
Dysbiosis in the Rhizosphere Microbiome of Standing Dead Korean Fir (Abies koreana)

 , , and
, , and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Metagenomic 16S rRNA Sequencing Results
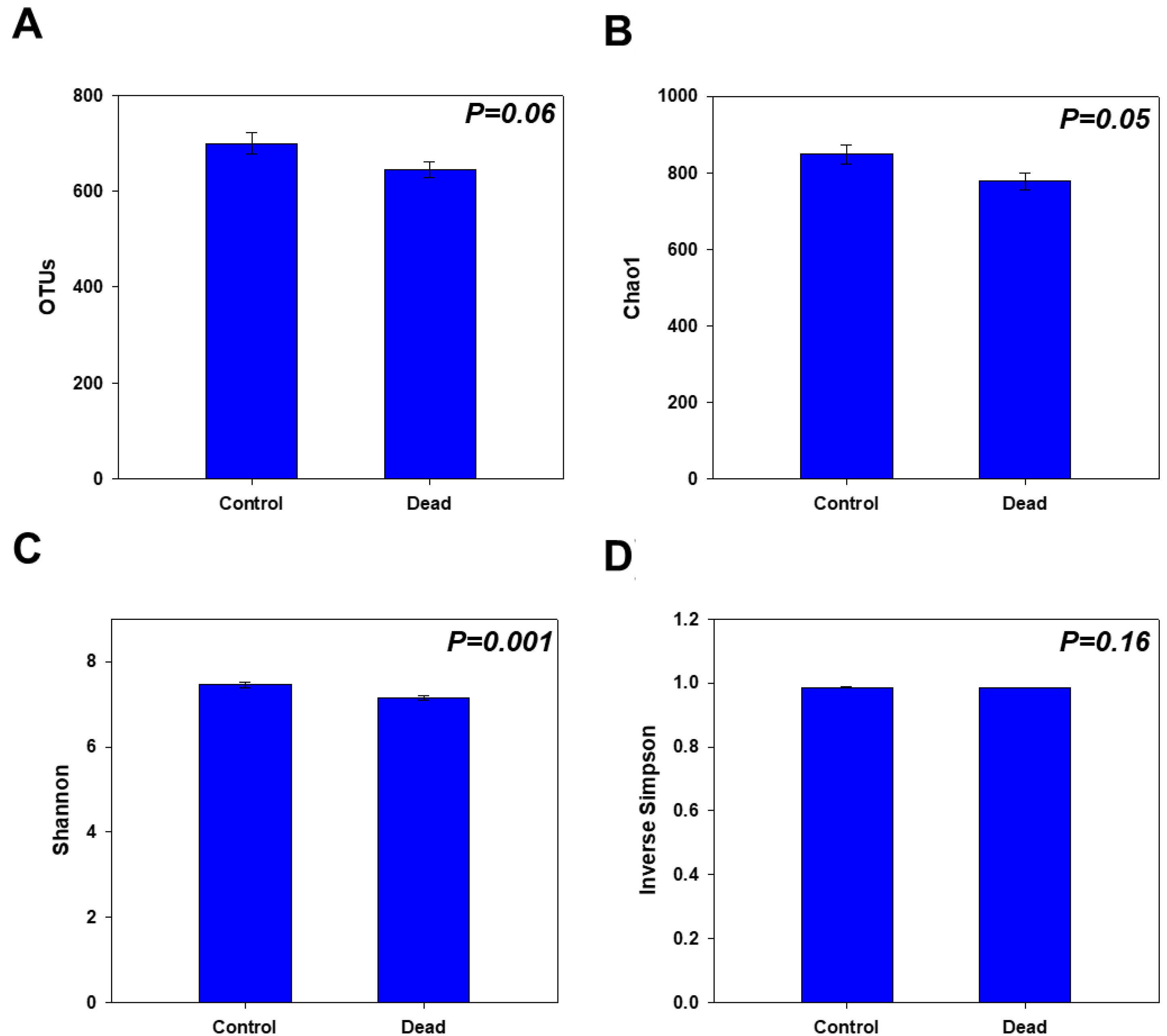
2.2. Rhizosphere Microbiome Diversity and Richness in Healthy and Dead Korean Fir Trees
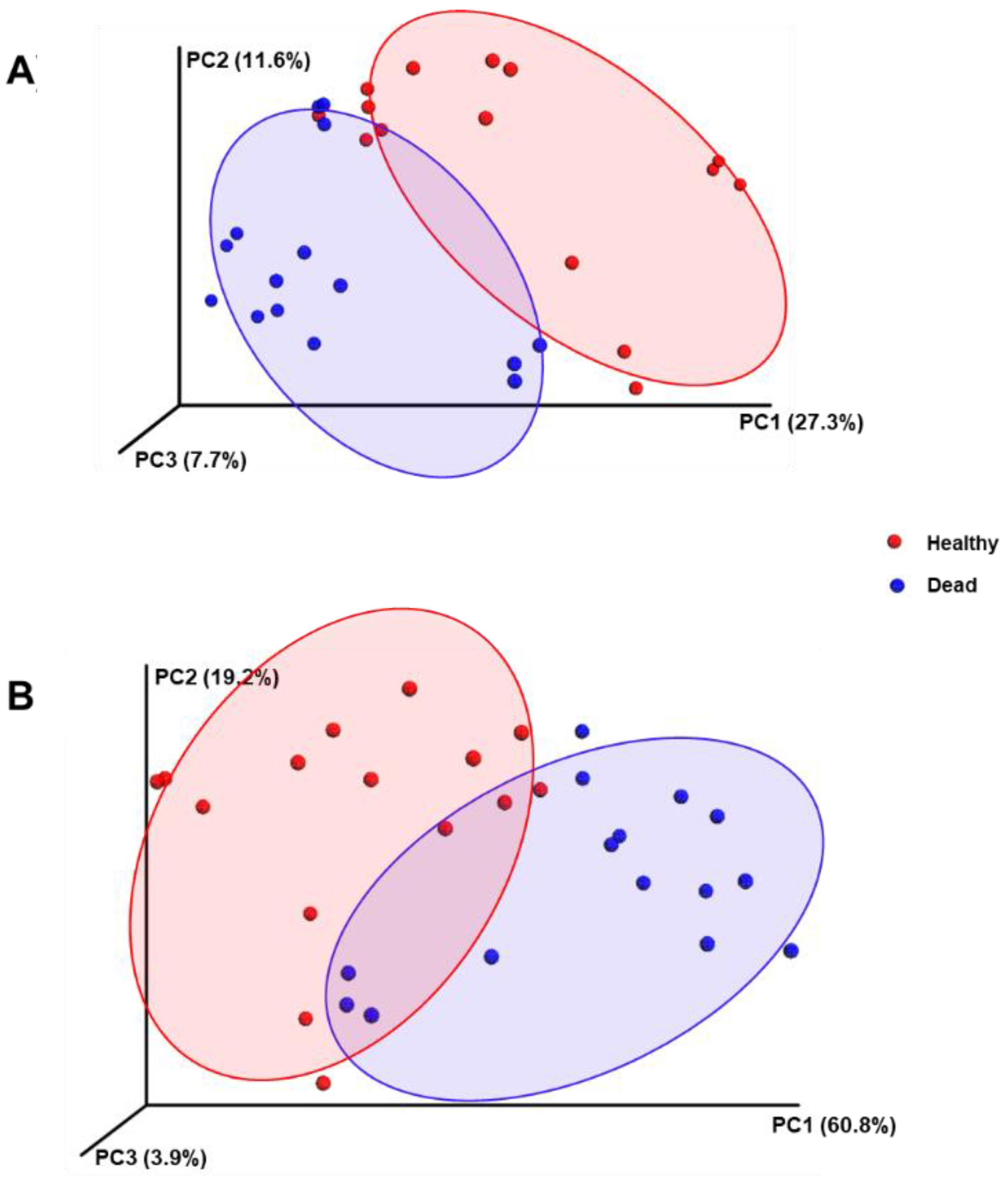
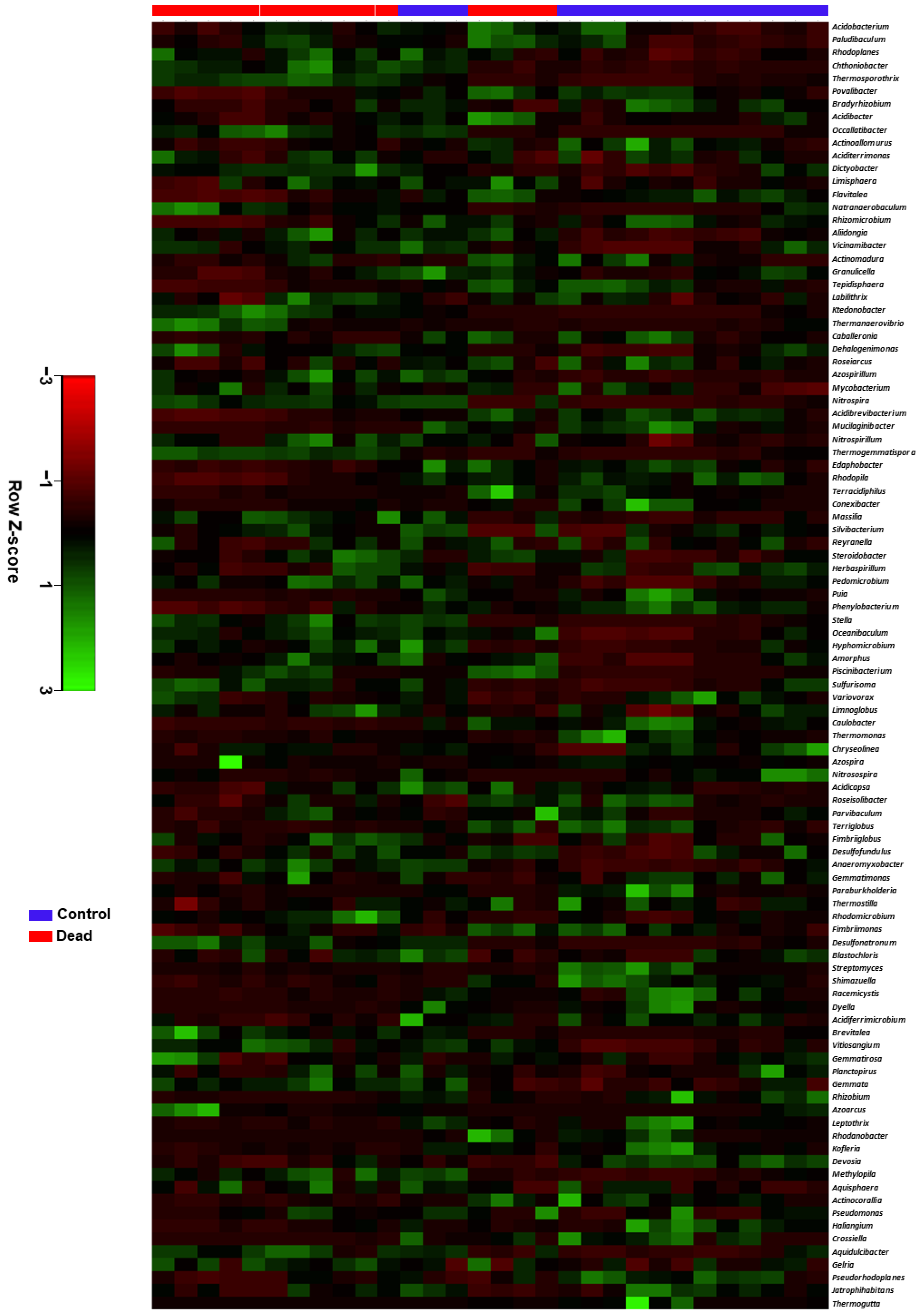
2.3. Taxonomic Characterisation and Comparative Analysis of the Rhizosphere Microbiome Structure between Healthy and Dead Korean Fir Trees
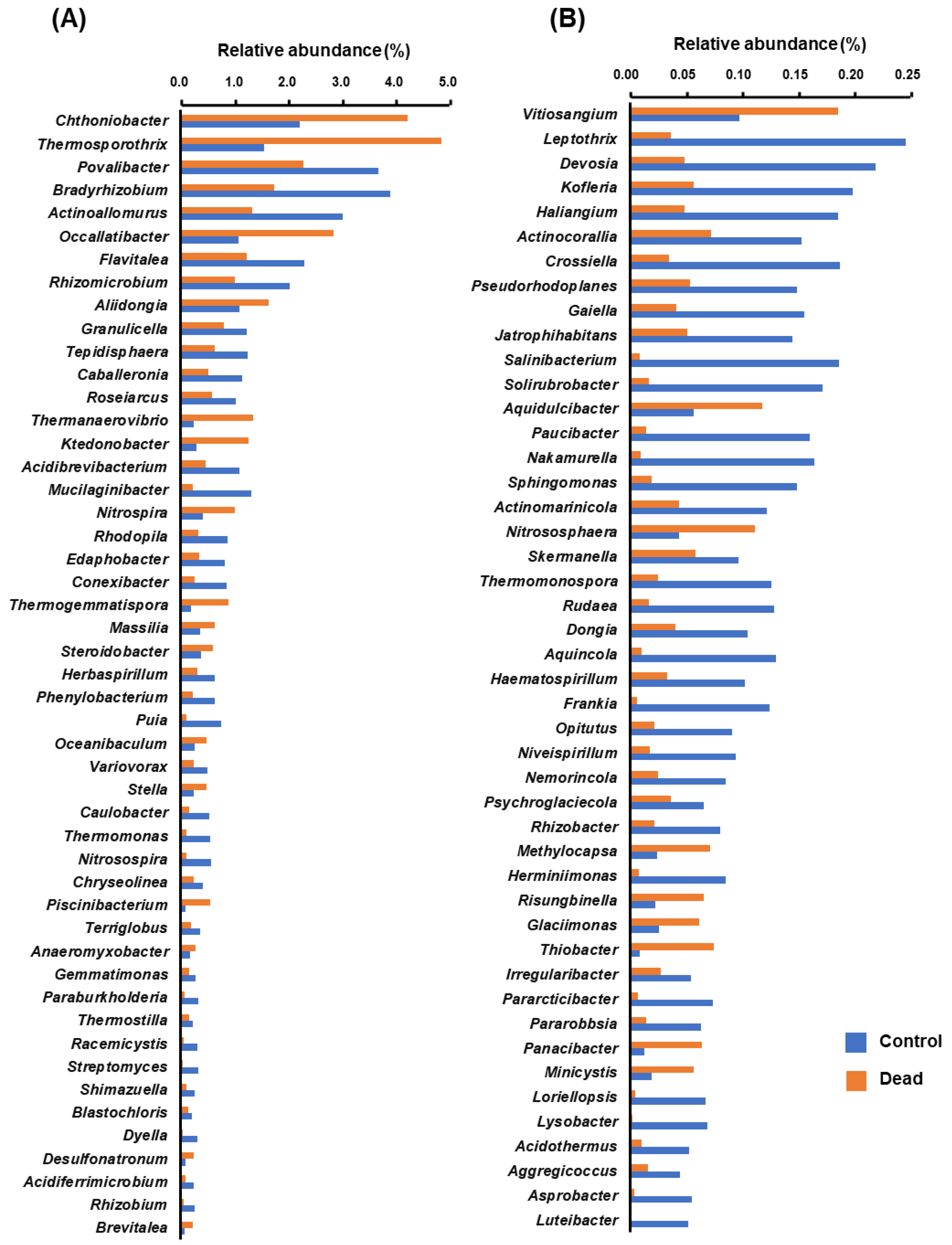
2.4. Differential Occurrence of Bacterial Genera in the Rhizosphere Microbiota between Healthy and Dead Korean Fir Trees
3. Discussion
4. Materials and Methods
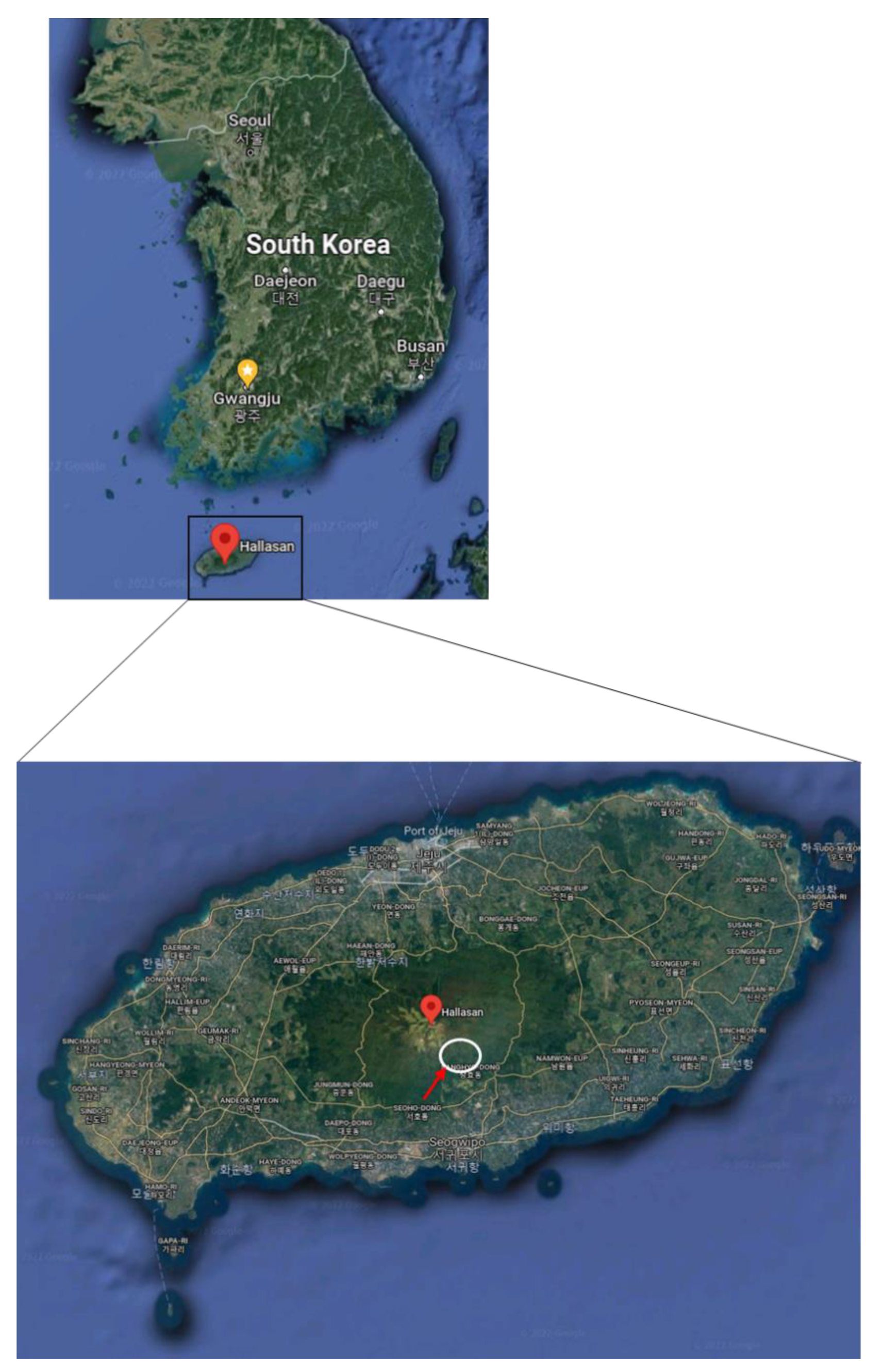
4.1. Study Site and Tree Selection
4.2. Sample Collection and Metagenomic DNA Extraction
4.3. Metagenomic 16S rRNA Amplification, Sequencing, and Microbiome Analysis
4.4. Statistical and Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wilson, E.H. Four new conifers from Korea. J. Arnold Arbor. 1920, 1, 186–190. [Google Scholar]
- Park, J.S.; Shin, H.S.; Choi, C.H.; Lee, J.; Kim, J. Hierarchical environmental factors affecting the distribution of Abies koreana on the Korean Peninsula. Forests 2018, 9, 777. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.B. Endemic plants and their distribution in Korea. J. Nat. Acad. Sci. 1982, xxi, 71–113. [Google Scholar]
- Kim, Y.-S.; Chang, C.-S.; Kim, C.-S.; Gardner, M. Abies koreana. The IUCN Red List of Threatened Species 2011: e.T31244A9618913. Available online: https://dx.doi.org/10.2305/IUCN.UK.2011-2.RLTS.T31244A9618913.en(accessed on 12 February 2022).
- Woo, S.Y. Forest decline of the world: A linkage with air pollution and global warming. Afr. J. Biotechnol. 2009, 8, 7409–7414. [Google Scholar]
- Koo, K.A.; Kong, W.S.; Park, S.U.; Lee, J.H.; Kim, J.; Jung, H. Sensitivity of Korean fir (Abies koreana Wils.), a threatened climate relict species, to increasing temperature at an island subalpine area. Ecol. Modell. 2017, 353, 5–16. [Google Scholar] [CrossRef]
