
6mA DNA Methylation on Genes in Plants Is Associated with Gene Complexity, Expression and Duplication


 and
and 
Abstract
:1. Introduction
2. Results
2.1. Genome Assemblies of Four Lotuses
2.2. Patterns of Genome-Wide Distribution of 6mA in Four Lotuses
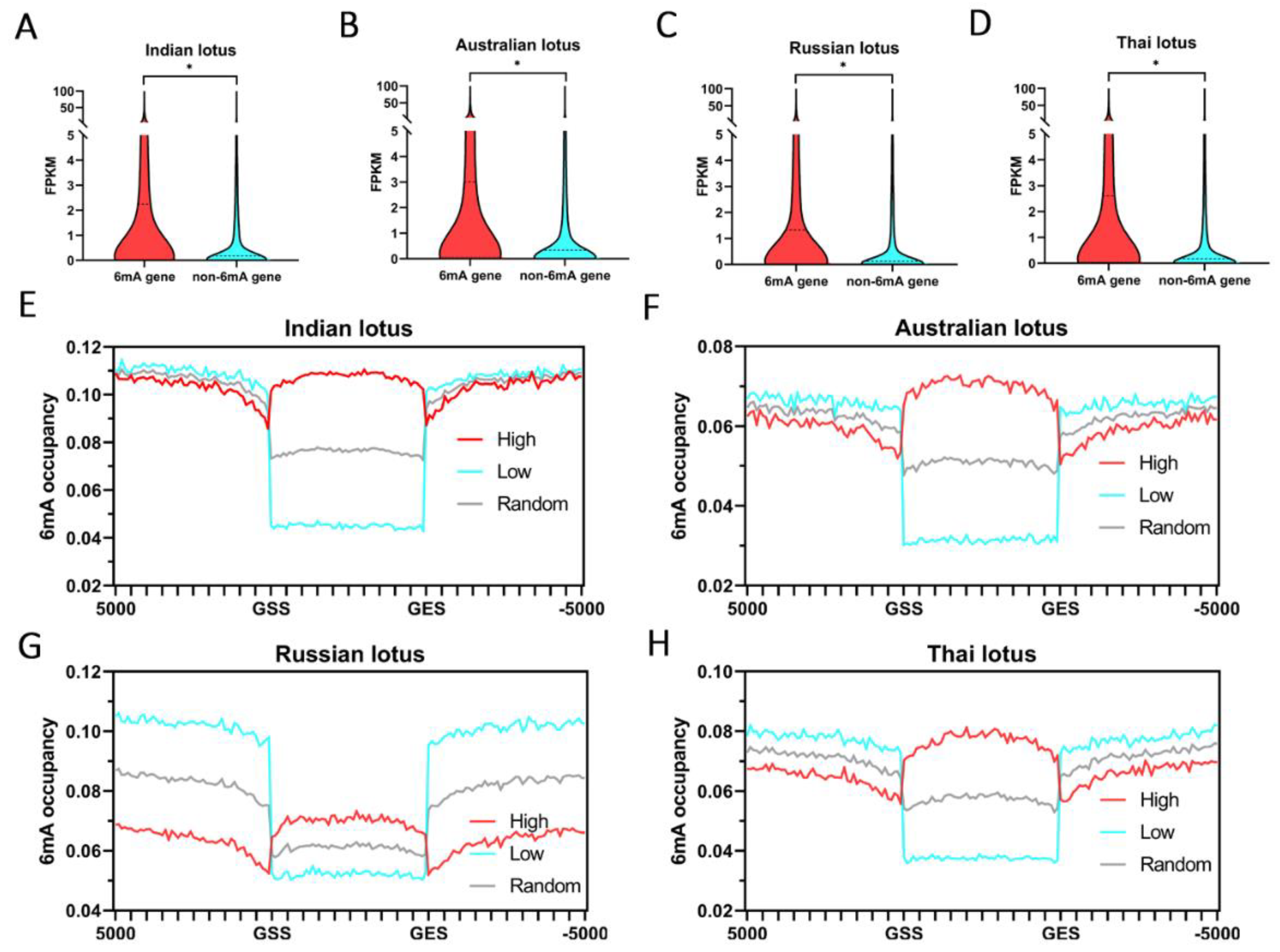
2.3. The Relationships between 6mA and Gene Expression in Lotus
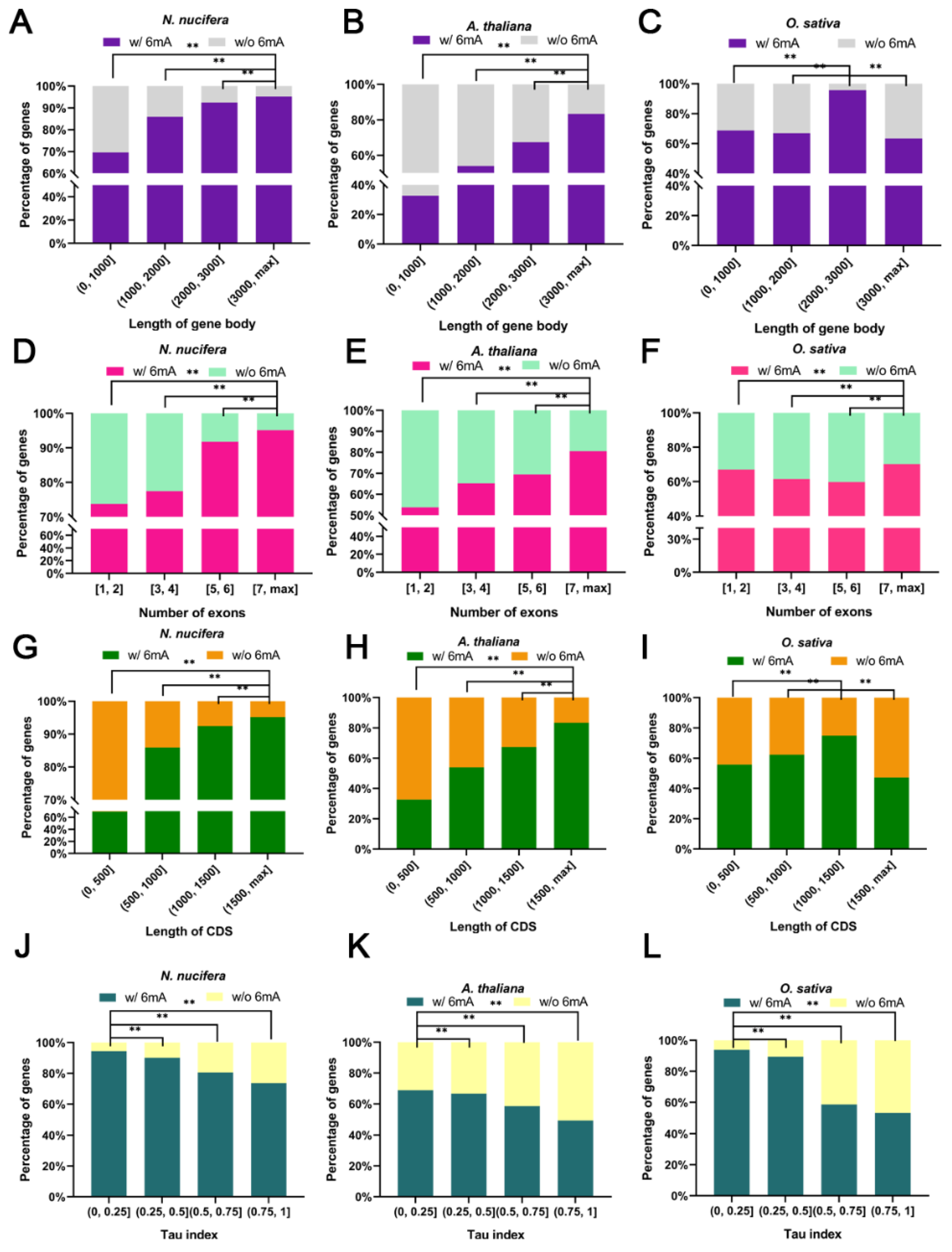
2.4. Higher Gene Structural Complexity, Expression Level, and Expression Breadth for 6mA-Methylated Genes in Plants
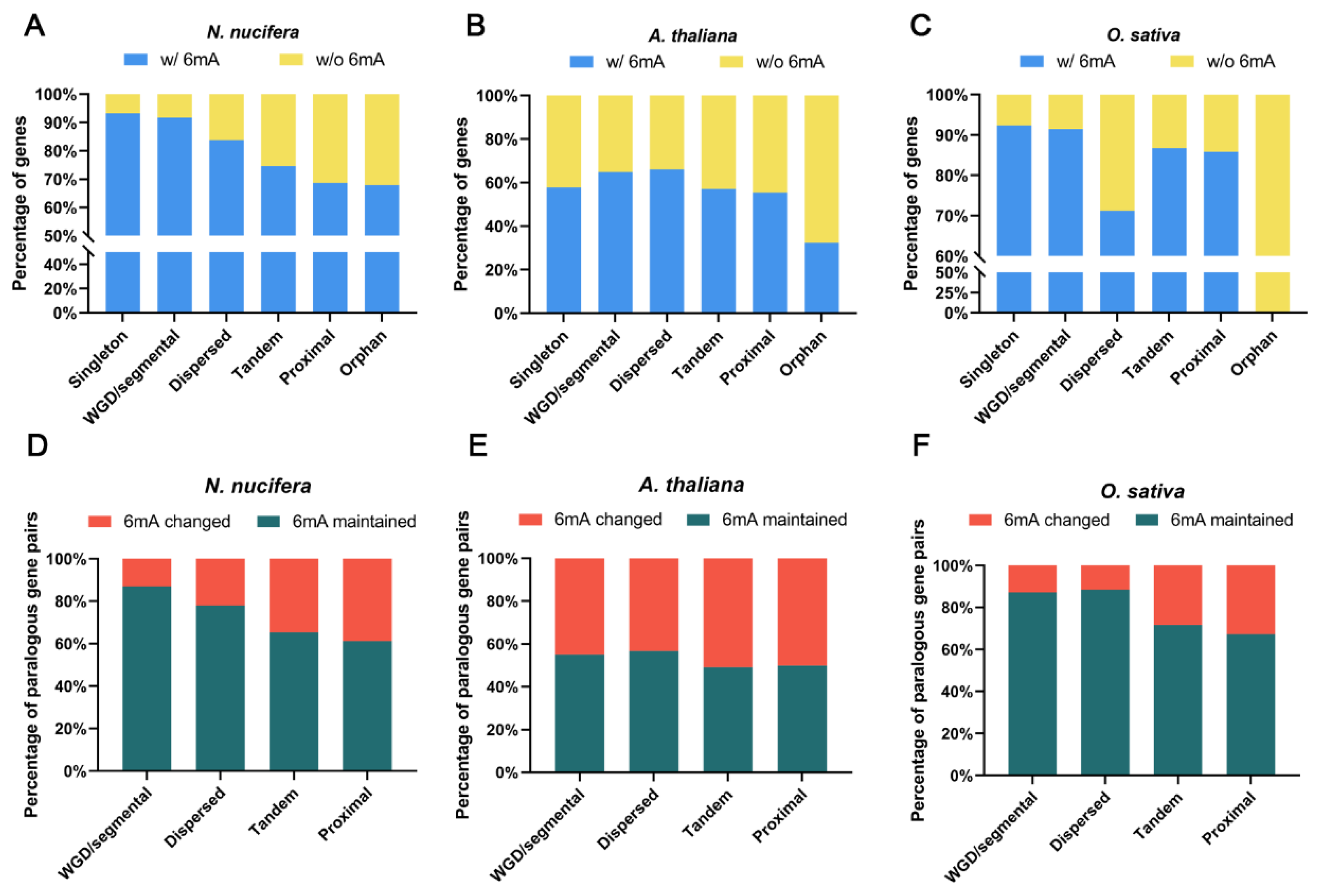
2.5. Evolution of 6mA Modification in Duplicated Genes from Different Origins
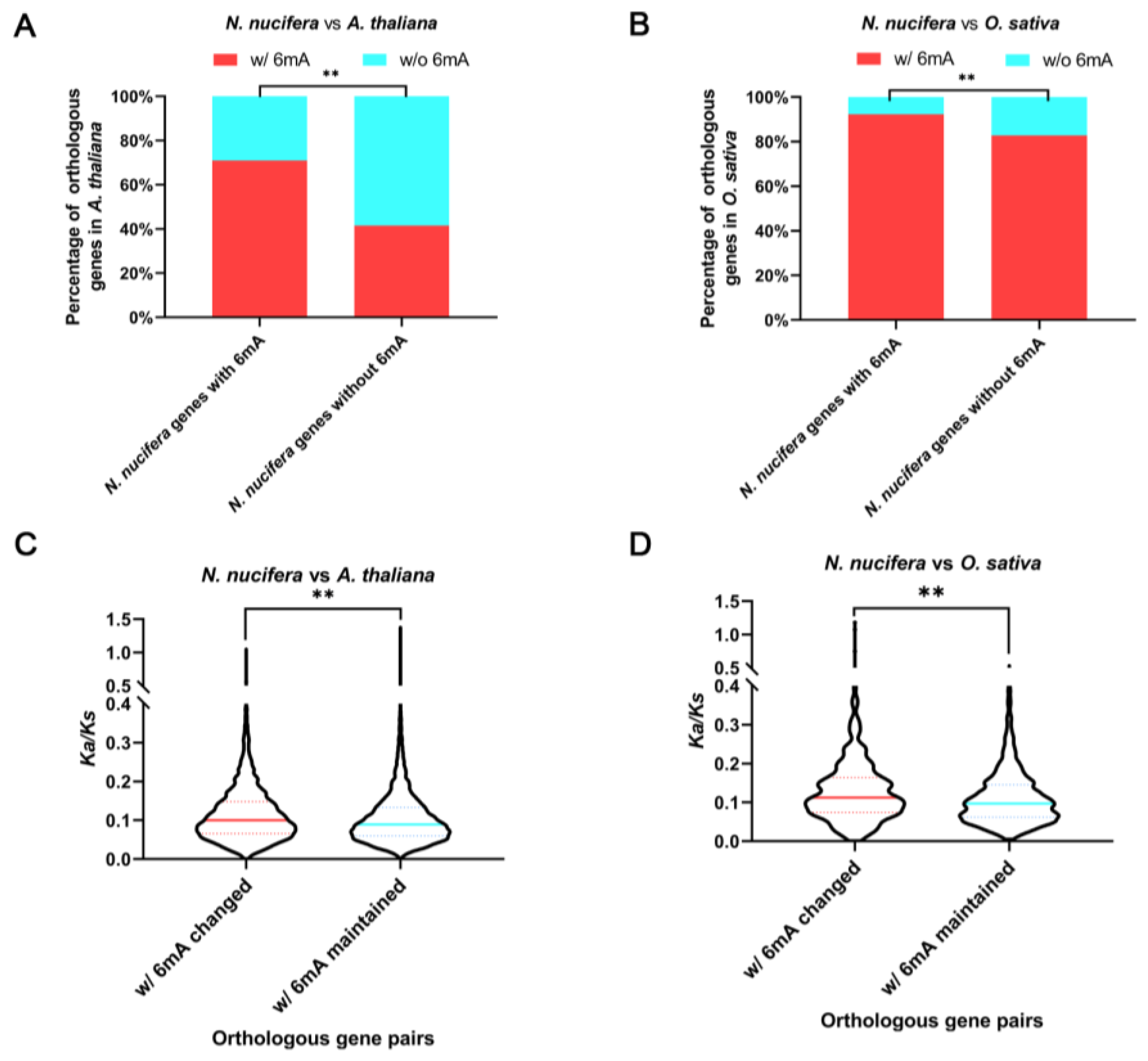
2.6. Evolution of 6mA Methylation on Orthologous Genes after Long-Term Species Divergence
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Plant Materials, Library Construction, and Genome Sequencing
5.2. Genome Assembly and Annotation
5.3. 6mA Modification Detection and Filtering
5.4. Whole-Genome 5mC Analysis
5.5. RNA-seq and Expression Analysis
5.6. Classification of Different Types of Duplicated Genes and Identification of Orthologous Genes
5.7. GO Enrichment Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lang, Z.; Zhu, J.K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 2018, 19, 489–506. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Chinnusamy, V.; Mohapatra, T. Epigenetics of Modified DNA Bases: 5-Methylcytosine and Beyond. Front. Genet. 2018, 9, 640. [Google Scholar] [CrossRef]
- Wion, D.; Casadesús, J. N6-methyl-adenine: An epigenetic signal for DNA-protein interactions. Nat. Rev. Microbiol. 2006, 4, 183–192. [Google Scholar] [CrossRef]
- Fu, Y.; Luo, G.Z.; Chen, K.; Deng, X.; Yu, M.; Han, D.; Hao, Z.; Liu, J.; Lu, X.; Dore, L.C.; et al. N6-methyldeoxyadenosine marks active transcription start sites in Chlamydomonas. Cell 2015, 161, 879–892. [Google Scholar] [CrossRef]
- Vanyushin, B.F.; Belozersky, A.N.; Kokurina, N.A.; Kadirova, D.X. 5-methylcytosine and 6-methylamino-purine in bacterial DNA. Nature 1968, 218, 1066–1067. [Google Scholar] [CrossRef]
- Campbell, J.L.; Kleckner, N. E. coli oriC and the dnaA gene promoter are sequestered from dam methyltransferase following the passage of the chromosomal replication fork. Cell 1990, 62, 967–979. [Google Scholar] [CrossRef]
- Collier, J.; McAdams, H.H.; Shapiro, L. A DNA methylation ratchet governs progression through a bacterial cell cycle. Proc. Natl. Acad. Sci. USA 2007, 104, 17111–17116. [Google Scholar] [CrossRef]
