
Comparative Analysis Reveals Different Evolutionary Fates and Biological Functions in Wheat Duplicated Genes (Triticum aestivum L.)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
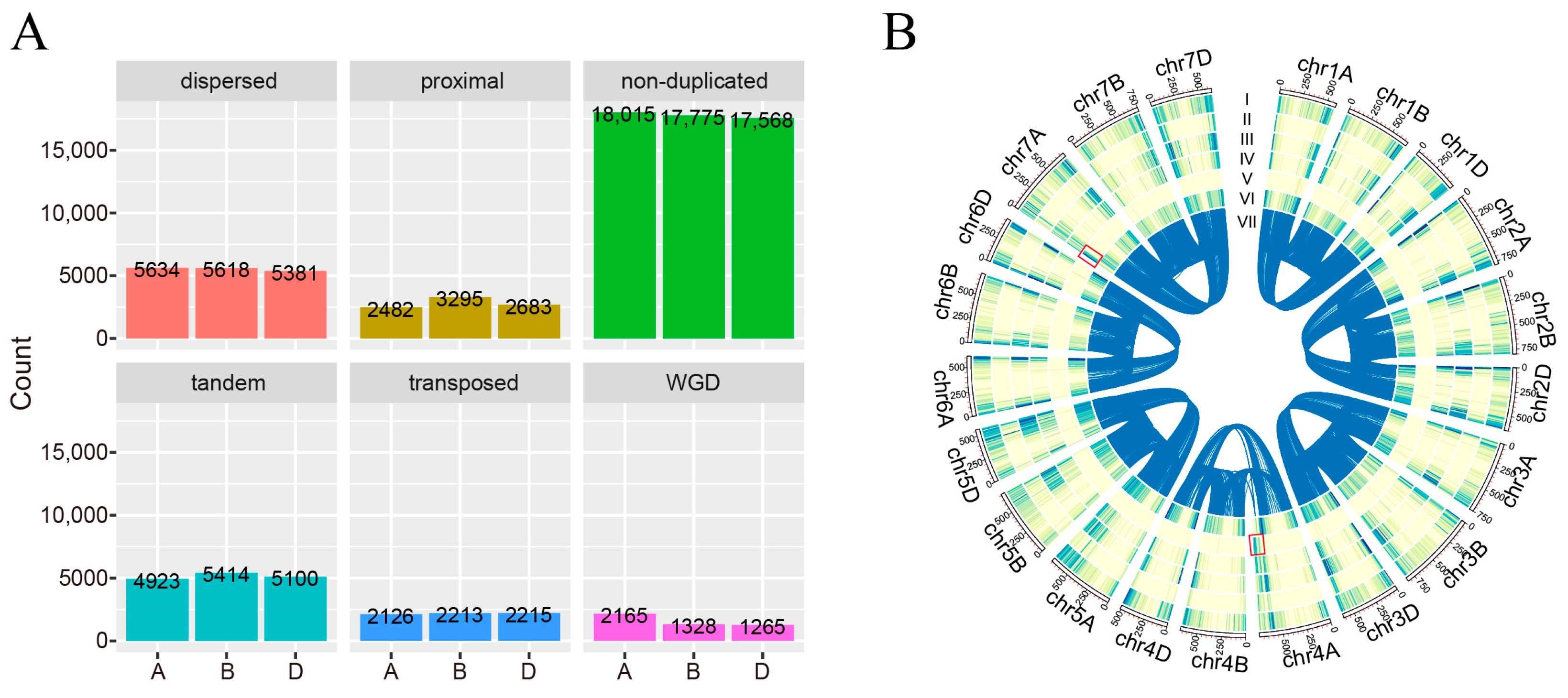
2.1. The Landscape of Gene Duplication in A, B, and D Subgenomes of T. aestivum
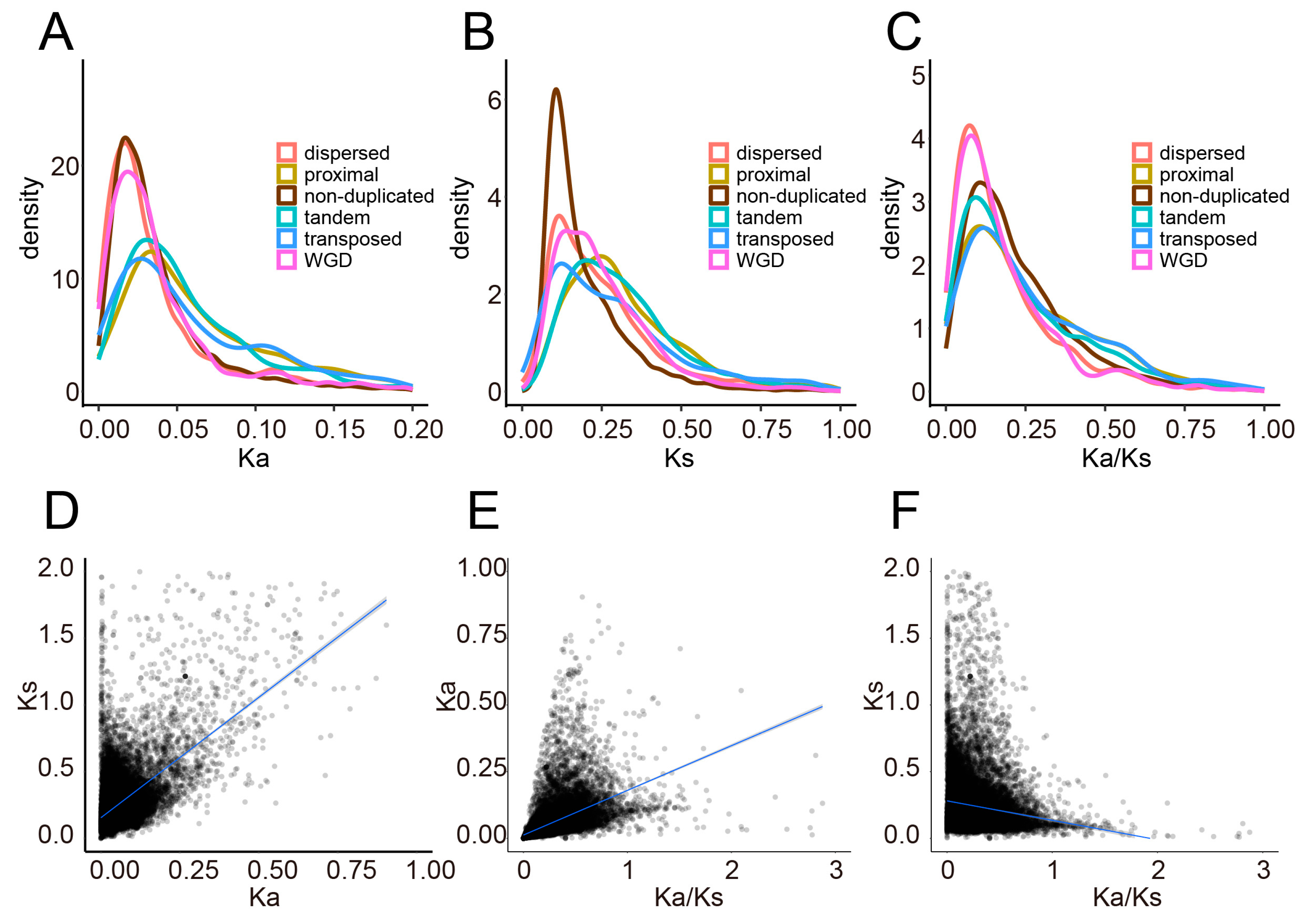
2.2. Genome-Wide Distribution and Correlation Analysis of Ka, Ks, and Ka/Ks
2.3. Comparison between Gene Properties of Different Modes of Duplicated Genes
2.4. Divergence in Codon Usage Bias of Different Modes of Duplicated Genes
2.5. Expression Levels and Tissue Specificity of Duplicated Genes
2.6. Functional Divergence of Different Types of Duplicated Genes
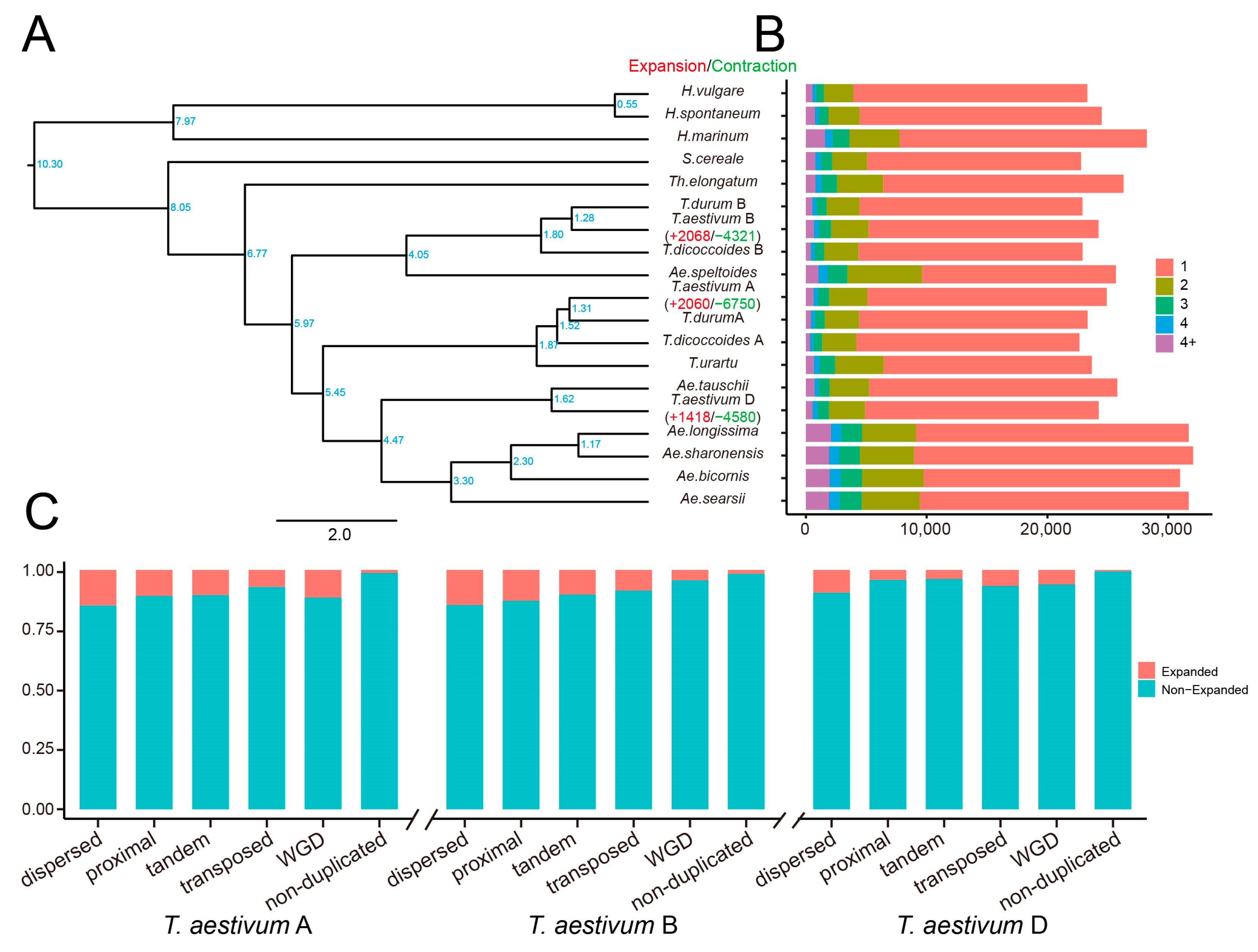
2.7. The Contribution of Duplicated Genes to the Expansion of Gene Families
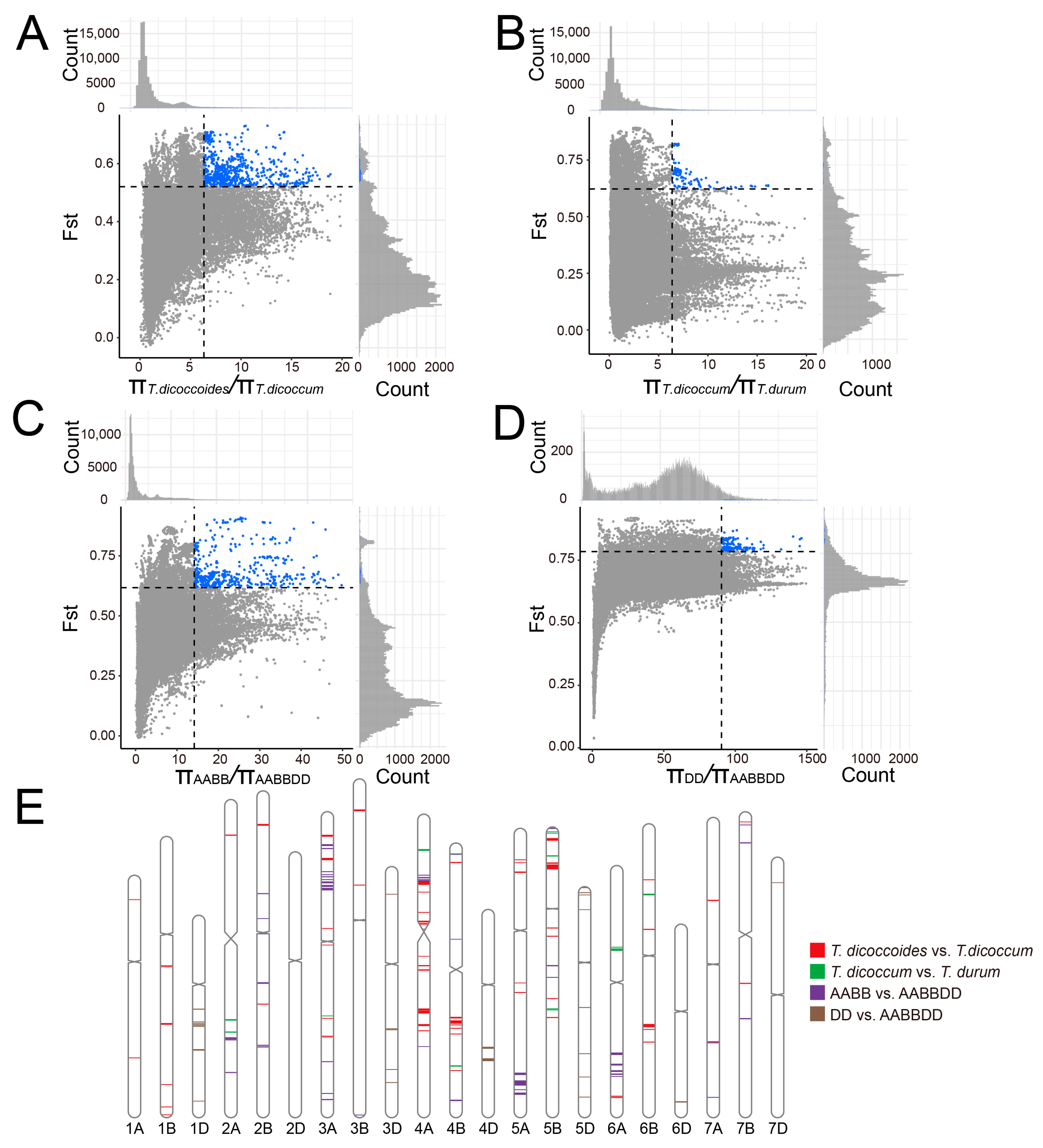
2.8. Nucleotide Variation and Genetic Bottleneck in the Domestication and Polyploidization of T. aestivum
2.9. Hotspots of Duplicated Genes during Wheat Domestication and Hexaploidization
3. Discussion
3.1. Evolutionary Rate, Gene Compactness, and Expression Patterns between Duplicated Genes and Non-Duplicated Genes
3.2. Divergence in Evolutionary Rate, Expression, and Function between Duplication Modes
3.3. The Asymmetrical Evolutionary Patterns of the A, B, and D Subgenomes in T. aestivum
3.4. Duplicated Genes Might Play Essential Roles in Various Plant Growth and Stress Response of T. aestivum
4. Materials and Methods
4.1. Identification of Gene Duplication Modes in T. aestivum
4.2. Evolutionary Rate Calculation
4.3. Codon Usage Bias and Gene Compactness Analysis
4.4. Enrichment Analysis of Different Modes of Duplicated Genes
4.5. Gene Family Expansion and Contraction Analysis
4.6. Expression Profile Analysis
4.7. Nucleotide Diversity, Phylogenetic Relationships, and Selective Sweep Analysis
4.8. Plotting and Statistical Tests
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Flagel, L.E.; Wendel, J.F. Gene duplication and evolutionary novelty in plants. New Phytol. 2009, 183, 557–564. [Google Scholar] [CrossRef]
- Panchy, N.; Lehti-Shiu, M.; Shiu, S.H. Evolution of gene duplication in plants. Plant Physiol. 2016, 171, 2294–2316. [Google Scholar] [CrossRef] [PubMed]
- Freeling, M. Bias in plant gene content following different sorts of duplication: Tandem, whole-genome, segmental, or by transposition. Annu. Rev. Plant Biol. 2009, 60, 433–453. [Google Scholar] [CrossRef] [PubMed]
