
Proteomic Investigation of Molecular Mechanisms in Response to PEG-Induced Drought Stress in Soybean Roots

 ,
,
Abstract

1. Introduction
2. Materials and Methods
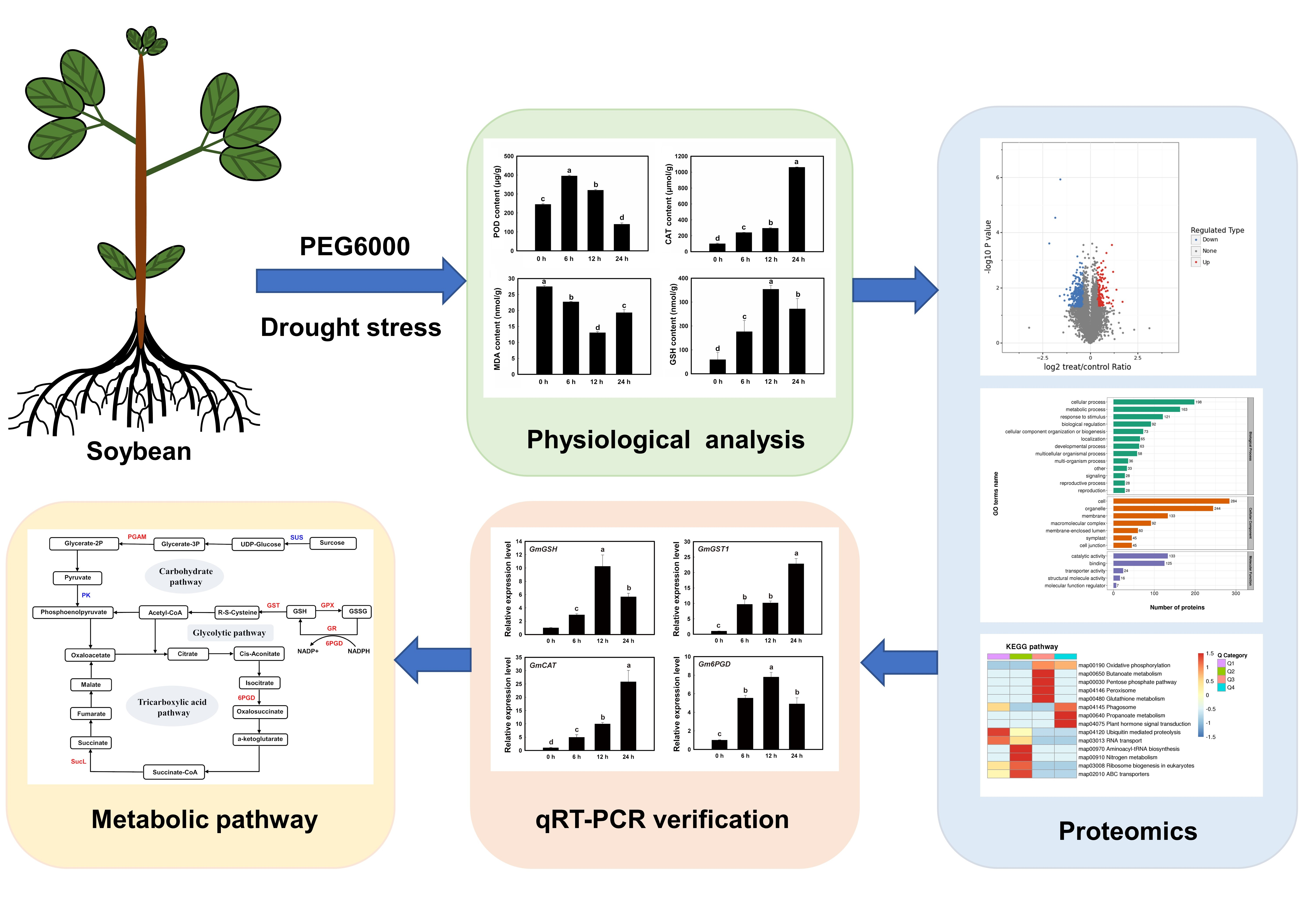
2.1. Soybean Plants and Physiological Variations in Soybean Roots under Drought Stress
2.2. Extraction, Tandem Mass Tag Labeling, and Identification of Proteins in Soybean Roots
2.3. LC-MS/MS Analysis and Protein Annotation
2.4. Functional Enrichment Analysis of Proteins Identified in Soybean Roots
2.5. Transcriptional Analysis of Differentially Expressed Genes Involved in Glutathione Metabolism
3. Results
3.1. Effect of Drought Stress on the Physiological Changes in Soybean Roots
3.2. Protein Profiling of Soybean Roots under Drought Stress
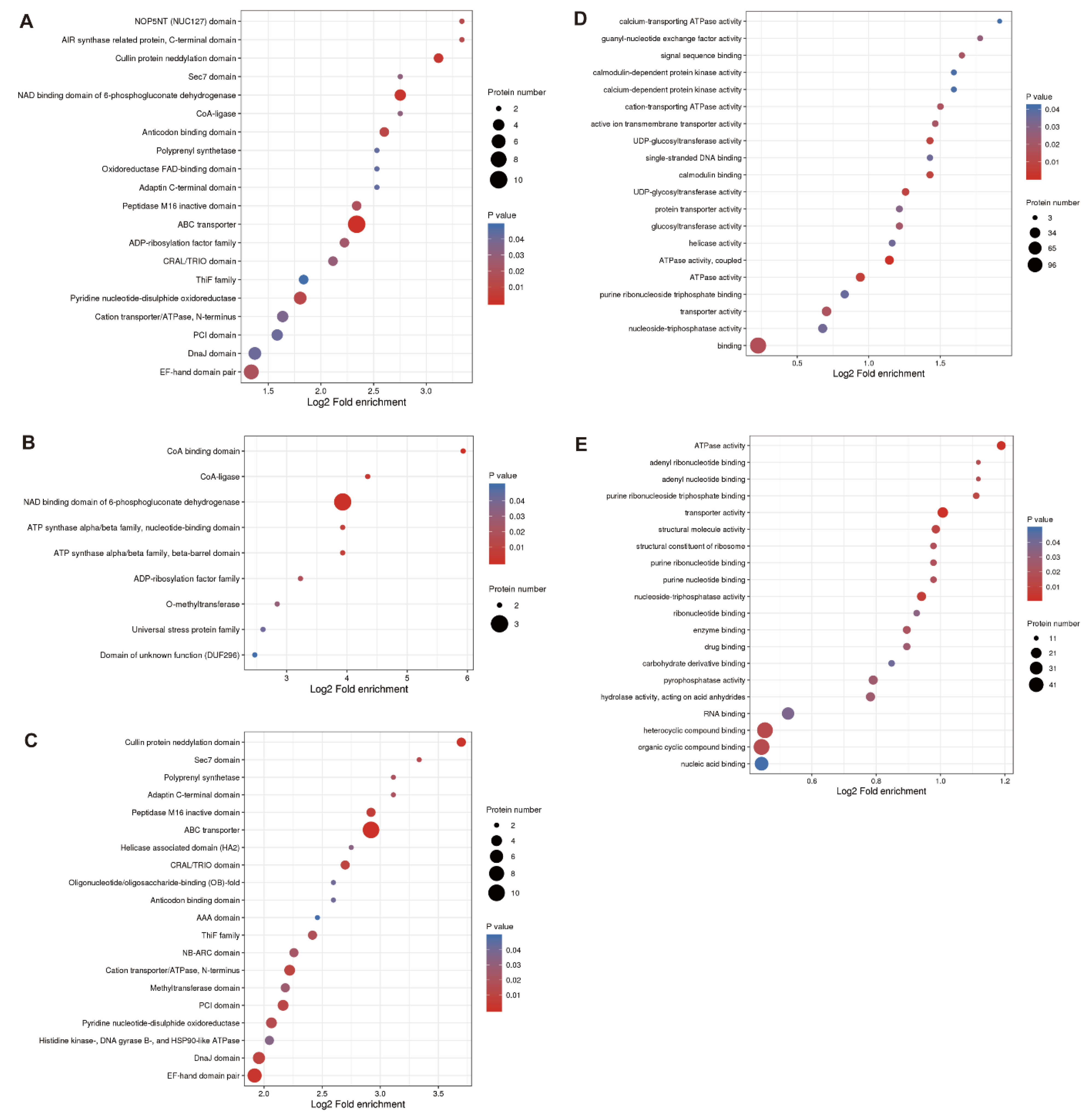
3.3. GO and COG Annotations of Proteins Induced by Drought Stress
3.4. Subcellular Localization of Proteins Induced by Drought Stress in Soybean Roots
3.5. KEGG Functional Annotation and Enrichment Analysis of Differentially Expressed Proteins Induced by Drought Stress in Soybean Roots
3.6. KEGG Pathway Enrichment Analysis of Proteins of Groups Q1 to Q4 Induced by Drought Stress in Soybean Roots
3.7. Glutathione Metabolism in Soybean Roots under Drought Stress
3.8. Validation of the Expression of Differentially Expressed Proteins in Soybean Roots by qRT-PCR
4. Discussion
4.1. Application of Proteomics in the Investigation of Molecular Response to Drought Stress in Soybean Roots
4.2. Drought-Induced Variations in the Proteomic Composition of Soybean Roots
4.3. Carbohydrate Metabolism in Soybean Roots in Response to Drought Stress
4.4. Metabolism of the Osmotic Regulation Substances in Soybean Roots
4.5. Enhanced Drought Tolerance by the Antioxidant Defense System in Soybean Roots
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gupta, A.; Rico-Medina, A.; Cao-Delgado, A.I. The physiology of plant responses to drought. Science 2020, 368, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Bechtold, U.; Field, B. Molecular mechanisms controlling plant growth during abiotic stress. J. Exp. Bot. 2018, 69, 2753–2758. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, L.; Gupta, S.; Mishra, S.K.; Pandey, G.; Kumar, S.; Chauhan, P.S.; Chakrabarty, D.; Nautiyal, C.S. Elucidation of Complex Nature of PEG Induced Drought-Stress Response in Rice Root Using Comparative Proteomics Approach. Front. Plant Sci. 2016, 7, 1466. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-H.; Xu, X.-F.; Sun, Y.-M.; Zhang, J.-L.; Li, C.-Z. Influence of drought hardening on the resistance physiology of potato seedlings under drought stress. J. Integr. Agric. 2018, 17, 336–347. [Google Scholar] [CrossRef]
