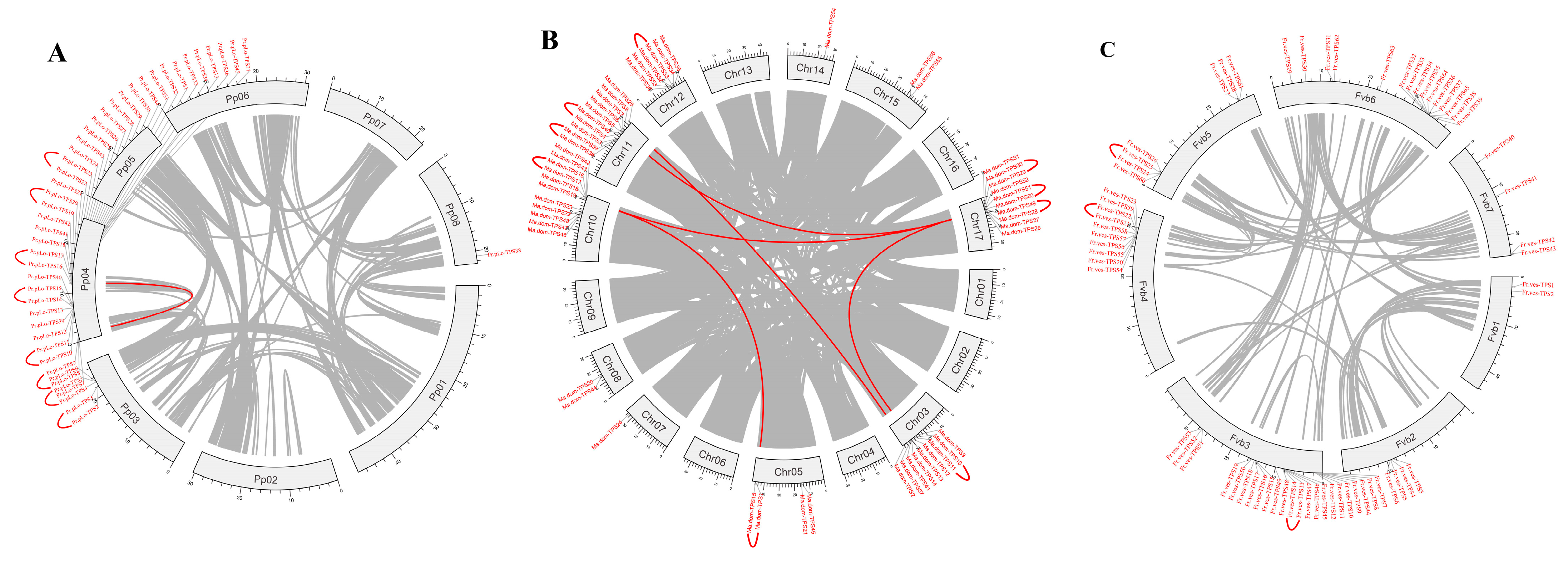
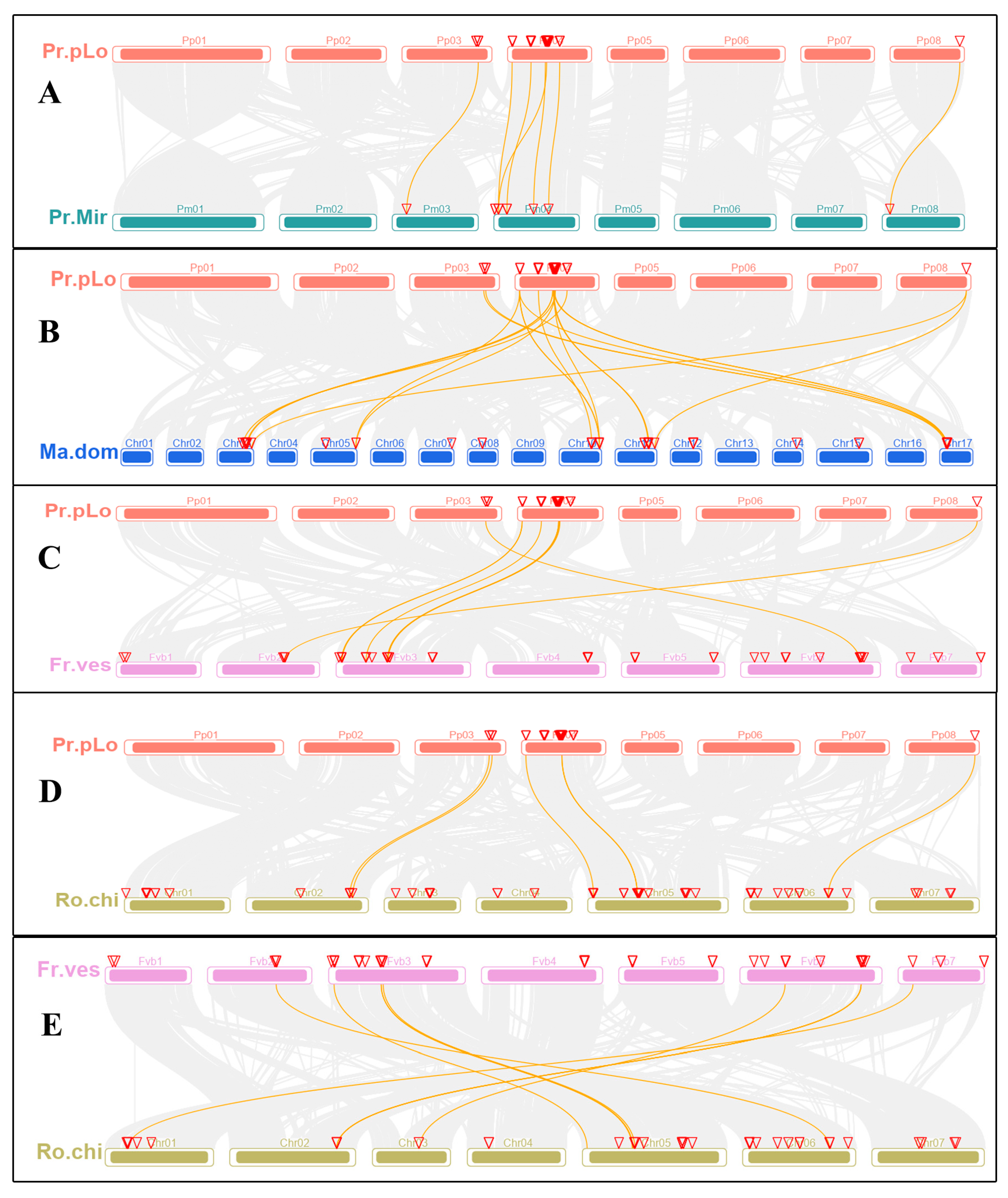
2.4. Chromosomal Location, Synteny Analysis of TPS Family Members in Rosaceae
2.5. Ka/Ks Ratios of TPS Family Members in Rosaceae
2.6. Function Diversity of TPSs in Rosaceae
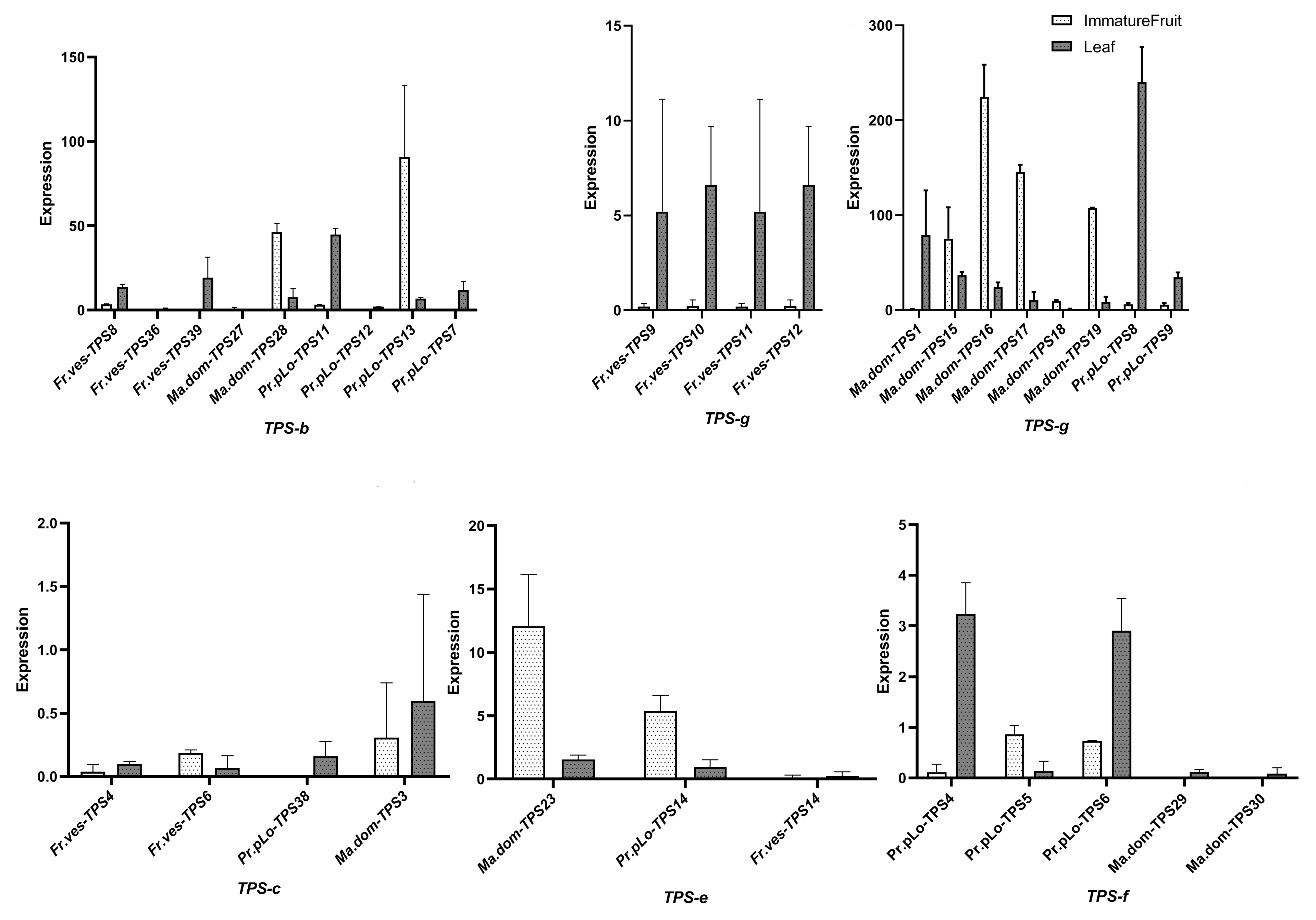
2.7. Tissue-Specific Expression of TPSs in Rosaceae
3. Discussion
4. Material and Methods
4.1. Identification of TPSs in Rosaceae
4.2. Motif Annotation, Subcellular Localization, and Physical Localization
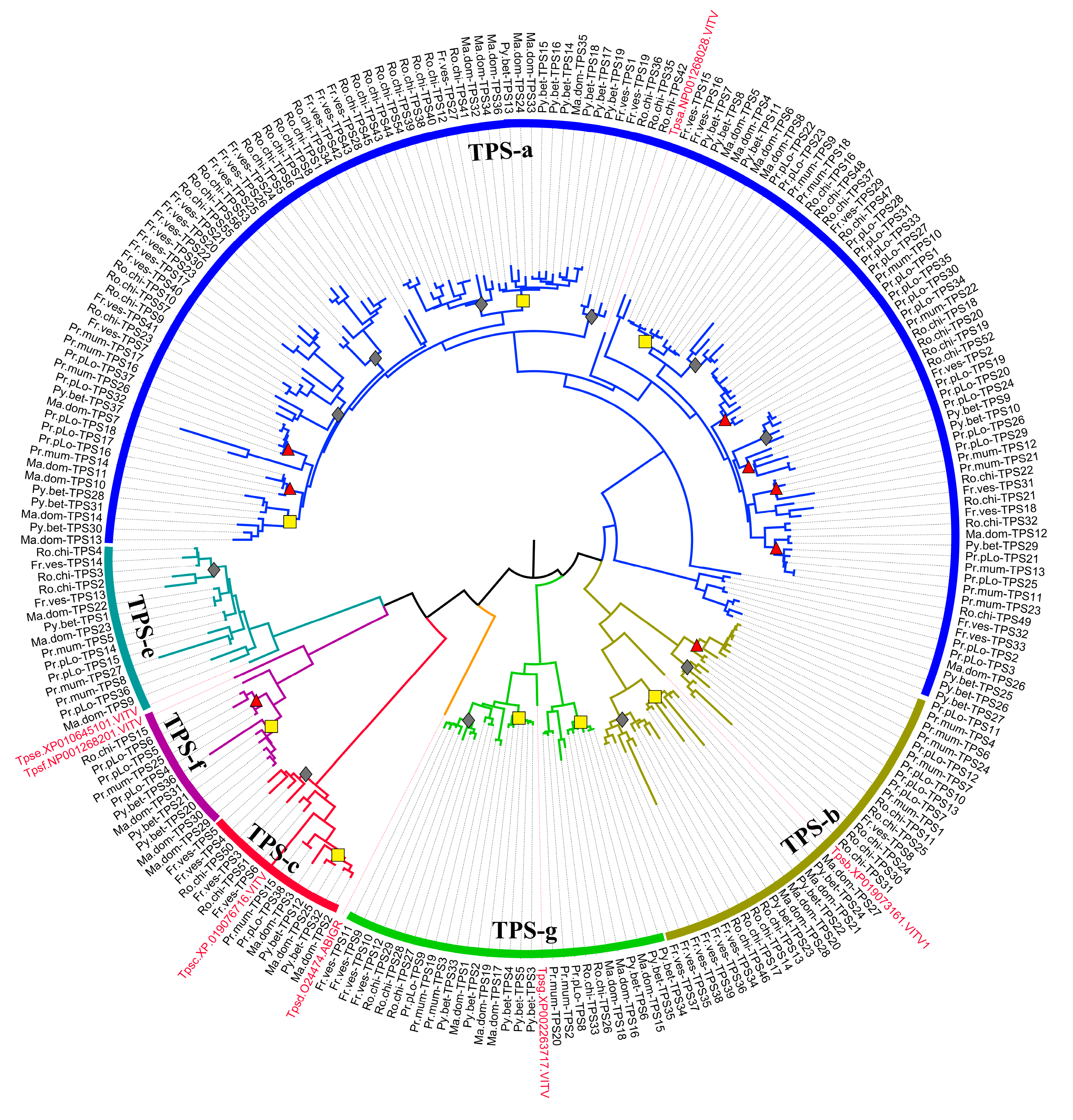
4.3. Phylogenetic Tree Construction and TPS Classification
4.4. Ka/Ks Analysis of TPS Family Members in Rosaceae
4.5. Synteny Analysis and Detection of Tandemly/Segmentally Duplicated TPSs
4.6. Expression Analysis of TPSs in Rosaceae
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HMM | Hidden Markove Model |
| TPSs | terpene synthase genes |
| GPP | geranyl diphosphate |
| FPP | farnesyl diphosphate |
| GGPP | geranylgeranyl diphosphate |
| VOCs | volatile organic compounds |
References
- Wang, Y.J.; Chen, F.; Fang, J.B.; Yang, C.X.; Zhao, J.B.; Jiang, Q.; Li, S.H. Effects of Germplasm Origin and Fruit Character on Volatile Composition of Peaches and Nectarines. Flavor Health Benefits Small Fruits 2010, 1035, 95–117. [Google Scholar]
- Wei, C.Y.; Liu, H.R.; Cao, X.M.; Zhang, M.L.; Li, X.; Chen, K.S.; Zhang, B. Synthesis of flavour-related linalool is regulated by PpbHLH1 and associated with changes in DNA methylation during peach fruit ripening. Plant Biotechnol. J. 2021, 19, 2082–2096. [Google Scholar] [CrossRef] [PubMed]