- Karrer, K.M.; VanNuland, T.A. Methylation of adenine in the nuclear DNA of Tetrahymena is internucleosomal and independent of histone H1. Nucleic Acids Res. 2002, 30, 1364–1370. [Google Scholar] [CrossRef]
- Touzain, F.; Petit, M.A.; Schbath, S.; El Karoui, M. DNA motifs that sculpt the bacterial chromosome. Nat. Rev. Microbiol. 2011, 9, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Shen, L.; Cui, X.; Bao, S.; Geng, Y.; Yu, G.; Liang, F.; Xie, S.; Lu, T.; Gu, X.; et al. DNA N(6)-Adenine Methylation in Arabidopsis thaliana. Dev. Cell 2018, 45, 406–416.e403. [Google Scholar] [CrossRef]
- Liu, J.; Zhu, Y.; Luo, G.Z.; Wang, X.; Yue, Y.; Wang, X.; Zong, X.; Chen, K.; Yin, H.; Fu, Y.; et al. Abundant DNA 6mA methylation during early embryogenesis of zebrafish and pig. Nat. Commun. 2016, 7, 13052. [Google Scholar] [CrossRef] [PubMed]
- Mondo, S.J.; Dannebaum, R.O.; Kuo, R.C.; Louie, K.B.; Bewick, A.J.; LaButti, K.; Haridas, S.; Kuo, A.; Salamov, A.; Ahrendt, S.R.; et al. Widespread adenine N6-methylation of active genes in fungi. Nat. Genet. 2017, 49, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.P.; Wang, T.; Seetin, M.G.; Lai, Y.; Zhu, S.; Lin, K.; Liu, Y.; Byrum, S.D.; Mackintosh, S.G.; Zhong, M.; et al. DNA methylation on N(6)-adenine in mammalian embryonic stem cells. Nature 2016, 532, 329–333. [Google Scholar] [CrossRef]
- Greer, E.L.; Blanco, M.A.; Gu, L.; Sendinc, E.; Liu, J.; Aristizabal-Corrales, D.; Hsu, C.H.; Aravind, L.; He, C.; Shi, Y. DNA Methylation on N6-Adenine in C. elegans. Cell 2015, 161, 868–878. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, Z.; Fu, T.; Hu, L.; Xu, C.; Gong, L.; Wendel, J.F.; Liu, B. Gene-body CG methylation and divergent expression of duplicate genes in rice. Sci. Rep. 2017, 7, 2675. [Google Scholar] [CrossRef]
- Zhang, Q.; Liang, Z.; Cui, X.; Ji, C.; Li, Y.; Zhang, P.; Liu, J.; Riaz, A.; Yao, P.; Liu, M.; et al. N(6)-Methyladenine DNA Methylation in Japonica and Indica Rice Genomes and Its Association with Gene Expression, Plant Development, and Stress Responses. Mol. Plant 2018, 11, 1492–1508. [Google Scholar] [CrossRef]
- Liu, Q.; Fang, L.; Yu, G.; Wang, D.; Xiao, C.L.; Wang, K. Detection of DNA base modifications by deep recurrent neural network on Oxford Nanopore sequencing data. Nat. Commun. 2019, 10, 2449. [Google Scholar] [CrossRef]
- McIntyre, A.B.R.; Alexander, N.; Grigorev, K.; Bezdan, D.; Sichtig, H.; Chiu, C.Y.; Mason, C.E. Single-molecule sequencing detection of N6-methyladenine in microbial reference materials. Nat. Commun. 2019, 10, 579. [Google Scholar] [CrossRef]