- Niu, J.; Zhang, S.; Liu, S.; Ma, H.; Chen, J.; Shen, Q.; Ge, C.; Zhang, X.; Pang, C.; Zhao, X. The compensation effects of physiology and yield in cotton after drought stress. J. Plant Physiol. 2018, 224-225, 30–48. [Google Scholar] [CrossRef]
- Osmond, C.B.; Grace, S.C. Perspectives on photoinhibition and photorespiration in the field: Quintessential inefficiencies of the light and dark reactions of photosynthesis. J. Exp. Bot. 1995, 46, 1351–1362. [Google Scholar] [CrossRef]
- Rivas-Ubach, A.; Gargallo-Garriga, A.; Sardans, J.; Oravec, M.; Mateu-Castell, L.; Pérez-Trujillo, M.; Parella, T.; Ogaya, R.; Urban, O.; Peñuelas, J. Drought enhances folivory by shifting foliar metabolomes in Quercus ilex trees. New Phytol. 2014, 202, 874–885. [Google Scholar] [CrossRef]
- Farooq, M.; Wahid, A.; Kobayashi, N.; Fujita, D.; Basra, S.M.A. Plant drought stress: Effects, mechanisms and management. Agron. Sustain. Dev. 2009, 29, 185–212. [Google Scholar] [CrossRef]
- Agurla, S.; Gahir, S.; Munemasa, S.; Munemasa, S.; Murata, Y.; Raghavendra, A.S. Mechanism of Stomatal Closure in Plants Exposed to Drought and Cold Stress. Adv. Exp. Med. Biol. 2018, 1081, 215–232. [Google Scholar]
- Chaves, M.M.; Flexas, J.; Pinheiro, C. Photosynthesis under drought and salt stress: Regulation mechanisms from whole plant to cell. Ann. Bot. 2009, 103, 551–560. [Google Scholar] [CrossRef]
- Ghatak, A.; Chaturvedi, P.; Bachmann, G.; Valledor, L.; Ramšak, Ž.; Bazargani, M.M.; Bajaj, P.; Jegadeesan, S.; Li, W.; Sun, X.; et al. Physiological and Proteomic Signatures Reveal Mechanisms of Superior Drought Resilience in Pearl Millet Compared to Wheat. Front. Plant Sci. 2021, 11, 600278. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Peng, Y.; Zhao, X.; Wu, B.; Chen, F.; Ren, B.; Zhuang, Z.; Gao, Q.; Ding, Y. Comparative Proteomics Analysis of the Seedling Root Response of Drought-sensitive and Drought-tolerant Maize Varieties to Drought Stress. Int. J. Mol. Sci. 2019, 20, 2793. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, S.; Wada, T.; Abalea, Y.; Nouri, M.Z.; Nanjo, Y.; Nakayama, N.; Shimamura, S.; Yamamoto, R.; Nakamura, T.; Furukawa, K. Analysis of plasma membrane proteome in soybean and application to flooding stress response. J. Proteome Res. 2009, 8, 4487–4499. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, P.; Rabara, R.C.; Reese, R.N.; Miller, M.A.; Rohila, J.S.; Subramanian, S.; Shen, Q.J.; Morandi, D.; Bücking, H.; Shulaev, V.; et al. A tool box of genes, proteins, metabolites and promoters for improving drought tolerance in soybean includes the metabolite coumestrol and stomatal development genes. BMC Genom. 2016, 17, 102. [Google Scholar] [CrossRef] [PubMed]