- Julca, I.; Marcet-Houben, M.; Cruz, F.; Vargas-Chavez, C.; Johnston, J.S.; Gómez-Garrido, J.; Frias, L.; Corvelo, A.; Loska, D.; Cámara, F.; et al. Phylogenomics identifies an ancestral burst of gene duplications predating the diversification of Aphidomorpha. Mol. Biol. Evol. 2020, 37, 730–756. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Wickett, N.J.; Ayyampalayam, S.; Chanderbali, A.S.; Landherr, L.; Ralph, P.E.; Tomsho, L.P.; Hu, Y.; Liang, H.; Soltis, P.S.; et al. Ancestral polyploidy in seed plants and angiosperms. Nature 2011, 473, 97–100. [Google Scholar] [CrossRef]
- Amborella Genome Project. The Amborella genome and the evolution of flowering plants. Science 2013, 342, 1241089. [Google Scholar] [CrossRef]
- Lynch, M.; Conery, J.S. The evolutionary fate and consequences of duplicate genes. Science 2000, 290, 1151–1155. [Google Scholar] [CrossRef]
- Soltis, P.S.; Marchant, D.B.; Van de Peer, Y.; Soltis, D.E. Polyploidy and genome evolution in plants. Curr. Opin. Genet. Dev. 2015, 35, 119–125. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Paterson, A.H. Genome and gene duplications and gene expression divergence: A view from plants. Ann. NY Acad. Sci. 2012, 1256, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, H.E.; Zhou, C.; Tang, H.; Khadke, P.P.; Das, S.; Lin, Y.R.; Ge, Z.; Clemente, T.; Upadhyaya, H.D.; Hash, C.T.; et al. The evolution of photoperiod-insensitive flowering in sorghum, a genomic model for panicoid grasses. Mol. Biol. Evol. 2016, 33, 2417–2428. [Google Scholar] [CrossRef]
- Hahn, M.W. Distinguishing among evolutionary models for the maintenance of gene duplicates. J. Hered. 2009, 100, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.P.; Si, Y.; Hanson, R.E.; Crane, C.F.; Price, H.J.; Stelly, D.M.; Wendel, J.F.; Paterson, A.H. Dispersed repetitive DNA has spread to new genomes since polyploid formation in cotton. Genome Res. 1998, 8, 479–492. [Google Scholar] [CrossRef]
- Freeling, M.; Lyons, E.; Pedersen, B.; Alam, M.; Ming, R.; Lisch, D. Many or most genes in Arabidopsis transposed after the origin of the order Brassicales. Genome Res. 2008, 18, 1924–1937. [Google Scholar] [CrossRef] [PubMed]
- Cusack, B.P.; Wolfe, K.H. Not born equal: Increased rate asymmetry in relocated and retrotransposed rodent gene duplicates. Mol. Biol. Evol. 2007, 24, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Ganko, E.W.; Meyers, B.C.; Vision, T.J. Divergence in expression between duplicated genes in Arabidopsis. Mol. Biol. Evol. 2007, 24, 2298–2309. [Google Scholar] [CrossRef]