- Ni, P.; Huang, N.; Zhang, Z.; Wang, D.P.; Liang, F.; Miao, Y.; Xiao, C.L.; Luo, F.; Wang, J. DeepSignal: Detecting DNA methylation state from Nanopore sequencing reads using deep-learning. Bioinformatics 2019, 35, 4586–4595. [Google Scholar] [CrossRef] [PubMed]
- Cheetham, S.W.; Kindlova, M.; Ewing, A.D. Methylartist: Tools for visualizing modified bases from nanopore sequence data. Bioinformatics 2022, 38, 3109–3112. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.A.; Martin, M.D.; Nguyen, M.; Mendelson, T.C. Epigenetic divergence as a potential first step in darter speciation. Mol. Ecol. 2016, 25, 1883–1894. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, K.; Tomioka, S.; Suzuki, Y.; Nakamura, R.; Doi, K.; Yoshimura, J.; Kumagai, M.; Inoue, Y.; Uchida, Y.; Irie, N.; et al. Centromere evolution and CpG methylation during vertebrate speciation. Nat. Commun. 2017, 8, 1833. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Tang, J.W.; Yang, Z.; Cao, Y.B.; Ren, J.L.; Ben-Abu, Y.; Li, K.; Chen, X.Q.; Du, J.Z.; Nevo, E. Adaptive methylation regulation of p53 pathway in sympatric speciation of blind mole rats, Spalax. Proc. Natl. Acad. Sci. USA 2016, 113, 2146–2151. [Google Scholar] [CrossRef]
- Takuno, S.; Gaut, B.S. Gene body methylation is conserved between plant orthologs and is of evolutionary consequence. Proc. Natl. Acad. Sci. USA 2013, 110, 1797–1802. [Google Scholar] [CrossRef]
- Clark, J.W.; Donoghue, P.C.J. Whole-Genome Duplication and Plant Macroevolution. Trends Plant Sci. 2018, 23, 933–945. [Google Scholar] [CrossRef]
- Qiao, X.; Li, Q.; Yin, H.; Qi, K.; Li, L.; Wang, R.; Zhang, S.; Paterson, A.H. Gene duplication and evolution in recurring polyploidization-diploidization cycles in plants. Genome Biol. 2019, 20, 38. [Google Scholar] [CrossRef]
- Lynch, M. Genomics. Gene duplication and evolution. Science 2002, 297, 945–947. [Google Scholar] [CrossRef]
- Zhang, J. Evolution by gene duplication: An update. Trends Ecol. Evol. 2003, 18, 292–298. [Google Scholar] [CrossRef]
- Shi, T.; Rahmani, R.S.; Gugger, P.F.; Wang, M.; Li, H.; Zhang, Y.; Li, Z.; Wang, Q.; Van de Peer, Y.; Marchal, K.; et al. Distinct Expression and Methylation Patterns for Genes with Different Fates following a Single Whole-Genome Duplication in Flowering Plants. Mol. Biol. Evol. 2020, 37, 2394–2413. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, X.; Lee, T.H.; Mansoor, S.; Paterson, A.H. Gene body methylation shows distinct patterns associated with different gene origins and duplication modes and has a heterogeneous relationship with gene expression in Oryza sativa (rice). New Phytol. 2013, 198, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xie, J.; Hu, J.; Lan, B.; You, C.; Li, F.; Wang, Z.; Wang, H. Comparative epigenomics reveals evolution of duplicated genes in potato and tomato. Plant J. Cell Mol. Biol. 2018, 93, 460–471. [Google Scholar] [CrossRef] [PubMed]
