
Small RNA-Seq to Unveil the miRNA Expression Patterns and Identify the Target Genes in Panax ginseng


Abstract
:1. Introduction
2. Results
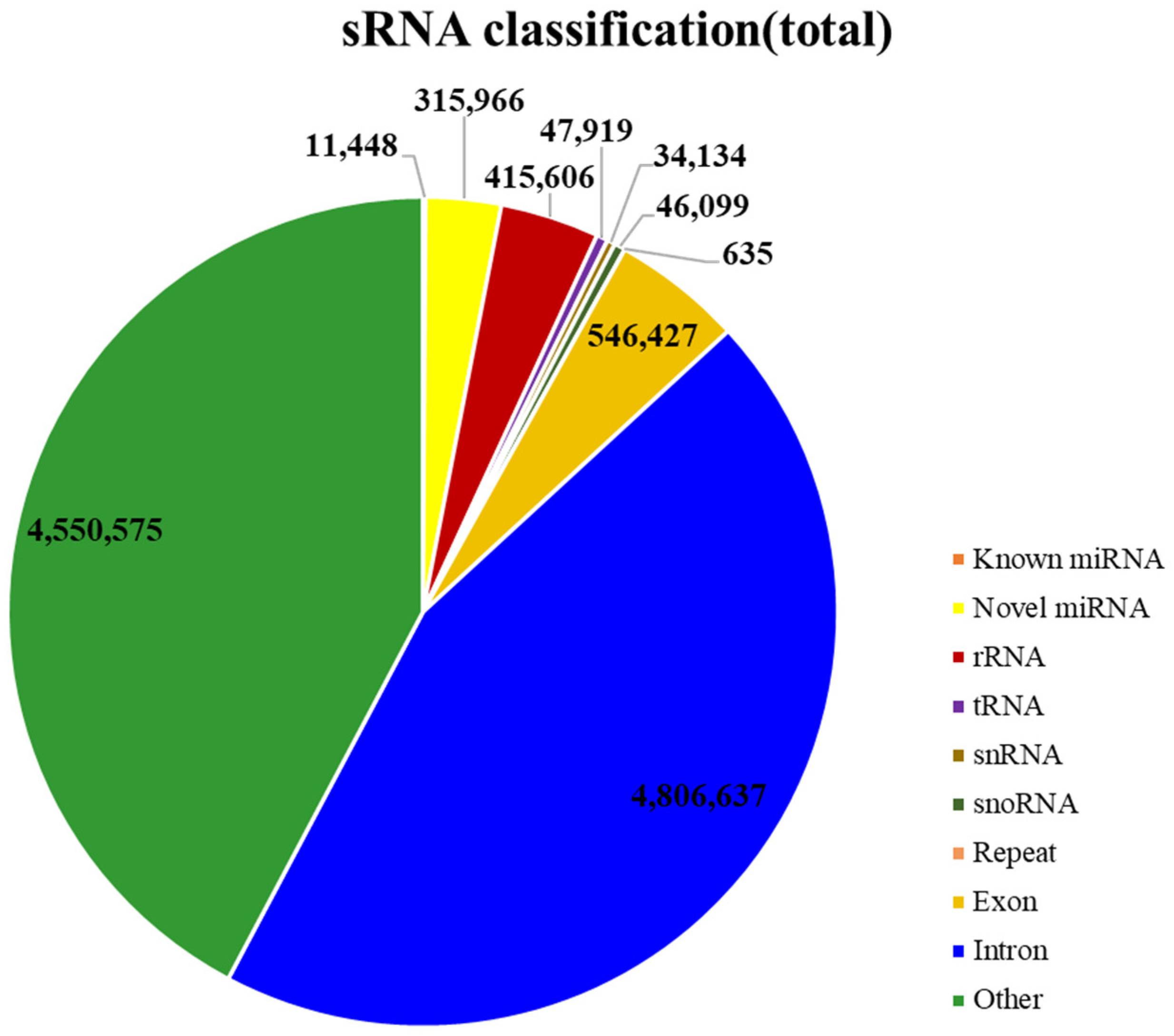
2.1. Features of miRNA Population in Panax ginseng
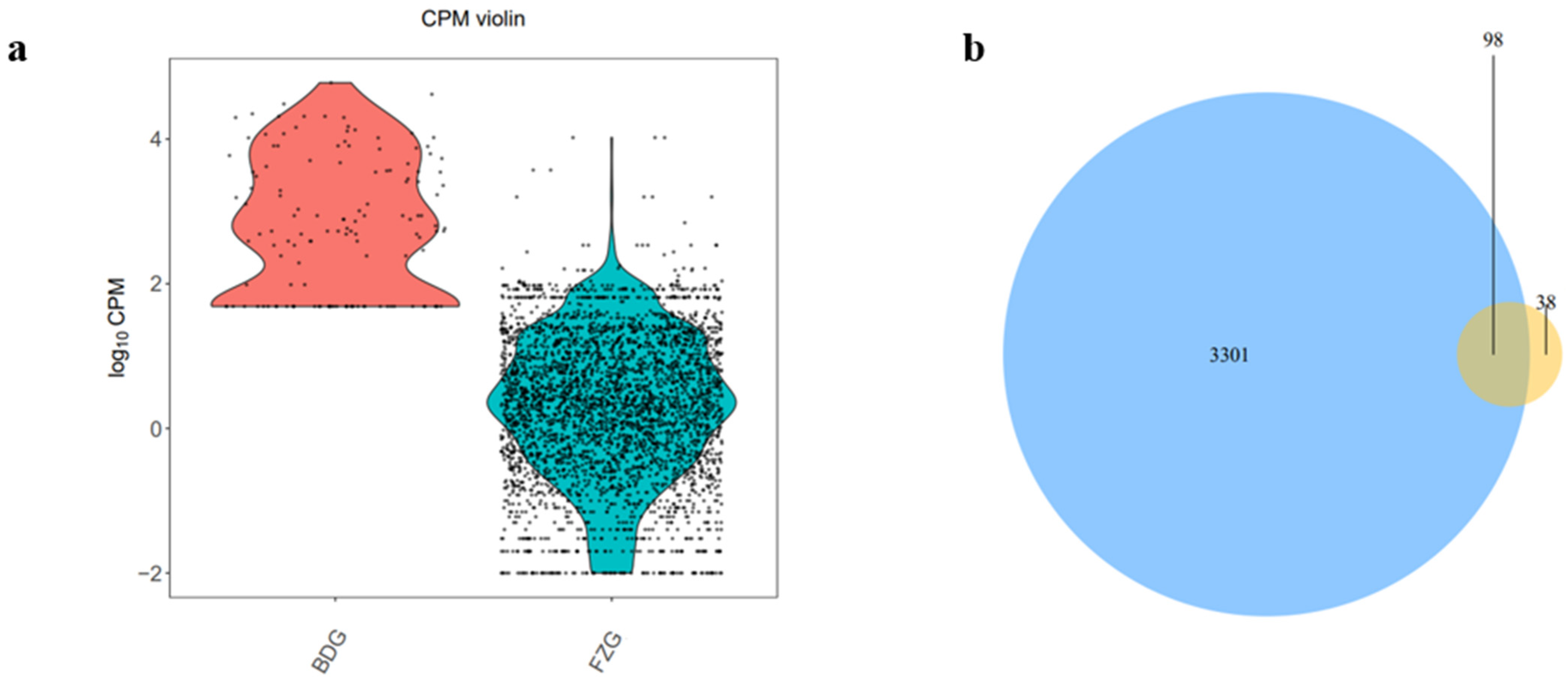
2.2. Identification of Known and Novel miRNAs
2.3. miRNAs Differentially Expressed
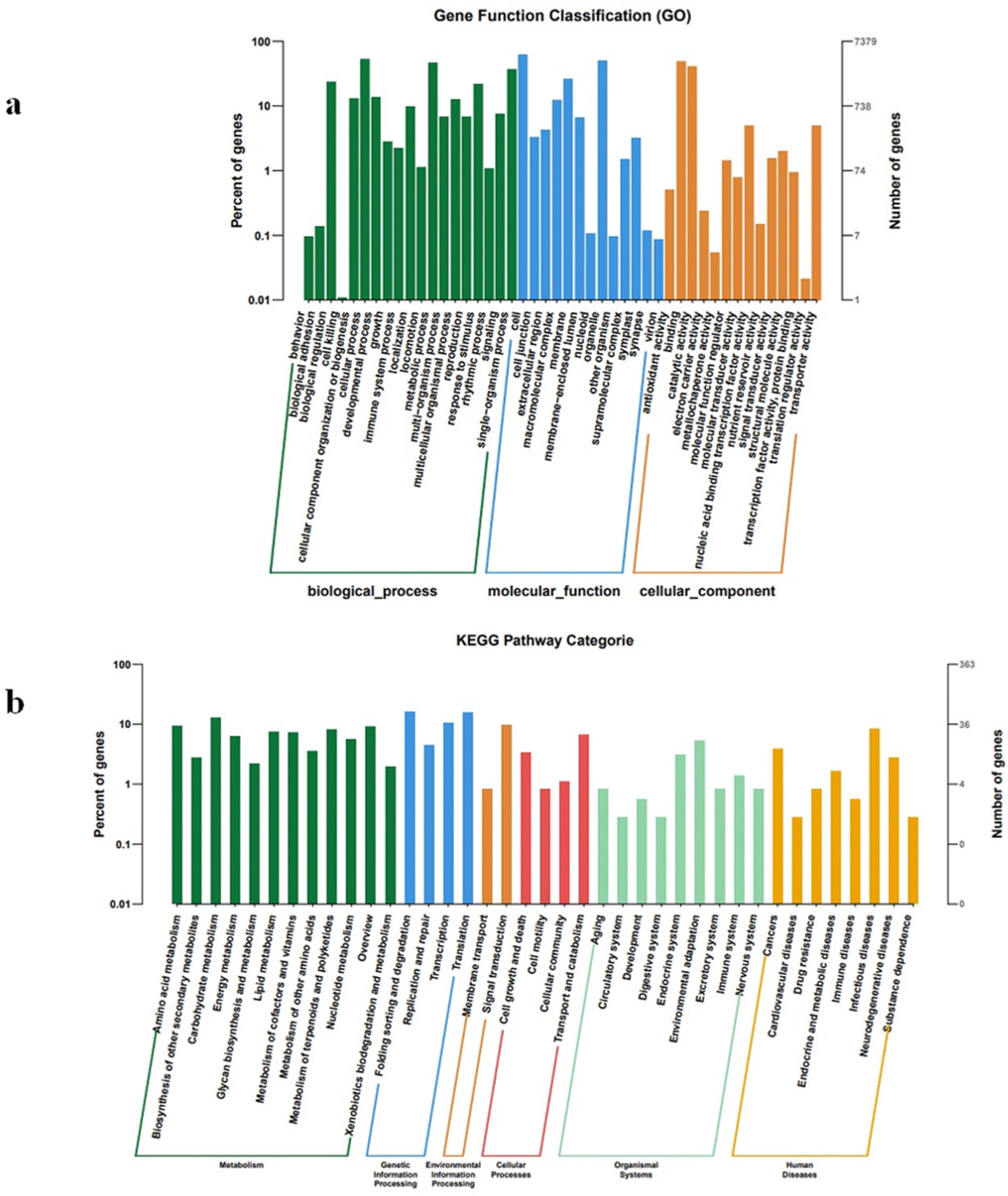
2.4. Target Gene Prediction and Functional Annotation
2.5. qRT-PCR Verification and Analysis
3. Discussion
4. Materials and Methods
4.1. Plant Materials in this Study
4.2. Library Construction and Sequencing of RNA in Ginseng
4.3. Quality Control of Sequencing Data
4.4. Identification of Known and Novel miRNAs in Ginseng Adventurous Roots and Ginseng Hairy Roots
4.5. The miRNA Expression Pattern Analysis
4.6. The miRNA Target Gene Prediction
4.7. Functional Annotation of miRNA Target Genes
4.8. The miRNA Genes Expression Analysis Using the qRT-PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Rubio-Somoza, I.; Weigel, D. MicroRNA Networks and Developmental Plasticity in Plants. Trends Plant Sci. 2011, 16, 258–264. [Google Scholar] [CrossRef]
- Flynt, A.S.; Lai, E.C. Biological Principles of MicroRNA-Mediated Regulation: Shared Themes Amid Diversity. Nat. Rev. Genet. 2008, 9, 831–842. [Google Scholar] [CrossRef]
- Bernardo, B.C.; Ooi, J.Y.; Lin, R.C.; McMullen, J.R. miRNA Therapeutics: A New Class of Drugs with Potential Therapeutic Applications in the Heart. Future Med. Chem. 2015, 7, 1771–1792. [Google Scholar] [CrossRef]
- Chen, L.; Heikkinen, L.; Wang, C.; Yang, Y.; Sun, H.; Wong, G. Trends in the Development of miRNA Bioinformatics Tools. Brief Bioinform. 2019, 20, 1836–1852. [Google Scholar] [CrossRef]
- Sun, M.; Xu, S.; Mei, Y.; Li, J.; Gu, Y.; Zhang, W.; Wang, J. MicroRNAs in Medicinal Plants. Int. J. Mol. Sci. 2022, 23, 10477. [Google Scholar] [CrossRef]
- Ma, X.; Tang, Z.; Qin, J.; Meng, Y. The Use of High-Throughput Sequencing Methods for Plant MicroRNA Research. RNA Biol. 2015, 12, 709–719. [Google Scholar] [CrossRef]