- Wang, Y.; Ficklin, S.P.; Wang, X.; Feltus, F.A.; Paterson, A.H. Large-scale gene relocations following an ancient genome triplication associated with the diversification of core eudicots. PLoS ONE 2016, 11, e0155637. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]
- Qiao, X.; Li, Q.; Yin, H.; Qi, K.; Li, L.; Wang, R.; Zhang, S.; Paterson, A.H. Gene duplication and evolution in recurring polyploidization-diploidization cycles in plants. Genome Biol. 2019, 20, 38. [Google Scholar] [CrossRef]
- Shiferaw, B.; Smale, M.; Braun, H.J.; Duveiller, E.; Muricho, G.J.F.S. Crops that feed the world 10. Past successes and future challenges to the role played by wheat in global food security. Food Secur. 2013, 5, 291–317. [Google Scholar] [CrossRef]
- Feldman, M.; Levy, A.A. Genome evolution due to allopolyploidization in wheat. Genetics 2012, 192, 763–774. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhao, X.; Li, Y.; Xu, J.; Bi, A.; Kang, L.; Xu, D.; Chen, H.; Wang, Y.; Wang, Y.G.; et al. Triticum population sequencing provides insights into wheat adaptation. Nat. Genet. 2020, 52, 1412–1422. [Google Scholar] [CrossRef]
- Li, L.F.; Zhang, Z.B.; Wang, Z.H.; Li, N.; Sha, Y.; Wang, X.F.; Ding, N.; Li, Y.; Zhao, J.; Wu, Y.; et al. Genome sequences of five Sitopsis species of Aegilops and the origin of polyploid wheat B subgenome. Mol. Plant 2022, 15, 488–503. [Google Scholar] [CrossRef]
- Avni, R.; Nave, M.; Barad, O.; Baruch, K.; Twardziok, S.O.; Gundlach, H.; Hale, I.; Mascher, M.; Spannagl, M.; Wiebe, K.; et al. Wild emmer genome architecture and diversity elucidate wheat evolution and domestication. Science 2017, 357, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Salamini, F.; Ozkan, H.; Brandolini, A.; Schäfer-Pregl, R.; Martin, W. Genetics and geography of wild cereal domestication in the near east. Nat. Rev. Genet. 2002, 3, 429–441. [Google Scholar] [CrossRef] [PubMed]
- International Wheat Genome Sequencing Consortium (IWGSC). Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, eaar7191. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Gaut, B.S. Factors that contribute to variation in evolutionary rate among Arabidopsis genes. Mol. Biol. Evol. 2011, 28, 2359–2369. [Google Scholar] [CrossRef]