- El Baidouri, M.; Kim, K.D.; Abernathy, B.; Li, Y.H.; Qiu, L.J.; Jackson, S.A. Genic C-Methylation in Soybean Is Associated with Gene Paralogs Relocated to Transposable Element-Rich Pericentromeres. Mol. Plant 2018, 11, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.D.; El Baidouri, M.; Abernathy, B.; Iwata-Otsubo, A.; Chavarro, C.; Gonzales, M.; Libault, M.; Grimwood, J.; Jackson, S.A. A Comparative Epigenomic Analysis of Polyploidy-Derived Genes in Soybean and Common Bean. Plant Physiol. 2015, 168, 1433–1447. [Google Scholar] [CrossRef]
- Xu, C.; Nadon, B.D.; Kim, K.D.; Jackson, S.A. Genetic and epigenetic divergence of duplicate genes in two legume species. Plant Cell Environ. 2018, 41, 2033–2044. [Google Scholar] [CrossRef]
- Hua, Z.; Pool, J.E.; Schmitz, R.J.; Schultz, M.D.; Shiu, S.H.; Ecker, J.R.; Vierstra, R.D. Epigenomic programming contributes to the genomic drift evolution of the F-Box protein superfamily in Arabidopsis. Proc. Natl. Acad. Sci. USA 2013, 110, 16927–16932. [Google Scholar] [CrossRef]
- Keller, T.E.; Yi, S.V. DNA methylation and evolution of duplicate genes. Proc. Natl. Acad. Sci. USA 2014, 111, 5932–5937. [Google Scholar] [CrossRef]
- Dyson, C.J.; Goodisman, M.A.D. Gene Duplication in the Honeybee: Patterns of DNA Methylation, Gene Expression, and Genomic Environment. Mol. Biol. Evol. 2020, 37, 2322–2331. [Google Scholar] [CrossRef]
- Wang, H.; Beyene, G.; Zhai, J.; Feng, S.; Fahlgren, N.; Taylor, N.J.; Bart, R.; Carrington, J.C.; Jacobsen, S.E.; Ausin, I. CG gene body DNA methylation changes and evolution of duplicated genes in cassava. Proc. Natl. Acad. Sci. USA 2015, 112, 13729–13734. [Google Scholar] [CrossRef]
- Shi, T.; Chen, J. A reappraisal of the phylogenetic placement of the Aquilegia whole-genome duplication. Genome Biol. 2020, 21, 295. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Nyong, A.T.; Shi, T.; Yang, P. The complexity of alternative splicing and landscape of tissue-specific expression in lotus (Nelumbo nucifera) unveiled by Illumina- and single-molecule real-time-based RNA-sequencing. DNA Res. Int. J. Rapid Publ. Rep. Genes Genomes 2019, 26, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, X.; Wang, Q.; Chen, J.; Shi, T. Distinct methylome patterns contribute to ecotypic differentiation in the growth of the storage organ of a flowering plant (sacred lotus). Mol. Ecol. 2021, 30, 2831–2845. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.T.; Workman, R.E.; Zuzarte, P.C.; David, M.; Dursi, L.J.; Timp, W. Detecting DNA cytosine methylation using nanopore sequencing. Nat. Methods 2017, 14, 407–410. [Google Scholar] [CrossRef]
- Yuan, D.H.; Xing, J.F.; Luan, M.W.; Ji, K.K.; Guo, J.; Xie, S.Q.; Zhang, Y.M. DNA N6-Methyladenine Modification in Wild and Cultivated Soybeans Reveals Different Patterns in Nucleus and Cytoplasm. Front. Genet. 2020, 11, 736. [Google Scholar] [CrossRef]
- Peterson, D.G.; Arick, M. Sequencing Plant Genomes. In Progress in Botany; Cánovas, F.M., Lüttge, U., Matyssek, R., Pretzsch, H., Eds.; Springer International Publishing: Cham, Switzerland, 2019; Volume 80, pp. 109–193. [Google Scholar]
- Van Bel, M.; Diels, T.; Vancaester, E.; Kreft, L.; Botzki, A.; Van de Peer, Y.; Coppens, F.; Vandepoele, K. PLAZA 4.0: An integrative resource for functional, evolutionary and comparative plant genomics. Nucleic Acids Res. 2018, 46, D1190–D1196. [Google Scholar] [CrossRef]
- Ratel, D.; Ravanat, J.L.; Berger, F.; Wion, D. N6-methyladenine: The other methylated base of DNA. BioEssays News Rev. Mol. Cell. Dev. Biol. 2006, 28, 309–315. [Google Scholar] [CrossRef]
- Luo, G.Z.; Blanco, M.A.; Greer, E.L.; He, C.; Shi, Y. DNA N(6)-methyladenine: A new epigenetic mark in eukaryotes? Nat. Rev. Mol. Cell Biol. 2015, 16, 705–710. [Google Scholar] [CrossRef]
- Song, C.; Liu, Y.; Song, A.; Dong, G.; Zhao, H.; Sun, W.; Ramakrishnan, S.; Wang, Y.; Wang, S.; Li, T.; et al. The Chrysanthemum nankingense Genome Provides Insights into the Evolution and Diversification of Chrysanthemum Flowers and Medicinal Traits. Mol. Plant 2018, 11, 1482–1491. [Google Scholar] [CrossRef]
- Stoiber, M.H.; Quick, J.; Egan, R.; Lee, J.E.; Celniker, S.E.; Neely, R.; Loman, N.; Pennacchio, L.; Brown, J.B. De novo identification of DNA modifications enabled by genome-guided nanopore signal processing. bioRxiv 2016, 094672. [Google Scholar] [CrossRef]
- Rand, A.C.; Jain, M.; Eizenga, J.M.; Musselman-Brown, A.; Olsen, H.E.; Akeson, M.; Paten, B. Mapping DNA methylation with high-throughput nanopore sequencing. Nat. Methods 2017, 14, 411–413. [Google Scholar] [CrossRef] [PubMed]