- Verma, P.; Singh, N.; Khan, S.A.; Mathur, A.K.; Sharma, A.; Jamal, F. TIAs Pathway Genes and Associated miRNA Identification in Vinca Minor: Supporting Aspidosperma and Eburnamine Alkaloids Linkage Via Transcriptomic Analysis. Physiol. Mol. Biol. Plants 2020, 26, 1695–1711. [Google Scholar] [CrossRef]
- Wang, L.; Du, H.; Wuyun, T.N. Genome-Wide Identification of MicroRNAs and Their Targets in the Leaves and Fruits of Eucommia ulmoides Using High-Throughput Sequencing. Front. Plant Sci. 2016, 7, 1632. [Google Scholar] [CrossRef]
- Singh, N.; Srivastava, S.; Shasany, A.K.; Sharma, A. Identification of miRNAs and Their Targets Involved in the Secondary Metabolic Pathways of Mentha spp. Comput. Biol. Chem. 2016, 64, 154–162. [Google Scholar] [CrossRef]
- Petijová, L.; Jurčacková, Z.; Čellárová, E. Computational Screening of miRNAs and Their Targets in Leaves of Hypericum spp. by Transcriptome-Mining: A Pilot Study. Planta 2020, 251, 49. [Google Scholar] [CrossRef]
- Abla, M.; Sun, H.; Li, Z.; Wei, C.; Gao, F.; Zhou, Y.; Feng, J. Identification of miRNAs and Their Response to Cold Stress in Astragalus Membranaceus. Biomolecules 2019, 9, 182. [Google Scholar] [CrossRef]
- Khan, S.; Ali, A.; Saifi, M.; Saxena, P.; Ahlawat, S.; Abdin, M.Z. Identification and the Potential Involvement of miRNAs in the Regulation of Artemisinin Biosynthesis in A. annua. Sci. Rep. 2020, 10, 13614. [Google Scholar] [CrossRef]
- Ye, J.; Zhang, X.; Tan, J.; Xu, F.; Cheng, S.; Chen, Z.; Zhang, W.; Liao, Y. Global Identification of Ginkgo Biloba microRNAs and Insight into Their Role in Metabolism Regulatory Network of Terpene Trilactones by High-Throughput Sequencing and Degradome Analysis. Ind. Crop. Prod. 2020, 148, 112289. [Google Scholar] [CrossRef]
- Zeng, S.; Liu, Y.; Pan, L.; Hayward, A.; Wang, Y. Identification and Characterization of miRNAs in Ripening Fruit of Lycium barbarum L. Using High-Throughput Sequencing. Front. Plant Sci. 2015, 6, 778. [Google Scholar] [CrossRef]
- Shen, E.M.; Singh, S.K.; Ghosh, J.S.; Patra, B.; Paul, P.; Yuan, L.; Pattanaik, S. The miRNAome of Catharanthus roseus: Identification, Expression Analysis, and Potential Roles of microRNAs in Regulation of Terpenoid Indole Alkaloid Biosynthesis. Sci. Rep. 2017, 7, 43027. [Google Scholar] [CrossRef]