- Hamanishi, E.T.; Barchet, G.L.; Dauwe, R.; Mansfield, S.D.; Campbell, M.M. Poplar trees reconfigure the transcriptome and metabolome in response to drought in a genotype- and time-of-day-dependent manner. BMC Genom. 2015, 16, 329. [Google Scholar] [CrossRef]
- Kosová, K.; Urban, M.O.; Vítámvás, P.; Prášil, I.T. Drought Stress Response in Common Wheat, Durum Wheat, and Barley: Transcriptomics, Proteomics, Metabolomics, Physiology, and Breeding for an Enhanced Drought Tolerance. Drought Stress Toler. Plants 2016, 2, 277–314. [Google Scholar]
- Gietler, M.; Nykiel, M.; Orzechowski, S.; Fettke, J.; Zagdańska, B. Protein carbonylation linked to wheat seedling tolerance to water deficiency. Environ. Exp. Bot. 2017, 137, 84–95. [Google Scholar] [CrossRef]
- Jiang, Z.; Jin, F.; Shan, X.; Li, Y. iTRAQ-Based Proteomic Analysis Reveals Several Strategies to Cope with Drought Stress in Maize Seedlings. Int. J. Mol. Sci. 2019, 20, 5956. [Google Scholar] [CrossRef]
- Cui, G.; Zhao, Y.; Zhang, J.; Chao, M.; Xie, K.; Zhang, C.; Sun, F.; Liu, S.; Xi, Y. Proteomic analysis of the similarities and differences of soil drought and polyethylene glycol stress responses in wheat (Triticum aestivum L.). Plant Mol. Biol. 2019, 100, 391–410. [Google Scholar] [CrossRef]
- Hossain, Z.; Khatoon, A.; Komatsu, S. Soybean Proteomics for Unraveling Abiotic Stress Response Mechanism. J. Proteome Res. 2013, 12, 4670–4684. [Google Scholar] [CrossRef]
- Lin, P.H.; Chao, Y.Y. Different Drought-Tolerant Mechanisms in Quinoa (Chenopodium quinoa Willd.) and Djulis (Chenopodium formosanum Koidz.) Based on Physiological Analysis. Plants 2021, 10, 2279. [Google Scholar] [CrossRef] [PubMed]
- Koramutla, M.K.; Negi, M.; Ayele, B.T. Roles of glutathione in mediating abscisic acid signaling and its regulation of seed dormancy and drought tolerance. Genes 2017, 12, 1620. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.P.; Wu, M.; Teng, Y.J.; Jia, S.H.; Yu, D.S.; Wei, T.; Chen, C.B.; Song, W.Q. Overexpression of the Glutathione Peroxidase 5 (RcGPX5) Gene From Rhodiola crenulata Increases Drought Tolerance in Salvia miltiorrhiza. Front. Plant Sci. 2019, 9, 1950. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.L.; Xu, Z.W.; Wei, Y.Y.; Wang, M.P. Overexpression of the Maize Sulfite Oxidase Increases Sulfate and GSH levels and Enhances Drought Tolerance in Transgenic Tobacco. Front. Plant Sci. 2018, 9, 298. [Google Scholar] [CrossRef]
- Du, Y.; Zhao, Q.; Chen, L.; Yao, X.; Zhang, W.; Zhang, B.; Xie, F. Effect of drought stress on sugar metabolism in leaves and roots of soybean seedlings. Plant Physiol. Biochem. 2020, 146, 1–12. [Google Scholar] [CrossRef]