- Michael, T.P.; Jupe, F.; Bemm, F.; Motley, S.T.; Sandoval, J.P.; Lanz, C.; Loudet, O.; Weigel, D.; Ecker, J.R. High contiguity Arabidopsis thaliana genome assembly with a single nanopore flow cell. Nat. Commun. 2018, 9, 541. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.H.; Vogel, A.; Denton, A.K.; Istace, B.; Wormit, A.; van de Geest, H.; Bolger, M.E.; Alseekh, S.; Maß, J.; Pfaff, C.; et al. De Novo Assembly of a New Solanum pennellii Accession Using Nanopore Sequencing. Plant Cell 2017, 29, 2336–2348. [Google Scholar] [CrossRef] [PubMed]
- Bewick, A.J.; Ji, L.; Niederhuth, C.E.; Willing, E.M.; Hofmeister, B.T.; Shi, X.; Wang, L.; Lu, Z.; Rohr, N.A.; Hartwig, B.; et al. On the origin and evolutionary consequences of gene body DNA methylation. Proc. Natl. Acad. Sci. USA 2016, 113, 9111–9116. [Google Scholar] [CrossRef] [PubMed]
- Bewick, A.J.; Niederhuth, C.E.; Ji, L.; Rohr, N.A.; Griffin, P.T.; Leebens-Mack, J.; Schmitz, R.J. The evolution of CHROMOMETHYLASES and gene body DNA methylation in plants. Genome Biol. 2017, 18, 65. [Google Scholar] [CrossRef]
- Niederhuth, C.E.; Bewick, A.J.; Ji, L.; Alabady, M.S.; Kim, K.D.; Li, Q.; Rohr, N.A.; Rambani, A.; Burke, J.M.; Udall, J.A.; et al. Widespread natural variation of DNA methylation within angiosperms. Genome Biol. 2016, 17, 194. [Google Scholar] [CrossRef]
- Wang, J.; Marowsky, N.C.; Fan, C. Divergence of gene body DNA methylation and evolution of plant duplicate genes. PLoS ONE 2014, 9, e110357. [Google Scholar] [CrossRef]
- Kawakatsu, T.; Huang, S.C.; Jupe, F.; Sasaki, E.; Schmitz, R.J.; Urich, M.A.; Castanon, R.; Nery, J.R.; Barragan, C.; He, Y.; et al. Epigenomic Diversity in a Global Collection of Arabidopsis thaliana Accessions. Cell 2016, 166, 492–505. [Google Scholar] [CrossRef]
- Seymour, D.K.; Koenig, D.; Hagmann, J.; Becker, C.; Weigel, D. Evolution of DNA methylation patterns in the Brassicaceae is driven by differences in genome organization. PLoS Genet. 2014, 10, e1004785. [Google Scholar] [CrossRef]
- Feng, S.; Cokus, S.J.; Zhang, X.; Chen, P.Y.; Bostick, M.; Goll, M.G.; Hetzel, J.; Jain, J.; Strauss, S.H.; Halpern, M.E.; et al. Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. USA 2010, 107, 8689–8694. [Google Scholar] [CrossRef]
- Kiefer, C.; Willing, E.M.; Jiao, W.B.; Sun, H.; Piednoël, M.; Hümann, U.; Hartwig, B.; Koch, M.A.; Schneeberger, K. Interspecies association mapping links reduced CG to TG substitution rates to the loss of gene-body methylation. Nat. Plants 2019, 5, 846–855. [Google Scholar] [CrossRef] [PubMed]