- Tian, M.; Li, L.N.; Zheng, R.R.; Yang, L.; Wang, Z.T. Advances on Hormone-like Activity of Panax ginseng and Ginsenosides. Chin. J. Nat. Med. 2020, 18, 526–535. [Google Scholar] [CrossRef]
- Fang, X.; Wang, M.; Zhou, X.; Wang, H.; Wang, H.; Xiao, H. Effects of Growth Years on Ginsenoside Biosynthesis of Wild Ginseng and Cultivated Ginseng. BMC Genom. 2022, 23, 325. [Google Scholar] [CrossRef]
- Baque, M.A.; Moh, S.H.; Lee, E.J.; Zhong, J.J.; Paek, K.Y. Production of Biomass and Useful Compounds from Adventitious Roots of High-Value Added Medicinal Plants Using Bioreactor. Biotechnol. Adv. 2012, 30, 1255–1267. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.J.; Li, C.Y.; Qian, Y.C.; Luo, X.P.; Zhang, X.; Wang, X.S.; Kang, B.Y. Induction of Hairy Roots of Panax ginseng and Studies on Suitable Culture Condition of Ginseng Hairy Roots. Sheng Wu Gong Cheng Xue Bao 2004, 20, 215–220. (In Chinese) [Google Scholar]
- Shanks, J.V.; Morgan, J. Plant ‘hairy root’ culture. Curr. Opin. Biotechnol. 1999, 10, 151–155. [Google Scholar] [CrossRef]
- Mallol, A.; Cusidó, R.M.; Palazón, J.; Bonfill, M.; Morales, C.; Piñol, M.T. Ginsenoside Production in Different Phenotypes of Panax ginseng Transformed Roots. Phytochemistry 2001, 57, 365–371. [Google Scholar] [CrossRef]
- Choi, P.S.; Kim, Y.D.; Choi, K.M.; Chung, H.J.; Choi, D.W.; Liu, J.R. Plant Regeneration from Hairy-root Cultures Transformed by Infection with Agrobacterium rhizogenes in Catharanthus roseus. Plant Cell Rep. 2004, 22, 828–831. [Google Scholar] [CrossRef]
- Wu, B.; Wang, M.; Ma, Y.; Yuan, L.; Lu, S. High-throughput Sequencing and Characterization of the Small RNA Transcriptome Reveal Features of Novel and Conserved microRNAs in Panax ginseng. PLoS ONE 2012, 7, e44385. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, Y.; Peng, M.; Yang, C.; Yang, Z.; Gong, M.; Yin, Y.; Zeng, Y. Identification of microRNA and Analysis of Target Genes in Panax ginseng. Chin. Herb. Med. 2023, 15, 69–75. [Google Scholar] [CrossRef]