- Dumont, S.; Rivoal, J. Consequences of Oxidative Stress on Plant Glycolytic and Respiratory Metabolism. Front. Plant Sci. 2019, 10, 166. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Q.; Yu, Y.; Li, X.; Tan, H. Integrated proteomics, metabolomics and physiological analyses for dissecting the toxic effects of halosulfuron-methyl on soybean seedlings (Glycine max Merr.). Plant Physiol. Biochem. 2020, 157, 303–315. [Google Scholar] [CrossRef]
- Wang, X.; Komatsu, S. Review: Proteomic Techniques for the Development of Flood-Tolerant Soybean. Int. J. Mol. Sci. 2020, 21, 7497. [Google Scholar] [CrossRef]
- Wang, X.; Komatsu, S. Proteomic Analysis of Calcium Effects on Soybean Root Tip under Flooding and Drought Stresses. Plant Cell Physiol. 2017, 58, 1405–1420. [Google Scholar] [CrossRef]
- Khan, M.N.; Komatsu, S. Proteomic analysis of soybean root including hypocotyl during recovery from drought stress. J. Proteom. 2016, 144, 39–50. [Google Scholar] [CrossRef]
- Alam, I.; Sharmin, S.A.; Kim, K.H.; Yang, J.K.; Choi, M.S.; Lee, B.H. Proteome analysis of soybean roots subjected to short-term drought stress. Plant Soil 2010, 333, 491–505. [Google Scholar] [CrossRef]
- Hossain, Z.; Komatsu, S. Potentiality of Soybean Proteomics in Untying the Mechanism of Flood and Drought Stress Tolerance. Proteomes 2014, 2, 107–127. [Google Scholar] [CrossRef] [PubMed]
- Yahoueian, S.H.; Bihamta, M.R.; Babaei, H.R.; Bazargani, M.M. Proteomic analysis of drought stress response mechanism in soybean (Glycine max L.) leaves. Food Sci. Nutr. 2021, 9, 2010–2020. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, P.P.; Moieni, A.; Hiraga, S.; Komatsu, S. Organ-specific proteomic analysis of drought-stressed soybean seedlings. J. Proteom. 2012, 75, 1906–1923. [Google Scholar] [CrossRef] [PubMed]
- Yigit, N.; Sevik, H.; Cetin, M.; Kaya, N. Determination of the effect of drought stress on the seed germination in some plant species. In Water Stress in Plants; Rahman, I.M.M., Begum, Z.A., Hasegawa, H., Eds.; IntechOpen: Rijeka, Croatia, 2016; Available online: https://www.intechopen.com/chapters/50584 (accessed on 24 April 2022). [CrossRef]
- Chen, Z.; Fang, X.; Yuan, X.; Zhang, Y.; Li, H.; Zhou, Y.; Cui, X. Overexpression of transcription factor gmtga15 enhances drought tolerance in transgenic soybean hairy roots and arabidopsis plants. Agronomy 2021, 11, 170. [Google Scholar] [CrossRef]
- Li, F.; Wang, Y.; Li, Y.; Yang, H.; Wang, H. Quantitative analysis of the global proteome in peripheral blood mononuclear cells from patients with new-onset psoriasis. Proteomics 2018, 18, e1800003. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Tang, K.; Zhang, Y.; Zhang, H.F.; Xu, P.W.; Liu, J.; Ma, J.W.; Lv, M.; Li, D.P.; Katirai, F.; Shen, G.X.; et al. Delivery of chemotherapeutic drugs in tumour cell-derived microparticles. Nat. Commun. 2012, 3, 1282. [Google Scholar] [CrossRef]