- Kim, D.W.; Jeong, D.Y.; Park, H.C. Identification of Molecular Markers for Population Diagnosis of Korean Fir (Abies koreana) Vulnerable to Climate Change. Proc. Nat. Inst. Ecol. Rep. Korea 2020, 1, 68–73. [Google Scholar]
- Ahn, U.S.; Yun, Y.S. Causes of Decline in the Korean Fir Based on Spatial Distribution in the Mt. Halla Region in Korea: A Meta-Analysis. Forests 2020, 11, 391. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.W.; Choi, E.B.; Park, J.H.; Kim, Y.J.; Lim, H.I. The Role of Aging and Wind in Inducing Death and/or Growth Reduction in Korean Fir (Abies Koreana Wilson) on Mt. Halla, Korea. Atmosphere 2021, 12, 1135. [Google Scholar] [CrossRef]
- Ahn, U.S.; Kim, D.S.; Yun, Y.S.; Ko, S.H.; Kim, K.S.; Cho, I.S. The inference about the cause of death of Korean Fir in Mt. Halla through the analysis of spatial dying pattern-Proposing the possibility of excess soil moisture by climate changes. Korean J. Agric. For. Meteorol. 2019, 21, 1–28. [Google Scholar]
- Kim, E.S.; Oh, C.H.; Park, H.C.; Lee, S.H.; Choi, J.; Lee, S.H.; Cho, H.B.; Lim, W.; Kim, H.; Yoon, Y.K. Disturbed regeneration of saplings of Korean fir (Abies koreana Wilson), an endemic tree species, in Hallasan National Park, a UNESCO Biosphere Reserve, Jeju Island, Korea. J. Mar. Island Cult. 2016, 5, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Vandenkoornhuyse, P.; Quaiser, A.; Duhamel, M.; Le Van, A.; Dufresne, A. The importance of the microbiome of the plant holobiont. New Phytol. 2015, 206, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Schlaeppi, K.; Bulgarelli, D. The plant microbiome at work. Mol. Plant-Microbe Interact. 2015, 28, 212–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohanram, S.; Kumar, P. Rhizosphere microbiome: Revisiting the synergy of plant-microbe interactions. Ann. Microbiol. 2019, 69, 307–320. [Google Scholar] [CrossRef]