- Wang, Y.; Peng, M.; Chen, Y.; Wang, W.; He, Z.; Yang, Z.; Lin, Z.; Gong, M.; Yin, Y.; Zeng, Y. Analysis of Panax ginseng miRNAs and Their Target Prediction Based on High-Throughput Sequencing. Planta Med. 2019, 85, 1168–1176. [Google Scholar] [CrossRef]
- Komatsu, S.; Kitai, H.; Suzuki, H.I. Network Regulation of microRNA Biogenesis and Target Interaction. Cells 2023, 12, 306. [Google Scholar] [CrossRef]
- Kuang, Z.; Zhao, Y.; Yang, X. Plant MicroRNA Identification and Annotation Using Deep Sequencing Data. Methods Mol. Biol. 2023, 2595, 239–250. [Google Scholar]
- Li, M.Y.; Wang, F.; Xu, Z.S.; Jiang, Q.; Ma, J.; Tan, G.F.; Xiong, A.S. High throughput Sequencing of Two Celery Varieties Small RNAs Identifies microRNAs Involved in Temperature Stress Response. BMC Genom. 2014, 15, 242. [Google Scholar] [CrossRef]
- Waheed, S.; Liang, F.; Zhang, M.; He, D.; Zeng, L. High-Throughput Sequencing Reveals Novel microRNAs Involved in the Continuous Flowering Trait of Longan (Dimocarpus longan Lour.). Int. J. Mol. Sci. 2022, 23, 15565. [Google Scholar] [CrossRef]
- Liu, N.; Jiang, Y.; Zhu, T.; Li, Z.; Sui, S. Small RNA and Degradome Sequencing in Floral Bud Reveal Roles of miRNAs in Dormancy Release of Chimonanthus praecox. Int. J. Mol. Sci. 2023, 24, 4210. [Google Scholar] [CrossRef]
- Qiu, D.; Pan, X.; Wilson, I.W.; Li, F.; Liu, M.; Teng, W.; Zhang, B. High Throughput Sequencing Technology Reveals that the Taxoid Elicitor Methyl Jasmonate Regulates microRNA Expression in Chinese yew (Taxus chinensis). Gene 2009, 436, 37–44. [Google Scholar] [CrossRef]
- Sunkar, R.; Zhou, X.; Zheng, Y.; Zhang, W.; Zhu, J.K. Identification of Novel and Candidate miRNAs in Rice by High Throughput Sequencing. BMC Plant Biol. 2008, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Mi, S.; Cai, T.; Hu, Y.; Chen, Y.; Hodges, E.; Ni, F.; Wu, L.; Li, S.; Zhou, H.; Long, C.; et al. Sorting of Small RNAs into Arabidopsis Argonaute Complexes is Directed by the 5′ Terminal Nucleotide. Cell 2008, 133, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Somoza, I.; Cuperus, J.T.; Weigel, D.; Carrington, J.C. Regulation and Functional Specialization of Small RNA-target Nodes During Plant Development. Curr. Opin. Plant Biol. 2009, 12, 622–627. [Google Scholar] [CrossRef]