- Nawade, B.; Yahyaa, M.; Reuveny, H.; Shaltiel-Harpaz, L.; Eisenbach, O.; Faigenboim, A.; Bar-Yaakov, I.; Holland, D.; Ibdah, M. Profiling of volatile terpenes from almond (Prunus dulcis) young fruits and characterization of seven terpene synthase genes. Plant Sci. 2019, 287, 110187. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuizen, N.J.; Green, S.A.; Chen, X.; Bailleul, E.J.; Matich, A.J.; Wang, M.Y.; Atkinson, R.G. Functional genomics reveals that a compact terpene synthase gene family can account for terpene volatile production in apple. Plant Physiol. 2013, 161, 787–804. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.J.; Hao, N.N.; Feng, R.F.; Meng, Z.P.; Li, Y.N.; Zhao, Z.Y. Transcriptome and metabolite profiling analyses provide insight into volatile compounds of the apple cultivar ‘Ruixue’ and its parents during fruit development. Bmc Plant Biol. 2021, 21, 1–16. [Google Scholar] [CrossRef]
- Klee, H.J.; Tieman, D.M. The genetics of fruit flavour preferences. Nat. Rev. Genet. 2018, 19, 347–356. [Google Scholar] [CrossRef]
- Espino-Diaz, M.; Sepulveda, D.R.; Gonzalez-Aguilar, G.; Olivas, G.I. Biochemistry of Apple Aroma: A Review. Food Technol. Biotechnol. 2016, 54, 375–397. [Google Scholar] [CrossRef]
- Zhang, X.; Niu, M.; Teixeira da Silva, J.A.; Zhang, Y.; Yuan, Y.; Jia, Y.; Xiao, Y.; Li, Y.; Fang, L.; Zeng, S.; et al. Identification and functional characterization of three new terpene synthase genes involved in chemical defense and abiotic stresses in Santalum album. BMC Plant Biol. 2019, 19, 115. [Google Scholar] [CrossRef] [Green Version]
- Block, A.K.; Vaughan, M.M.; Schmelz, E.A.; Christensen, S.A. Biosynthesis and function of terpenoid defense compounds in maize (Zea mays). Planta 2019, 249, 21–30. [Google Scholar] [CrossRef]