- Mendes, R.; Garbeva, P.; Raaijmakers, J.M. The rhizosphere microbiome: Significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. FEMS Microbiol. Rev. 2013, 37, 634–663. [Google Scholar] [CrossRef]
- Raaijmakers, J.M.; Paulitz, T.C.; Steinberg, C.; Alabouvette, C.; Moënne-Loccoz, Y. The rhizosphere: A playground and battlefield for soilborne pathogens and beneficial microorganisms. Plant Soil 2009, 321, 341–361. [Google Scholar] [CrossRef] [Green Version]
- Mercado-Blanco, J.; Abrantes, I.; Barra Caracciolo, A.; Bevivino, A.; Ciancio, A.; Grenni, P.; Hrynkiewicz, K.; Kredics, L.; Proença, D.N. Belowground microbiota and the health of tree crops. Front. Microbiol. 2018, 9, 1006. [Google Scholar] [CrossRef] [Green Version]
- Fierer, N.; Ladau, J.; Clemente, J.C.; Leff, J.W.; Owens, S.M.; Pollard, K.S.; Knight, R.; Gilbert, J.A.; McCulley, R.L. Reconstructing the microbial diversity and function of pre-agricultural tallgrass prairie soils in the United States. Science 2013, 342, 621–624. [Google Scholar] [CrossRef] [Green Version]
- Maghnia, F.Z.; Abbas, Y.; Mahé, F.; Prin, Y.; El Ghachtouli, N.; Duponnois, R.; Sanguin, H. The rhizosphere microbiome: A key component of sustainable cork oak forests in trouble. For. Ecol. Manag. 2019, 434, 29–39. [Google Scholar] [CrossRef]
- Yan, Y.; Li, B.; Huang, Z.; Zhang, H.; Wu, X.; Farooq, T.H.; Wu, P.; Li, M.; Ma, X. Characteristics and Driving Factors of Rhizosphere Bacterial Communities of Chinese Fir Provenances. Forests 2021, 12, 1362. [Google Scholar] [CrossRef]
- Kim, C.S.; Jo, J.W.; Lee, H.; Kwag, Y.N.; Cho, S.E.; Oh, S.H. Comparison of soil higher fungal communities between dead and living Abies koreana in Mt. Halla, the Republic of Korea. Mycobiology 2020, 48, 364–372. [Google Scholar] [CrossRef]
- Modi, D.; Simard, S.; Lavkulich, L.; Hamelin, R.C.; Grayston, S.J. Stump removal and tree species composition promote a bacterial microbiome that may be beneficial in the suppression of root disease. FEMS Microbiol. Ecol. 2021, 97, fiaa213. [Google Scholar] [CrossRef]