- Czech, B.; Hannon, G.J. Small RNA sorting: Matchmaking for Argonautes. Nat. Rev. Genet 2011, 12, 19–31. [Google Scholar] [CrossRef]
- Qiang, B.; Miao, J.; Phillips, N.; Wei, K.; Gao, Y. Recent Advances in the Tissue Culture of American Ginseng (Panax quinquefolius). Chem. Biodivers 2020, 17, e2000366. [Google Scholar] [CrossRef]
- Stepanova, A.Y.; Malunova, M.V.; Gladkov, E.A.; Evsyukov, S.V.; Tereshonok, D.V.; Solov’Eva, A.I. Collection of Hairy Roots as a Basis for Fundamental and Applied Research. Molecules 2022, 27, 8040. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, S.; Verma, S.; Chakraborty, A.; Bhatia, S. Identification and Molecular Characterization of miRNAs and Their Target Genes Associated with Seed Development Through Small RNA Sequencing in Chickpea. Funct. Integr. Genomics 2021, 21, 283–298. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, K.; Chai, Z.; Song, Y.; Wang, X.; Duan, Y.; Zhang, M. Identification of miRNAs and Target Genes at Key Stages of Sexual Differentiation in Androdioecious Osmanthus fragrans. Int. J. Mol. Sci. 2022, 23, 10386. [Google Scholar] [CrossRef]
- Juarez, M.T.; Twigg, R.W.; Timmermans, M.C. Specification of Adaxial Cell Fate During Maize Leaf Development. Development 2004, 131, 4533–4544. [Google Scholar] [CrossRef]
- Ding, N.; Huertas, R.; Torres-Jerez, I.; Liu, W.; Watson, B.; Scheible, W.R.; Udvardi, M. Transcriptional, Metabolic, Physiological and Developmental Responses of Switchgrass to Phosphorus Limitation. Plant Cell Environ. 2021, 44, 186–202. [Google Scholar] [CrossRef]
- Liu, M.; Thomas, P.D. GO Functional Similarity Clustering Depends on Similarity Measure, Clustering Method, and Annotation Completeness. BMC Bioinform. 2019, 20, 155. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chen, Z.; Zhang, M.; Jia, S. KEGG_Extractor: An Effective Extraction Tool for KEGG Orthologs. Genes 2023, 14, 386. [Google Scholar] [CrossRef] [PubMed]