- Du, J.; Tian, Z.; Sui, Y.; Zhao, M.; Song, Q.; Cannon, S.B.; Cregan, P.; Ma, J. Pericentromeric effects shape the patterns of divergence, retention, and expression of duplicated genes in the paleopolyploid soybean. Plant Cell 2012, 24, 21–32. [Google Scholar] [CrossRef]
- Cao, Y.; Jiang, L.; Wang, L.; Cai, Y. Evolutionary rate heterogeneity and functional divergence of orthologous genes in Pyrus. Biomolecules 2019, 9, 490. [Google Scholar] [CrossRef]
- Schrider, D.R.; Kern, A.D. Soft sweeps are the dominant mode of adaptation in the human genome. Mol. Biol. Evol. 2017, 34, 1863–1877. [Google Scholar] [CrossRef]
- Ikemura, T. Codon usage and tRNA content in unicellular and multicellular organisms. Mol. Biol. Evol. 1985, 2, 13–34. [Google Scholar]
- Dong, H.; Nilsson, L.; Kurland, C.G. Co-variation of tRNA abundance and codon usage in Escherichia coli at different growth rates. J. Mol. Biol. 1996, 260, 649–663. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Li, W.H. The codon adaptation index—A measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef] [PubMed]
- Bennetzen, J.L.; Hall, B.D. Codon selection in yeast. J. Biol. Chem. 1982, 257, 3026–3031. [Google Scholar] [CrossRef] [PubMed]
- Ikemura, T. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes: A proposal for a synonymous codon choice that is optimal for the E. coli translational system. J. Mol. Biol. 1981, 151, 389–409. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Sun, J.; Yang, G. Comparative analysis of selection mode reveals different evolutionary rate and expression pattern in Arachis duranensis and Arachis ipaënsis duplicated genes. Plant Mol. Biol. 2018, 98, 349–361. [Google Scholar] [CrossRef]
- Guo, Y.; Liu, J.; Zhang, J.; Liu, S.; Du, J. Selective modes determine evolutionary rates, gene compactness and expression patterns in Brassica. Plant J. 2017, 91, 34–44. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, F.; Yu, Y.; Feng, L.; Jia, J.; Liu, B.; Li, B.; Guo, H.; Zhai, J. A comprehensive online database for exploring ∼20,000 public Arabidopsis RNA-Seq libraries. Mol. Plant 2020, 13, 1231–1233. [Google Scholar] [CrossRef]
- Glémin, S.; Scornavacca, C.; Dainat, J.; Burgarella, C.; Viader, V.; Ardisson, M.; Sarah, G.; Santoni, S.; David, J.; Ranwez, V. Pervasive hybridizations in the history of wheat relatives. Sci. Adv. 2019, 5, eaav9188. [Google Scholar] [CrossRef]