- Durán, P.; Thiergart, T.; Garrido-Oter, R.; Agler, M.; Kemen, E.; Schulze-Lefert, P.; Hacquard, S. Microbial interkingdom interactions in roots promote Arabidopsis survival. Cell 2018, 175, 973–983. [Google Scholar] [CrossRef]
- van Elsas, J.D.; Chiurazzi, M.; Mallon, C.A.; Elhottovā, D.; Krištůfek, V.; Salles, J.F. Microbial diversity determines the invasion of soil by a bacterial pathogen. Proc. Nat. Acad. Sci. USA 2012, 109, 1159–1164. [Google Scholar] [CrossRef] [Green Version]
- Garbeva, P.V.; Van Veen, J.A.; Van Elsas, J.D. Microbial diversity in soil: Selection of microbial populations by plant and soil type and implications for disease suppressiveness. Annu. Rev. Phytopathol. 2004, 42, 243–270. [Google Scholar] [CrossRef]
- Hu, J.; Wei, Z.; Friman, V.P.; Gu, S.H.; Wang, X.F.; Eisenhauer, N.; Yang, T.J.; Ma, J.; Shen, Q.R.; Xu, Y.C.; et al. Probiotic diversity enhances rhizosphere microbiome function and plant disease suppression. MBio 2016, 7, e01790-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Zhang, Y.; Mao, W.; Wang, C.; Yin, S. Functional potential differences between Firmicutes and Proteobacteria in response to manure amendment in a reclaimed soil. Can. J. Microbiol. 2020, 66, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Fierer, N.; Bradford, M.A.; Jackson, R.B. Toward an ecological classification of soil bacteria. Ecology 2007, 88, 1354–1364. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.N.; Verma, P.; Kumar, S.; Kumar, V.; Kumar, M.; Sugitha, T.C.K.; Singh, B.P.; Saxena, A.K.; Dhaliwal, H.S. Actinobacteria from Rhizosphere: Molecular Diversity, Distributions, And Potential Biotechnological Applications. In New and Future Developments in Microbial Biotechnology and Bioengineering; Elsevier: Amsterdam, The Netherlands, 2018; pp. 13–41. [Google Scholar]
- Schrey, S.D.; Tarkka, M.T. Friends and foes: Streptomycetes as modulators of plant disease and symbiosis. Antonie Van Leeuwenhoek 2008, 94, 11–19. [Google Scholar] [CrossRef]
- Bhatti, A.A.; Haq, S.; Bhat, R.A. Actinomycetes benefaction role in soil and plant health. Microb. Pathog. 2017, 111, 458–467. [Google Scholar] [CrossRef]