- Huang, X.Z.; Xiao, Y.T.; Kollner, T.G.; Jing, W.X.; Kou, J.F.; Chen, J.Y.; Liu, D.F.; Gu, S.H.; Wu, J.X.; Zhang, Y.J.; et al. The terpene synthase gene family in Gossypium hirsutum harbors a linalool synthase GhTPS12 implicated in direct defence responses against herbivores. Plant Cell Environ. 2018, 41, 261–274. [Google Scholar] [CrossRef]
- Zhou, F.; Pichersky, E. More is better: The diversity of terpene metabolism in plants. Curr. Opin. Plant Biol. 2020, 55, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kulheim, C.; Padovan, A.; Hefer, C.; Krause, S.T.; Kollner, T.G.; Myburg, A.A.; Degenhardt, J.; Foley, W.J. The Eucalyptus terpene synthase gene family. BMC Genom. 2015, 16, 450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Tholl, D.; Bohlmann, J.; Pichersky, E. The family of terpene synthases in plants: A mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J. 2011, 66, 212–229. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Honzatko, R.B.; Peters, R.J. Terpenoid synthase structures: A so far incomplete view of complex catalysis. Nat. Prod. Rep. 2012, 29, 1153–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillwig, M.L.; Xu, M.; Toyomasu, T.; Tiernan, M.S.; Wei, G.; Cui, G.; Huang, L.; Peters, R.J. Domain loss has independently occurred multiple times in plant terpene synthase evolution. Plant J. 2011, 68, 1051–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.Y.; Jin, J.; Sarojam, R.; Ramachandran, S. A Comprehensive Survey on the Terpene Synthase Gene Family Provides New Insight into Its Evolutionary Patterns. Genome Biol. Evol. 2019, 11, 2078–2098. [Google Scholar] [CrossRef] [Green Version]
- Karunanithi, P.S.; Zerbe, P. Terpene Synthases as Metabolic Gatekeepers in the Evolution of Plant Terpenoid Chemical Diversity. Front. Plant Sci. 2019, 10, 1166. [Google Scholar] [CrossRef] [Green Version]