- Yang, Y.; Cui, L.; Lu, Z.; Li, G.; Yang, Z.; Zhao, G.; Kong, C.; Li, D.; Chen, Y.; Xie, Z.; et al. Genome sequencing of Sitopsis species provides insights into their contribution to the B subgenome of bread wheat. Plant Commun. 2023, 4, 100567. [Google Scholar] [CrossRef]
- Hufford, M.B.; Xu, X.; van Heerwaarden, J.; Pyhäjärvi, T.; Chia, J.M.; Cartwright, R.A.; Elshire, R.J.; Glaubitz, J.C.; Guill, K.E.; Kaeppler, S.M.; et al. Comparative population genomics of maize domestication and improvement. Nat. Genet. 2012, 44, 808–811. [Google Scholar] [CrossRef]
- Cheng, H.; Liu, J.; Wen, J.; Nie, X.; Xu, L.; Chen, N.; Li, Z.; Wang, Q.; Zheng, Z.; Li, M.; et al. Frequent intra- and inter-species introgression shapes the landscape of genetic variation in bread wheat. Genome Biol. 2019, 20, 136. [Google Scholar] [CrossRef] [PubMed]
- Akhunov, E.D.; Akhunova, A.R.; Anderson, O.D.; Anderson, J.A.; Blake, N.; Clegg, M.T.; Coleman-Derr, D.; Conley, E.J.; Crossman, C.C.; Deal, K.R.; et al. Nucleotide diversity maps reveal variation in diversity among wheat genomes and chromosomes. BMC Genom. 2010, 11, 702. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Pasam, R.; Shi, F.; Kant, S.; Keeble-Gagnere, G.; Kay, P.; Forrest, K.; Fritz, A.; Hucl, P.; Wiebe, K.; et al. Exome sequencing highlights the role of wild-relative introgression in shaping the adaptive landscape of the wheat genome. Nat. Genet 2019, 51, 896–904. [Google Scholar] [CrossRef]
- Mace, E.S.; Tai, S.; Gilding, E.K.; Li, Y.; Prentis, P.J.; Bian, L.; Campbell, B.C.; Hu, W.; Innes, D.J.; Han, X.; et al. Whole-genome sequencing reveals untapped genetic potential in Africa’s indigenous cereal crop sorghum. Nat. Commun. 2013, 4, 2320. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Jiang, Y.; Wang, Z.; Gou, Z.; Lyu, J.; Li, W.; Yu, Y.; Shu, L.; Zhao, Y.; Ma, Y.; et al. Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat. Biotechnol. 2015, 33, 408–414. [Google Scholar] [CrossRef]
- Zhao, X.; Guo, Y.; Kang, L.; Yin, C.; Bi, A.; Xu, D.; Zhang, Z.; Zhang, J.; Yang, X.; Xu, J.; et al. Population genomics unravels the Holocene history of bread wheat and its relatives. Nat. Plants 2023, 9, 403–419. [Google Scholar] [CrossRef]
- Ibarra, I.L.; Hollmann, N.M.; Klaus, B.; Augsten, S.; Velten, B.; Hennig, J.; Zaugg, J.B. Mechanistic insights into transcription factor cooperativity and its impact on protein-phenotype interactions. Nat. Commun. 2020, 11, 124. [Google Scholar] [CrossRef]