- Kavar, T.; Maras, M.; Kidrič, M.; Šuštar-Vozlič, J.; Meglič, V. Identification of genes involved in the response of leaves of Phaseolus vulgaris to drought stress. Mol. Breed. 2007, 21, 159–172. [Google Scholar] [CrossRef]
- Aebersold, R.; Mann, M. Mass spectrometry-based proteomics. Nature 2003, 422, 198–207. [Google Scholar] [CrossRef]
- Bantscheff, M.; Lemeer, S.; Savitski, M.M.; Kuster, B. Quantitative mass spectrometry in proteomics: Critical review update from 2007 to the present. Anal. Bioanal. Chem. 2012, 404, 939–965. [Google Scholar] [CrossRef] [PubMed]
- Dreger, M. Emerging strategies in mass-spectrometry based proteomics. Eur. J. Biochem. 2003, 270, 569. [Google Scholar] [CrossRef] [PubMed]
- Larance, M.; Lomond, A.I. Multidimensional proteomics for cell biology. Nat. Rev. Mol. Cell Biol. 2015, 16, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Vadivel, A.K.A. Gel-based proteomics in plants: Time to move on from the tradition. Front. Plant Sci. 2015, 6, 369. [Google Scholar]
- Yu, X.; James, A.T.; Yang, A.; Jones, A.; Mendoza-Porras, O.; Bétrix, C.A.; Ma, H.; Colgrave, M.L. A comparative proteomic study of drought-tolerant and drought-sensitive soybean seedlings under drought stress. Crop Pas. Sci. 2016, 67, 528–540. [Google Scholar] [CrossRef]
- Komatsu, S.; Yamamoto, R.; Nanjo, Y.; Mikami, Y.; Yunokawa, H.; Sakata, K. A comprehensive analysis of the soybean genes and proteins expressed under flooding stress using transcriptome and proteome techniques. J. Proteome Res. 2009, 8, 4766–4778. [Google Scholar] [CrossRef]
- Nouri, M.Z.; Komatsu, S. Comparative analysis of soybean plasma membrane proteins under osmotic stress using gel-based and LC MS/MS-based proteomics approaches. Proteomics 2010, 10, 1930–1945. [Google Scholar] [CrossRef]
- Das, A.; Eldakak, M.; Paudel, B.; Kim, D.; Hemmati, H.; Basu, C.; Rohila, J.S.E. Leaf proteome analysis reveals prospective drought and heat stress response mechanisms in soybean. BioMed Res. Int. 2016, 2016, 6021047. [Google Scholar] [CrossRef]
- Dugas, D.V.; Monaco, M.K.; Olsen, A.; Klein, R.R.; Kumari, S.; Ware, D.; Klein, P.E. Functional annotation of the transcriptome of Sorghum bicolor in response to osmotic stress and abscisic acid. BMC Genom. 2011, 12, 514. [Google Scholar] [CrossRef]
- Johnson, S.M.; Lim, F.L.; Finkler, A.; Fromm, H.; Slabas, A.R.; Knight, M.R. Transcriptomic analysis of Sorghum bicolor responding to combined heat and drought stress. BMC Genom. 2014, 15, 456. [Google Scholar] [CrossRef]