- Ruelland, E. and Zachowski, A. How plants sense temperature. Environ. Exp. Bot. 2010, 69, 225–232. [Google Scholar] [CrossRef]
- Lohani, N.; Jain, D.; Singh, M.B.; Bhalla, P.L. Engineering multiple abiotic stress tolerance in canola. Brassica Napus. Front. Plant Sci. 2020, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Sandrini, M.; Nerva, L.; Sillo, F.; Balestrini, R.; Chitarra, W.; Zampieri, E. Abiotic stress and belowground microbiome: The potential of omics approaches. Int. J. Mol. Sci. 2022, 23, 1091. [Google Scholar] [CrossRef]
- Nephali, L.; Piater, L.A.; Dubery, I.A.; Patterson, V.; Huyser, J.; Burgess, K.; Tugizimana, F. Biostimulants for plant growth and mitigation of abiotic stresses: A metabolomics perspective. Metabolites 2020, 10, 505. [Google Scholar] [CrossRef] [PubMed]
- Briglia, N.; Petrozza, A.; Hoeberichts, F.A.; Verhoef, N.; Povero, G. Investigating the impact of biostimulants on the row crops corn and soybean using high-efficiency phenotyping and next generation sequencing. Agronomy 2019, 9, 761. [Google Scholar] [CrossRef] [Green Version]
- Grover, M.; Bodhankar, S.; Maheswari, M.; Srinivasarao, C. Actinomycetes as Mitigators of Climate Change and Abiotic Stress. In Plant Growth Promoting Actinobacteria; Subramaniam, G., Arumugam, S., Rajendran, V., Eds.; Springer: Singapore, 2016; pp. 203–212. [Google Scholar]
- Chukwuneme, C.F.; Babalola, O.O.; Kutu, F.R.; Ojuederie, O.B. Characterization of actinomycetes isolates for plant growth promoting traits and their effects on drought tolerance in maize. J. Plant Interact. 2020, 15, 93–105. [Google Scholar] [CrossRef]
- Guglielmetti, S.; Basilico, R.; Taverniti, V.; Arioli, S.; Piagnani, C.; Bernacchi, A. Luteibacter rhizovicinus MIMR1 promotes root development in barley (Hordeum vulgare L.) under laboratory conditions. World J. Microbiol. Biotechnol. 2013, 29, 2025–2032. [Google Scholar] [CrossRef] [PubMed]