- Kumar, Y.; Khan, F.; Rastogi, S.; Shasany, A.K. Genome-wide detection of terpene synthase genes in holy basil (Ocimum sanctum L.). PLoS ONE 2018, 13, e0207097. [Google Scholar] [CrossRef]
- Muchlinski, A.; Ibdah, M.; Ellison, S.; Yahyaa, M.; Nawade, B.; Laliberte, S.; Senalik, D.; Simon, P.; Whitehead, S.R.; Tholl, D. Diversity and function of terpene synthases in the production of carrot aroma and flavor compounds. Sci. Rep. 2020, 10, 9989. [Google Scholar] [CrossRef]
- Zhou, H.C.; Shamala, L.F.; Yi, X.K.; Yan, Z.; Wei, S. Analysis of Terpene Synthase Family Genes in Camellia sinensis with an Emphasis on Abiotic Stress Conditions. Sci. Rep. 2020, 10, 933. [Google Scholar] [CrossRef]
- Wang, Y.J.; Yang, C.X.; Li, S.H.; Yang, L.; Wang, Y.N.; Zhao, J.B.; Jiang, Q. Volatile characteristics of 50 peaches and nectarines evaluated by HP-SPME with GC-MS. Food Chem. 2009, 116, 356–364. [Google Scholar] [CrossRef]
- Sonnhammer, E.L.; Koonin, E.V. Orthology, paralogy and proposed classification for paralog subtypes. Trends Genet. 2002, 18, 619–620. [Google Scholar] [CrossRef]
- Boos, F.; Muhlhaus, T.; Herrmann, J.M. Detection of Internal Matrix Targeting Signal-like Sequences (iMTS-Ls) in Mitochondrial Precursor Proteins Using the TargetP Prediction Tool. Bio-Protocol 2018, 8, e2474. [Google Scholar] [CrossRef]
- Lee, G.W.; Lee, S.; Chung, M.S.; Jeong, Y.S.; Chung, B.Y. Rice terpene synthase 20 (OsTPS20) plays an important role in producing terpene volatiles in response to abiotic stresses. Protoplasma 2015, 252, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Yoshitomi, K.; Taniguchi, S.; Tanaka, K.; Uji, Y.; Akimitsu, K.; Gomi, K. Rice terpene synthase 24 (OsTPS24) encodes a jasmonate-responsive monoterpene synthase that produces an antibacterial gamma-terpinene against rice pathogen. J. Plant Physiol. 