- Jin, Y.; Yang, H.; Wei, Z.; Ma, H.; Ge, X. Rice male development under drought stress: Phenotypic changes and stage-dependent transcriptomic reprogramming. Mol. Plant 2013, 6, 1630–1645. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.L.; Wang, Y.; Xie, H. Drought stress triggers proteomic changes involving lignin, flavonoids and fatty acids in tea plants. Sci. Rep. 2020, 10, 15504. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Shi, S. Physiological and Proteomic Responses of Contrasting Alfalfa (Medicago sativa L.) Varieties to PEG-Induced Osmotic Stress. Front. Plant Sci. 2018, 9, 242. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, D.; Kariyat, D.; Marriboina, S.; Reddy, A.R. Pod-wall proteomics provide novel insights into soybean seed-filling process under chemical-induced terminal drought stress. J. Sci. Food Agric. 2019, 99, 2481–2493. [Google Scholar] [CrossRef]
- Sharma, M.; Gupta, S.K.; Majumder, B.; Maurya, V.K.; Pandey, V. Proteomics unravel the regulating role of salicylic acid in soybean under yield limiting drought stress. Plant Physiol. Biochem. 2018, 130, 529–541. [Google Scholar] [CrossRef]
- Su, X.H.; Wei, F.J.; Huo, Y.J.; Xia, Z.L. Comparative physiological and molecular analyses of two contrasting flue-cured tobacco genotypes under progressive drought stress. Front. Plant Sci. 2017, 8, 827. [Google Scholar] [CrossRef]
- Chang, L.; Wang, L.; Peng, C.; Tong, Z.; Wang, X. The chloroplast proteome response to drought stress in cassava leaves. Plant Physiol. Biochem. 2019, 142, 351–362. [Google Scholar] [CrossRef]
- Vishwakarma, K.; Mishra, M.; Patil, G.; Mulkey, S.; Sharma, S.; Ramawat, N.; Singh, V.P.; Deshmukh, R.; Tripathi, D.K.; Nguyen, H.T. Avenues of the membrane transport system in adaptation of plants to abiotic stresses. Crit. Rev. Biotechnol. 2019, 39, 861–883. [Google Scholar] [CrossRef]
- Abdula, S.E.; Lee, H.J.; Kim, J.; Nio, M.C.; Cho, Y.G. Bruge1 transgenic rice showed improved growth performance with enhanced drought tolerance. Breed. Sci. 2016, 66, 226–233. [Google Scholar] [CrossRef][Green Version]
- Yao, K.; Wu, Y.Y. Phosphofructokinase and glucose-6-phosphate dehydrogenase in response to drought and bicarbonate stress at transcriptional and functional levels in mulberry. Russ. J. Plant Physiol. 2016, 63, 250–257. [Google Scholar] [CrossRef]
- Pinheiro, C.; Chaves, M.M.; Ricardo, C.P. Alterations in carbon and nitrogen metabolism induced by water deficit in stem and leaves of Lupinus albus (L.). J. Exp. Bot. 2001, 52, 1063–1070. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Zhao, Q.; Chen, L.; Yao, X.; Xie, F. Effect of drought stress during soybean r2–r6 growth stages on sucrose metabolism in leaf and seed. Int. J. Mol. Sci. 2020, 21, 618. [Google Scholar] [CrossRef] [PubMed]
- Yancey, P.H. Organic osmolytes as compatible, metabolic and counteracting cytoprotectants in high osmolarity and other stresses. J. Exp. Biol. 2005, 208, 2819–2830. [Google Scholar] [CrossRef] [PubMed]