- Rivas, R.; Velázquez, E.; Willems, A.; Vizcaíno, N.; Subba-Rao, N.S.; Mateos, P.F.; Gillis, M.; Dazzo, F.B.; Martínez-Molina, E. A new species of Devosia that forms a unique nitrogen-fixing root-nodule symbiosis with the aquatic legume Neptunia natans (Lf) Druce. Appl. Environ. Microbiol. 2002, 68, 5217–5222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peix, A.; Ramírez-Bahena, M.H.; Flores-Félix, J.D.; de la Vega, P.A.; Rivas, R.; Mateos, P.F.; Igual, J.M.; Martínez-Molina, E.; Trujillo, M.E.; Velázquez, E. Revision of the taxonomic status of the species Rhizobium lupini and reclassification as Bradyrhizobium lupini comb. nov. Int. J. Syst. Evol. Microbiol. 2015, 65, 1213–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palaniappan, P.; Chauhan, P.S.; Saravanan, V.S.; Anandham, R.; Sa, T. Isolation and characterization of plant growth promoting endophytic bacterial isolates from root nodule of Lespedeza sp. Biol. Fertil. Soils 2010, 46, 807–816. [Google Scholar] [CrossRef]
- Luo, D.; Langendries, S.; Mendez, S.G.; De Ryck, J.; Liu, D.; Beirinckx, S.; Willems, A.; Russinova, E.; Debode, J.; Goormachtig, S. Plant growth promotion driven by a novel Caulobacter strain. Mol. Plant Microbe Interact. 2019, 32, 1162–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, L.J.; Nicol, G.W.; Smith, Z.; Fear, J.; Prosser, J.I.; Baggs, E.M. Nitrosospira spp. can produce nitrous oxide via a nitrifier denitrification pathway. Environ. Microbiol. 2006, 8, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Tapia-García, E.Y.; Hernández-Trejo, V.; Guevara-Luna, J.; Rojas-Rojas, F.U.; Arroyo-Herrera, I.; Meza-Radilla, G.; Vásquez-Murrieta, M.S.; Estrada-de Los Santos, P. Plant growth-promoting bacteria isolated from wild legume nodules and nodules of Phaseolus vulgaris L. trap plants in central and southern Mexico. Microbiol. Res. 2020, 239, 126522. [Google Scholar] [CrossRef]
- Turan, M.; Sain, F. Role of plant growth promoting rhizobacter strain reduce application rates mineral fertilizer in barley. ProEnvironment Promediu 2013, 6, 324–331. [Google Scholar]