2016, 191, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Li, Y.; Liu, X.; Zhao, Y.; Li, B.; Ingvarsson, P.K.; Zhang, D. Evolutionary Origins of Pseudogenes and Their Association with Regulatory Sequences in Plants. Plant Cell 2019, 31, 563–578. [Google Scholar] [CrossRef] [Green Version]
- Mascagni, F.; Usai, G.; Cavallini, A.; Porceddu, A. Structural characterization and duplication modes of pseudogenes in plants. Sci. Rep. 2021, 11, 5292. [Google Scholar] [CrossRef]
- Zhang, A.; Zhou, H.; Jiang, X.; Han, Y.; Zhang, X. The Draft Genome of a Flat Peach (Prunus persica L. cv. ‘124 Pan’) Provides Insights into Its Good Fruit Flavor Traits. Plants 2021, 10, 538. [Google Scholar] [CrossRef]
- Potter, S.C.; Luciani, A.; Eddy, S.R.; Park, Y.; Lopez, R.; Finn, R.D. HMMER web server: 2018 update. Nucleic Acids Res. 2018, 46, W200–W204. [Google Scholar] [CrossRef] [Green Version]
- Li, W.Z.; Cowley, A.; Uludag, M.; Gur, T.; McWilliam, H.; Squizzato, S.; Park, Y.M.; Buso, N.; Lopez, R. The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res. 2015, 43, W580–W584. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef] [Green Version]
- Emanuelsson, O.; Brunak, S.; von Heijne, G.; Nielsen, H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2007, 2, 953–971. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanc, G.; Wolfe, K.H. Functional divergence of duplicated genes formed by polyploidy during Arabidopsis evolution. Plant Cell 2004, 16, 1679–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Li, J.; Paterson, A.H. MCScanX-transposed: Detecting transposed gene duplications based on multiple colinearity scans. Bioinformatics 2013, 29, 1458–1460. [Google Scholar] [CrossRef] [Green Version]