- Szabados, L.; Savoure, A. Proline: A multifunctional amino acid. Trends Plant Sci. 2010, 15, 89–97. [Google Scholar] [CrossRef]
- Adamipour, N.; Khosh-khui, M.; Salehi, H.; Razi, H.; Karami, A.; Moghadam, A. Metabolic and genes expression analyses involved in proline metabolism of two rose species under drought stress. Plant Physiol. Biochem. 2020, 155, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Dalal, V.K.; Tripathy, B.C. Modulation of chlorophyll biosynthesis by water stress in rice seedlings during chloroplast biogenesis. Plant Cell Environ. 2012, 35, 1685–1703. [Google Scholar] [CrossRef]
- Yang, D.; Ni, R.; Yang, S.H.; Pu, Y.N.; Qian, M.; Yang, Y.Q.; Yang, Y.P. Functional Characterization of the Stipa purpurea P5CS Gene under Drought Stress Conditions. Int. J. Mol. Sci. 2021, 22, 9599. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.; Ma, X.; Shah, S.; Wu, X.; Shaheen, A.; Xiao, L.; Wu, Y.; Wang, S. Drought-hardening improves drought tolerance in Nicotiana tabacum at physiological, biochemical, and molecular levels. BMC Plant Biol. 2020, 20, 486. [Google Scholar] [CrossRef] [PubMed]
- Ullah, A.; Sun, H.; Yang, X.Y.; Zhang, X.L. Drought coping strategies in cotton: Increased crop per drop. Plant Biotechnol. J. 2017, 15, 271–284. [Google Scholar] [CrossRef]
- Zhu, Y.M.; Luo, X.G.; Nawaz, G.; Yin, J.J.; Yang, J.N. Physiological and biochemical responses of four cassava cultivars to drought stress. Sci. Rep. 2020, 10, 6968. [Google Scholar] [CrossRef]
- Wang, T.Z.; Zhang, D.; Chen, L.; Wang, J.; Zhang, W.H. Genome-wide analysis of the glutathione S-transferase family in wild Medicago ruthenica and drought-tolerant breeding application of mrugstu39 gene in cultivated alfalfa. Theor. Appl. Genet. 2022, 135, 853–864. [Google Scholar] [CrossRef] [PubMed]
- Shukla, P.S.; Shotton, K.; Norman, E.; Neily, W.; Crithley, A.T. and Prithiviraj, B. Seaweed extract improve drought tolerance of soybean by regulating stress-response genes. AoB Plants 2017, 10, plx051. [Google Scholar] [PubMed]
- Zhang, Y.H.; He, J.Y.; Xiao, Y.Z.; Zhang, Y.G.; Liu, Y.Q.; Wan, S.Q.; Liu, L.; Dong, Y.; Liu, H.; Yu, Y.B. CsGSTU8, a glutathione S-transferase from Camellia sinensis, is regulated by CsWRKY48 and plays a positive role in drought tolerance. Front. Plant Sci. 2021, 12, 795919. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, D.; Singh, S.; Kumar, V.; Romero, R.; Prasad, R.; Singh, J. Antioxidant enzymes regulation in plants in reference to reactive oxygen species (ROS) and reactive nitrogen species (RNS). Plant Gene 2019, 19, 100182. [Google Scholar] [CrossRef]