- Gómez Expósito, R.; Postma, J.; Raaijmakers, J.M.; De Bruijn, I. Diversity and activity of Lysobacter species from disease suppressive soils. Front. Microbiol. 2015, 6, 1243. [Google Scholar] [CrossRef] [Green Version]
- South, K.A.; Nordstedt, N.P.; Jones, M.L. Identification of plant growth promoting rhizobacteria that improve the performance of greenhouse-grown petunias under low fertility conditions. Plants 2021, 10, 1410. [Google Scholar] [CrossRef] [PubMed]
- Fan, D.; Smith, D.L. Characterization of Selected Plant Growth-Promoting Rhizobacteria and Their Non-Host Growth Promotion Effects. Microbiol. Spectr. 2021, 9, e00279-21. [Google Scholar] [CrossRef] [PubMed]
- Muthukumarasamy, R.; Revathi, G.; Seshadri, S.; Lakshminarasimhan, C. Gluconacetobacter diazotrophicus (syn. Acetobacter diazotrophicus), a promising diazotrophic endophyte in tropics. Curr. Sci. 2002, 83, 137–145. [Google Scholar]
- Elbeltagy, A.; Nishioka, K.; Sato, T.; Suzuki, H.; Ye, B.; Hamada, T.; Isawa, T.; Mitsui, H.; Minamisawa, K. Endophytic colonization and in planta nitrogen fixation by a Herbaspirillum sp. isolated from wild rice species. Appl. Environ. Microbiol. 2001, 67, 5285–5293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.I.; Choi, H.K.; Lee, S.W.; Orwin, P.M.; Kim, J.; LaRoe, S.L.; Kim, T.G.; O′Neil, J.; Leadbetter, J.R.; Lee, S.Y.; et al. Complete genome sequence of the metabolically versatile plant growth-promoting endophyte Variovorax paradoxus S110. J. Bacteriol. 2011, 193, 1183–1190. [Google Scholar] [CrossRef] [Green Version]
- Berrios, L. The genus Caulobacter and its role in plant microbiomes. World J. Microbiol. Biotechnol. 2022, 38, 43. [Google Scholar] [CrossRef] [PubMed]
- Qian, G.L.; Hu, B.S.; Jiang, Y.H.; Liu, F.Q. Identification and characterization of Lysobacter enzymogenes as a biological control agent against some fungal pathogens. Agric. Sci. China 2009, 8, 68–75. [Google Scholar] [CrossRef]