- Jacquemin, J.; Ammiraju, J.S.; Haberer, G.; Billheimer, D.D.; Yu, Y.; Liu, L.C.; Rivera, L.F.; Mayer, K.; Chen, M.; Wing, R.A. Fifteen million years of evolution in the Oryza genus shows extensive gene family expansion. Mol. Plant 2014, 7, 642–656. [Google Scholar] [CrossRef]
- Bergthorsson, U.; Andersson, D.I.; Roth, J.R. Ohno’s dilemma: Evolution of new genes under continuous selection. Proc. Natl. Acad. Sci. USA 2007, 104, 17004–17009. [Google Scholar] [CrossRef]
- O’Toole, Á.N.; Hurst, L.D.; McLysaght, A. Faster evolving primate genes are more likely to duplicate. Mol. Biol. Evol. 2018, 35, 107–118. [Google Scholar] [CrossRef]
- Vance, Z.; Niezabitowski, L.; Hurst, L.D.; McLysaght, A. Evidence from Drosophila supports higher duplicability of faster evolving genes. Genome Biol. Evol. 2022, 14, evac003. [Google Scholar] [CrossRef] [PubMed]
- Echave, J.; Spielman, S.J.; Wilke, C.O. Causes of evolutionary rate variation among protein sites. Nat. Rev. Genet. 2016, 17, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, J.R. Determinants of the rate of protein sequence evolution. Nat. Rev. Genet. 2015, 16, 409–420. [Google Scholar] [CrossRef]
- Ren, L.L.; Liu, Y.J.; Liu, H.J.; Qian, T.T.; Qi, L.W.; Wang, X.R.; Zeng, Q.Y. Subcellular relocalization and positive selection play key roles in the retention of duplicate genes of Populus class III peroxidase family. Plant Cell 2014, 26, 2404–2419. [Google Scholar] [CrossRef] [PubMed]
- Cardoso-Moreira, M.; Arguello, J.R.; Gottipati, S.; Harshman, L.G.; Grenier, J.K.; Clark, A.G. Evidence for the fixation of gene duplications by positive selection in Drosophila. Genome Res. 2016, 26, 787–798. [Google Scholar] [CrossRef]
- Lan, X.; Pritchard, J.K. Coregulation of tandem duplicate genes slows evolution of subfunctionalization in mammals. Science 2016, 352, 1009–1013. [Google Scholar] [CrossRef]
- Ghanbarian, A.T.; Hurst, L.D. Neighboring genes show correlated evolution in gene expression. Mol. Biol. Evol. 2015, 32, 1748–1766. [Google Scholar] [CrossRef]
- Makino, T.; McLysaght, A. Positionally biased gene loss after whole genome duplication: Evidence from human, yeast, and plant. Genome Res. 2012, 22, 2427–2435. [Google Scholar] [CrossRef]
- Liu, S.; Liu, Y.; Yang, X.; Tong, C.; Edwards, D.; Parkin, I.A.; Zhao, M.; Ma, J.; Yu, J.; Huang, S.; et al. The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 2014, 5, 3930. [Google Scholar] [CrossRef]