- Lu, P.; Yu, S.; Zhu, N.; Chen, Y.R.; Zhou, B.; Pan, Y.; Tzeng, D.; Fabi, J.P.; Argyris, J.; Garcia-Mas, J.; et al. Genome encode analyses reveal the basis of convergent evolution of fleshy fruit ripening. Nat. Plants 2018, 4, 784–791. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subfamily | Species | Release Version * | The Prefix of Gene Symbol | Total Genes | Numbers of TPSs | Numbers of TPSs with Both Domains | Percent of TPSs with Both Domains | Numbers of Already Know TPSs |
|---|---|---|---|---|---|---|---|---|
| Prunoideae | Prunus persica | GDR, v2.0 | Pr.pLo | 26,873 | 45 | 38 | 84.44% | 38 (both domains) [2] |
| Prunus mira | GDR, v1.0 | Pr.mir | 26,958 | 10 | 9 | 90.00% | ||
| Prunus mume | NCBI, v1.0 | Pr.mum | 28,638 | 30 | 27 | 90.00% | ||
| Maloideae | Pyrus betulifolia | GDR, v1.0 | Py.bet | 59,552 | 48 | 37 | 77.08% | |
| Malus x domestica | GDR, HFTH1 v1.0 | Ma.dom | 44,677 | 56 | 36 | 64.29% | 55 (all) [4] | |
| Malus baccata | CNGB, v1.0 | Ma.bac | 45,931 | 48 | 33 | 68.75% | ||
| Rosoideae | Fragaria vesca | GDR, v4.0 | Fr.ves | 35,914 | 65 | 43 | 66.15% | |
| Rosa chinensis | GDR, v1.0 | Ro.chi | 39,669 | 76 | 57 | 75.00% |
| TPSs Clade | TPS-a | TPS-b | TPS-g | TPS-c | TPS-d | TPS-e | TPS-f | Total |
|---|---|---|---|---|---|---|---|---|
| Motif | DDXYD | DDVYD | DDIFD | DIDDT | DXDD, DXXD | DDFFD | DDFFD | |
| Prunus persica | 24 | 5 | 2 | 1 | 0 | 3 | 3 | 38 |
| Prunus mira | 0 | 0 | 4 | 1 | 0 | 3 | 1 | 9 |
| Prunus mume | 13 | 5 | 4 | 1 | 0 | 3 | 1 | 27 |