- Belimov, A.A.; Dodd, I.C.; Hontzeas, N.; Theobald, J.C.; Safronova, V.I.; Davies, W.J. Rhizosphere bacteria containing 1-aminocyclopropane-1-carboxylate deaminase increase yield of plants grown in drying soil via both local and systemic hormone signalling. New Phytol. 2009, 181, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Klein, E.; Ofek, M.; Katan, J.; Minz, D.; Gamliel, A. Soil suppressiveness to Fusarium disease: Shifts in root microbiome associated with reduction of pathogen root colonization. Phytopathology 2013, 103, 23–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.; Miyamoto, T.; Takahashi, K.; Hong, S.; Kim, J. Damage to Abies koreana seeds by soil-borne fungi on Mount Halla, Korea. Can. J. For. Res. 2007, 37, 371–382. [Google Scholar] [CrossRef] [Green Version]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef]
- Li, W.; Chang, Y. CD-HIT-OTU-MiSeq, an improved approach for clustering and analyzing paired end MiSeq 16S rRNA sequences. BioRxiv 2017, 153783. [Google Scholar]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Hammer, O.; Harper, D.A.; Ryan, P.D. PAST: Palaeontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 9. [Google Scholar]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, G.; Mannaa, M.; Jeon, H.; Jung, H.; Kim, J.-C.; Park, A.R.; Seo, Y.-S. Dysbiosis in the Rhizosphere Microbiome of Standing Dead Korean Fir (Abies koreana). Plants 2022, 11, 990. https://doi.org/10.3390/plants11070990
Han G, Mannaa M, Jeon H, Jung H, Kim J-C, Park AR, Seo Y-S. Dysbiosis in the Rhizosphere Microbiome of Standing Dead Korean Fir (Abies koreana). Plants. 2022; 11(7):990. https://doi.org/10.3390/plants11070990
Chicago/Turabian StyleHan, Gil, Mohamed Mannaa, Hyoseong Jeon, Hyejung Jung, Jin-Cheol Kim, Ae Ran Park, and Young-Su Seo. 2022. "Dysbiosis in the Rhizosphere Microbiome of Standing Dead Korean Fir (Abies koreana)" Plants 11, no. 7: 990. https://doi.org/10.3390/plants11070990
APA StyleHan, G., Mannaa, M., Jeon, H., Jung, H., Kim, J.-C., Park, A. R., & Seo, Y.-S. (2022). Dysbiosis in the Rhizosphere Microbiome of Standing Dead Korean Fir (Abies koreana). Plants, 11(7), 990. https://doi.org/10.3390/plants11070990