- Kenchanmane Raju, S.K. Epigenomic atlas in wheat reveals regulatory elements specifying subgenome divergence. Plant Cell 2021, 33, 783–785. [Google Scholar] [CrossRef]
- Ramírez-González, R.H.; Borrill, P.; Lang, D.; Harrington, S.A.; Brinton, J.; Venturini, L.; Davey, M.; Jacobs, J.; van Ex, F.; Pasha, A.; et al. The transcriptional landscape of polyploid wheat. Science 2018, 361, eaar6089. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Li, Z.; Zhang, Y.; Zhang, Y.; Xie, Y.; Ye, L.; Zhuang, Y.; Lin, K.; Zhao, F.; Guo, J.; et al. An atlas of wheat epigenetic regulatory elements reveals subgenome divergence in the regulation of development and stress responses. Plant Cell 2021, 33, 865–881. [Google Scholar] [CrossRef]
- Pei, H.; Li, Y.; Liu, Y.; Liu, P.; Zhang, J.; Ren, X.; Lu, Z. Chromatin accessibility landscapes revealed the subgenome-divergent regulation networks during wheat grain development. aBIOTECH 2023, 4, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Hao, C.; Jiao, C.; Hou, J.; Li, T.; Liu, H.; Wang, Y.; Zheng, J.; Liu, H.; Bi, Z.; Xu, F.; et al. Resequencing of 145 landmark cultivars reveals asymmetric sub-genome selection and strong founder genotype effects on wheat breeding in China. Mol. Plant 2020, 13, 1733–1751. [Google Scholar] [CrossRef]
- Yin, D.; Ji, C.; Song, Q.; Zhang, W.; Zhang, X.; Zhao, K.; Chen, C.Y.; Wang, C.; He, G.; Liang, Z.; et al. Comparison of Arachis monticola with diploid and cultivated tetraploid genomes reveals asymmetric subgenome evolution and improvement of peanut. Adv. Sci. 2020, 7, 1901672. [Google Scholar] [CrossRef]
- Wang, M.; Tu, L.; Lin, M.; Lin, Z.; Wang, P.; Yang, Q.; Ye, Z.; Shen, C.; Li, J.; Zhang, L.; et al. Asymmetric subgenome selection and cis-regulatory divergence during cotton domestication. Nat. Genet. 2017, 49, 579–587. [Google Scholar] [CrossRef]
- Soltis, P.S.; Soltis, D.E. Ancient WGD events as drivers of key innovations in angiosperms. Curr. Opin. Plant Biol. 2016, 30, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y. Double the genome, double the fun: Genome duplications in angiosperms. Mol. Plant 2018, 11, 357–358. [Google Scholar] [CrossRef]
- Innan, H. Population genetic models of duplicated genes. Genetica 2009, 137, 19–37. [Google Scholar] [CrossRef]
- Mao, Y.; Satoh, N. A likely ancient genome duplication in the speciose reef-building coral genus, Acropora. iScience 2019, 13, 20–32. [Google Scholar] [CrossRef]