- Yao, B.; Cheng, Y.; Wang, Z.; Li, Y.; Chen, L.; Huang, L.; Zhang, W.; Chen, D.; Wu, H.; Tang, B.; et al. DNA N6-methyladenine is dynamically regulated in the mouse brain following environmental stress. Nat. Commun. 2017, 8, 1122. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.C.; Rogers, S.W. Comparison of the effects of N6-methyldeoxyadenosine and N5-methyldeoxycytosine on transcription from nuclear gene promoters in barley. Plant J. Cell Mol. Biol. 1995, 7, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Chothia, C.; Finkelstein, A.V. The classification and origins of protein folding patterns. Annu. Rev. Biochem. 1990, 59, 1007–1039. [Google Scholar] [CrossRef]
- Chothia, C.; Gough, J.; Vogel, C.; Teichmann, S.A. Evolution of the protein repertoire. Science 2003, 300, 1701–1703. [Google Scholar] [CrossRef]
- Lee, H.S.; Chen, Z.J. Protein-coding genes are epigenetically regulated in Arabidopsis polyploids. Proc. Natl. Acad. Sci. USA 2001, 98, 6753–6758. [Google Scholar] [CrossRef]
- Chang, A.Y.; Liao, B.Y. DNA methylation rebalances gene dosage after mammalian gene duplications. Mol. Biol. Evol. 2012, 29, 133–144. [Google Scholar] [CrossRef]
- Vasu, K.; Nagaraja, V. Diverse functions of restriction-modification systems in addition to cellular defense. Microbiol. Mol. Biol. Rev. MMBR 2013, 77, 53–72. [Google Scholar] [CrossRef]
- Ye, C.; Ma, Z.S.; Cannon, C.H.; Pop, M.; Yu, D.W. Exploiting sparseness in de novo genome assembly. BMC Bioinform. 2012, 13, S1. [Google Scholar] [CrossRef]
- Ye, C.; Hill, C.M.; Wu, S.; Ruan, J.; Ma, Z.S. DBG2OLC: Efficient Assembly of Large Genomes Using Long Erroneous Reads of the Third Generation Sequencing Technologies. Sci. Rep. 2016, 6, 31900. [Google Scholar] [CrossRef]
- Li, H.; Yang, X.; Zhang, Y.; Gao, Z.; Liang, Y.; Chen, J.; Shi, T. Nelumbo genome database, an integrative resource for gene expression and variants of Nelumbo nucifera. Sci. Data 2021, 8, 38. [Google Scholar] [CrossRef] [PubMed]
- Marcais, G.; Delcher, A.L.; Phillippy, A.M.; Coston, R.; Salzberg, S.L.; Zimin, A. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Klepikova, A.V.; Kasianov, A.S.; Gerasimov, E.S.; Logacheva, M.D.; Penin, A.A. A high resolution map of the Arabidopsis thaliana developmental transcriptome based on RNA-seq profiling. Plant J. Cell Mol. Biol. 2016, 88, 1058–1070. [Google Scholar] [CrossRef]
- Xia, L.; Zou, D.; Sang, J.; Xu, X.; Yin, H.; Li, M.; Wu, S.; Hu, S.; Hao, L.; Zhang, Z. Rice Expression Database (RED): An integrated RNA-Seq-derived gene expression database for rice. J. Genet. Genom. = Yi Chuan Xue Bao 2017, 44, 235–241. [Google Scholar] [CrossRef]