| Pyrus betulifolia | 20 | 3 | 8 | 2 | 0 | 1 | 3 | 37 |
| Malus x domestica | 17 | 4 | 6 | 3 | 0 | 3 | 3 | 36 |
| Rosa chinensis | 37 | 9 | 5 | 2 | 0 | 3 | 1 | 57 |
| Fragaria vesca | 26 | 7 | 4 | 4 | 0 | 2 | 0 | 43 |
| TPSs Clade | TPS-a | TPS-b | TPS-g | TPS-c | TPS-e | TPS-f |
|---|---|---|---|---|---|---|
| Catalytic type | a. monoTPS 1, (−)-alpha-pinene synthase. C/P 2, S-linalool synthase b. SesquiTPS 1, (−)-germacrene D synthase. C/P 2, (3S, 6E)-nerolidol synthase. P 3, (E, E)-alpha-farnesene synthase. P c. DiTPS 1, Ent-kaur-16-ene synthase 2, Ent-copalyl diphosphate synthase. P 3, Copal-8-ol diphosphate hydratase. P | a. monoTPS 1, (−)-alpha-pinene synthase. C/P 3, Tricyclene synthase EBOS. C/P b. SesquiTPS 1, (−)-germacrene D synthase. C/P 2, (3S, 6E)-nerolidol synthase. P 3, (E, E)-alpha-farnesene synthase. P | a. monoTPS 1, (−)-alpha-pinene synthase. C/P 2, S-linalool synthase 3, Tricyclene synthase EBOS. C/P b. SesquiTPS 2, (3S, 6E)-nerolidol synthase. P c. DiTPS 2, Ent-copalyl diphosphate synthase. P | a. monoTPS 1, (−)-alpha-pinene synthase. C/P 3, Tricyclene synthase EBOS. C/P b. SesquiTPS 1, (−)-germacrene D synthase. C/P 2, (3S, 6E)-nerolidol synthase. P c. DiTPS 2, Ent-copalyl diphosphate synthase. P 3, Copal-8-ol diphosphate hydratase. P | a. monoTPS 1, (−)-alpha-pinene synthase. C/P b. SesquiTPS 2, (3S, 6E)-nerolidol synthase. P 3, (E, E)-alpha-farnesene synthase. P c. DiTPS 1, Ent-kaur-16-ene synthase | a. monoTPS 2, S-linalool synthase 3, Tricyclene synthase EBOS. C/P b. SesquiTPS 1, (−)-germacrene D synthase. C/P c. DiTPS 1, Ent-kaur-16-ene synthase |
| Prunus persica | a1 (19) *; b1 (5); | b3 (5); | b2 (2); | c2 (1); | c1 (3); | a2 (3); |