- Conant, G.C.; Birchler, J.A.; Pires, J.C. Dosage, duplication, and diploidization: Clarifying the interplay of multiple models for duplicate gene evolution over time. Curr. Opin. Plant Biol. 2014, 19, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Sonnhammer, E.L.; Östlund, G. InParanoid 8: Orthology analysis between 273 proteomes, mostly eukaryotic. Nucleic Acids Res. 2015, 43, D234–D239. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Villanueva-Cañas, J.L.; Laurie, S.; Albà, M.M. Improving genome-wide scans of positive selection by using protein isoforms of similar length. Genome Biol. Evol. 2013, 5, 457–467. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Jin, J.; Tian, F.; Yang, D.C.; Meng, Y.Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017, 45, D1040–D1045. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Sanderson, M.J. r8s: Inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 2003, 19, 301–302. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A resource for timelines, timetrees, and divergence times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- De Bie, T.; Cristianini, N.; Demuth, J.P.; Hahn, M.W. CAFE: A computational tool for the study of gene family evolution. Bioinformatics 2006, 22, 1269–1271. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Yanai, I.; Benjamin, H.; Shmoish, M.; Chalifa-Caspi, V.; Shklar, M.; Ophir, R.; Bar-Even, A.; Horn-Saban, S.; Safran, M.; Domany, E.; et al. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics 2005, 21, 650–659. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, L.; Cheng, H.; Yang, Z.; Xia, C.; Zhang, L.; Kong, X. Comparative Analysis Reveals Different Evolutionary Fates and Biological Functions in Wheat Duplicated Genes (Triticum aestivum L.). Plants 2023, 12, 3021. https://doi.org/10.3390/plants12173021
Cui L, Cheng H, Yang Z, Xia C, Zhang L, Kong X. Comparative Analysis Reveals Different Evolutionary Fates and Biological Functions in Wheat Duplicated Genes (Triticum aestivum L.). Plants. 2023; 12(17):3021. https://doi.org/10.3390/plants12173021
Chicago/Turabian StyleCui, Licao, Hao Cheng, Zhe Yang, Chuan Xia, Lichao Zhang, and Xiuying Kong. 2023. "Comparative Analysis Reveals Different Evolutionary Fates and Biological Functions in Wheat Duplicated Genes (Triticum aestivum L.)" Plants 12, no. 17: 3021. https://doi.org/10.3390/plants12173021