- Kryuchkova-Mostacci, N.; Robinson-Rechavi, M. A benchmark of gene expression tissue-specificity metrics. Brief. Bioinform. 2017, 18, 205–214. [Google Scholar] [CrossRef]
- Tautz, D.; Domazet-Lošo, T. The evolutionary origin of orphan genes. Nat. Rev. Genet. 2011, 12, 692–702. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, J.; Zhao, X.Q.; Wang, J.; Wong, G.K.; Yu, J. KaKs_Calculator: Calculating Ka and Ks through model selection and model averaging. Genom. Proteom. Bioinform. 2006, 4, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.Z.; You, L.J.; Yang, F.; Wang, L.N.; Guo, X.Q.; Gao, F.; Hua, C.; Tan, C.; Fang, L.; Shan, R.Q.; et al. CNGBdb: China National GeneBank DataBase. Yi Chuan 2020, 42, 799–809. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Indian Lotus | Australian Lotus | Russian Lotus | Thai Lotus |
|---|---|---|---|---|
| Genome size (bp) | 801,742,539 | 807,059,836 | 828,573,050 | 845,764,567 |
| Contig number | 421 | 471 | 1608 | 1117 |
| Longest contig (bp) | 40,391,228 | 30,603,528 | 14,404,078 | 23,450,770 |
| Average contig size (bp) | 1,903,041 | 1,708,757 | 513,269 | 756,434 |
| N50 | 9,768,644 | 9,418,673 | 2,477,834 | 4,066,382 |
| GC (%) | 38.86 | 38.88 | 38.89 | 38.87 |
| BUSCO (%) | 91.8 | 91.6 | 91.1 | 90.9 |
| Gene number | 34,966 | 35,093 | 37,129 | 36,314 |
| Exon number | 149,316 | 151,500 | 160,545 | 166,936 |
| Intron number | 114,350 | 116,407 | 123,416 | 130,622 |
| Average CDS length (bp) | 1457.19 | 1458.5 | 1458.81 | 1443.17 |
| High-confidence gene (%) | 80.15 | 80.20 | 79.79 | 79.93 |
| Repetitive elements (%) | 56.03 | 56.19 | 55.74 | 56.36 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Zhang, Q.; Yang, X.; Gu, X.; Chen, J.; Shi, T. 6mA DNA Methylation on Genes in Plants Is Associated with Gene Complexity, Expression and Duplication. Plants 2023, 12, 1949. https://doi.org/10.3390/plants12101949
Zhang Y, Zhang Q, Yang X, Gu X, Chen J, Shi T. 6mA DNA Methylation on Genes in Plants Is Associated with Gene Complexity, Expression and Duplication. Plants. 2023; 12(10):1949. https://doi.org/10.3390/plants12101949
Chicago/Turabian StyleZhang, Yue, Qian Zhang, Xingyu Yang, Xiaofeng Gu, Jinming Chen, and Tao Shi. 2023. "6mA DNA Methylation on Genes in Plants Is Associated with Gene Complexity, Expression and Duplication" Plants 12, no. 10: 1949. https://doi.org/10.3390/plants12101949
APA StyleZhang, Y., Zhang, Q., Yang, X., Gu, X., Chen, J., & Shi, T. (2023). 6mA DNA Methylation on Genes in Plants Is Associated with Gene Complexity, Expression and Duplication. Plants, 12(10), 1949. https://doi.org/10.3390/plants12101949