- Bela, K.; Horváth, E.; Gallé, Á.; Szabados, L.; Tari, I.; Csiszár, J. Plant glutathione peroxidases: Emerging role of the antioxidant enzymes in plant development and stress responses. J. Plant Physiol. 2015, 176, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Hasanuzzaman, M.; Nahar, K.; Anee, T.I.; Fujita, M. Glutathione in plants: Biosynthesis and physiological role in environmental stress tolerance. Physiol. Mol. Biol. Plants 2020, 23, 249–268. [Google Scholar] [CrossRef]
- Cheng, M.C.; Ko, K.; Chang, W.L.; Kuo, W.C.; Chen, G.H.; Lin, T.P. Increased glutathione contributes to stress tolerance and global translational changes in Arabidopsis. Plant J. 2015, 8, 926–939. [Google Scholar] [CrossRef]
- Noctor, G.; Mhamdi, A.; Chaouch, S.; Han, Y.; Neukermans, J.; Marquez-Garcia, B.; Queval, G.; Foyer, C.H. Glutathione in plants: An integrated overview. Plant Cell Environ. 2012, 35, 454–484. [Google Scholar] [CrossRef]
- Raja, V.; Qadir, S.U.; Alyemeni, M.N.; Ahmad, P. Impact of drought and heat stress individually and in combination on physio-biochemical parameters, antioxidant responses, and gene expression in Solanum lycopersicum. 3 Biotech 2020, 10, 208. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence |
|---|---|
| GmGSH | F: TGCAGTGTTCAGCATTCCAC R: CCCTCAACCAATACCACAGTCA |
| GmGST1 | F: CTGTGATCAAGGAGGGCCTG R: CCCTCAACCAATACCACAGTCA |
| GmGST2 | F: TCCACAAGAAAGTTCCAGTGCT R: CCCATACAGCTAACACACACTTC |
| GmCAT | F:GAGTGCTGGAGGCTTTTTGG R: AGTAACTTCTGGATATCCTTCTCAA |
| Gm6PGD | F: ACTGATCAACCTGTAGACAAGAAA R: GGCCAGTTCACCCAACTTCA |
| Tubulin | F: GGAAGGCTTTCTTGCATTGGTA R: AGTGGCATCCTGGTACTGC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Li, H.; Chen, H.; Yang, X.; Yu, T.; Wang, Y.; Wang, Y.; Jiang, K.; Wang, Y.; Chen, Z.; et al. Proteomic Investigation of Molecular Mechanisms in Response to PEG-Induced Drought Stress in Soybean Roots. Plants 2022, 11, 1173. https://doi.org/10.3390/plants11091173
Zhou Y, Li H, Chen H, Yang X, Yu T, Wang Y, Wang Y, Jiang K, Wang Y, Chen Z, et al. Proteomic Investigation of Molecular Mechanisms in Response to PEG-Induced Drought Stress in Soybean Roots. Plants. 2022; 11(9):1173. https://doi.org/10.3390/plants11091173
Chicago/Turabian StyleZhou, Ying, Huiying Li, Haoran Chen, Xiaoqin Yang, Tingting Yu, Yushuang Wang, Yujue Wang, Keting Jiang, Yan Wang, Zhanyu Chen, and et al. 2022. "Proteomic Investigation of Molecular Mechanisms in Response to PEG-Induced Drought Stress in Soybean Roots" Plants 11, no. 9: 1173. https://doi.org/10.3390/plants11091173
APA StyleZhou, Y., Li, H., Chen, H., Yang, X., Yu, T., Wang, Y., Wang, Y., Jiang, K., Wang, Y., Chen, Z., & Cui, X. (2022). Proteomic Investigation of Molecular Mechanisms in Response to PEG-Induced Drought Stress in Soybean Roots. Plants, 11(9), 1173. https://doi.org/10.3390/plants11091173