| Prunus mume | a1 (6); b1 (4); | b3 (5); | b2 (4); | c2 (1); | c1 (3); | a2 (1); |
| Prunus mira | b1 (1); | / | b2 (4); | c2 (1); | c1 (3); | a2 (1); |
| Fragaria vesca | a1 (8); b1 (18); | a3 (6); b3 (1); | b2 (4); | c2 (3), c3 (1); | c1 (2); | / |
| Rosa chinensis | a1 (10); b1 (16), b2 (5), b3 (5); c1 (1); | a1 (2), a3 (2); b1 (3), b3 (2); | a1 (3), a2 (1); a3 (1); | b1 (2); | c1 (3); | b1 (1); |
| Pyrus betulifolia | a1 (8), a2 (3); b1 (8), b2 (2), b3 (2); c2 (2), c3 (1); | b1 (3); | a1 (2), a3 (1); b2 (5), | a1(1), a3 (1); | c1 (1); | a3 (1); |
| Malus x domestica | a1 (4); b1 (4), b2 (2); c1 (5), c2 (2); | a3 (2); b2 (1), b3 (1); | a1 (1); b1 (3), b2 (1); c2 (1); | b1 (1), b2 (1); c2 (1); | a1 (1); b2 (1), b3 (1); | a3 (1); c1 (1); |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, A.; Xiong, Y.; Fang, J.; Jiang, X.; Wang, T.; Liu, K.; Peng, H.; Zhang, X. Diversity and Functional Evolution of Terpene Synthases in Rosaceae. Plants 2022, 11, 736. https://doi.org/10.3390/plants11060736
Zhang A, Xiong Y, Fang J, Jiang X, Wang T, Liu K, Peng H, Zhang X. Diversity and Functional Evolution of Terpene Synthases in Rosaceae. Plants. 2022; 11(6):736. https://doi.org/10.3390/plants11060736
Chicago/Turabian StyleZhang, Aidi, Yuhong Xiong, Jing Fang, Xiaohan Jiang, Tengfei Wang, Kangchen Liu, Huixiang Peng, and Xiujun Zhang. 2022. "Diversity and Functional Evolution of Terpene Synthases in Rosaceae" Plants 11, no. 6: 736. https://doi.org/10.3390/plants11060736
APA StyleZhang, A., Xiong, Y., Fang, J., Jiang, X., Wang, T., Liu, K., Peng, H., & Zhang, X. (2022). Diversity and Functional Evolution of Terpene Synthases in Rosaceae. Plants, 11(6), 736. https://doi.org/10.3390/plants11060736