- Everaert, C.; Verwilt, J.; Verniers, K.; Vandamme, N.; Marcos, R.A.; Vandesompele, J.; Mestdagh, P. Blocking Abundant RNA Transcripts by High-Affinity Oligonucleotides during Transcriptome Library Preparation. Biol. Proced. Online 2023, 25, 7. [Google Scholar] [CrossRef]
- Davies, J.; Denyer, T.; Hadfield, J. Bioanalyzer Chips Can be Used Interchangeably for Many Analyses of DNA or RNA. Biotechniques 2016, 60, 197–199. [Google Scholar] [CrossRef]
- Jeon, S.A.; Park, J.L.; Park, S.J.; Kim, J.H.; Goh, S.H.; Han, J.Y.; Kim, S.Y. Comparison between MGI and Illumina Sequencing Platforms for Whole Genome Sequencing. Genes Genom. 2021, 43, 713–724. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B. Aligning Short Sequencing Reads with Bowtie. Curr. Protoc. Bioinform. 2010, 11, 11–17. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA Sequences to Function. Nucleic. Acids. Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- Mackowiak, S.D. Identification of Novel and Known miRNAs in Deep-sequencing Data with miRDeep2. Curr. Protoc. Bioinform. 2011, 12, 10–12. [Google Scholar] [CrossRef]
- Hu, K. Become Competent within One Day in Generating Boxplots and Violin Plots for a Novice without Prior R Experience. Methods Protoc. 2020, 3, 64. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.H.; Yu, G.; Cai, P. ggVennDiagram: An Intuitive, Easy-to-Use, and Highly Customizable R Package to Generate Venn Diagram. Front. Genet. 2021, 12, 706907. [Google Scholar] [CrossRef]
- Bo, X.; Wang, S. TargetFinder: A Software for Antisense Oligonucleotide Target Site Selection Based on MAST and Secondary Structures of Target mRNA. Bioinformatics 2005, 21, 1401–1402. [Google Scholar] [CrossRef] [PubMed]
- Fahlgren, N.; Carrington, J.C. miRNA Target Prediction in Plants. Methods Mol. Biol. 2010, 592, 51–57. [Google Scholar] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A Universal Tool for Annotation, Visualization and Analysis in Functional Genomics Research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Ye, J.; Zhang, Y.; Cui, H.; Liu, J.; Wu, Y.; Cheng, Y.; Xu, H.; Huang, X.; Li, S.; Zhou, A.; et al. WEGO 2.0, A Web Tool for Analyzing and Plotting GO Annotations. Nucleic. Acids. Res. 2018, 46, W71–W75. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for Taxonomy-Based Analysis of Pathways and Genomes. Nucleic. Acids. Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A New Mathematical Model for Relative Quantification in Real-Time RT-PCR. Nucleic. Acids. Res. 2001, 29, e45. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA and Reference Gene | Stem-Loop Primers | Forward Primer | Reverse Primer |
|---|---|---|---|
| miRNA166 | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACGGGGAA | GCGTCGGACCAGGCTTCA | AGTGCAGGGTCCGAGGTATT |
| miRNA396 | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACCAGTTC | CGCGTTCCACAGCTTTCTT | AGTGCAGGGTCCGAGGTATT |
| miRNA156 | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACGTGCTC | CGCGCGTGACAGAAGAGAGT | AGTGCAGGGTCCGAGGTATT |
| miRNA399 | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAAGGGC | GCGCGCCAAAGGAGAGTT | AGTGCAGGGTCCGAGGTATT |
| miRNA482 | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACGGAATG | GCGTCTTGCCAATTCCTCC | AGTGCAGGGTCCGAGGTATT |
| miRNA157 | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACGTGCTC | CGCGCGTTGACAGAAGATAGA | AGTGCAGGGTCCGAGGTATT |
| miRNA172 | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACATGCAG | GCGCGAGAATCTTGATGATG | AGTGCAGGGTCCGAGGTATT |
| U6 | TTGTCTGACGACGAGAGAGAGCACG | GTGCAGGGTCCGAGGTTTGGACCATTTCTAGAT | TTGTCTGACGACGAGAGAGAGCACG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, C.; Jiang, Y.; Yun, Z.; Zhang, K.; Zhao, M.; Wang, Y.; Zhang, M.; Tian, Z.; Wang, K. Small RNA-Seq to Unveil the miRNA Expression Patterns and Identify the Target Genes in Panax ginseng. Plants 2023, 12, 3070. https://doi.org/10.3390/plants12173070
Liu C, Jiang Y, Yun Z, Zhang K, Zhao M, Wang Y, Zhang M, Tian Z, Wang K. Small RNA-Seq to Unveil the miRNA Expression Patterns and Identify the Target Genes in Panax ginseng. Plants. 2023; 12(17):3070. https://doi.org/10.3390/plants12173070
Chicago/Turabian StyleLiu, Chang, Yang Jiang, Ziyi Yun, Kexin Zhang, Mingzhu Zhao, Yi Wang, Meiping Zhang, Zhuo Tian, and Kangyu Wang. 2023. "Small RNA-Seq to Unveil the miRNA Expression Patterns and Identify the Target Genes in Panax ginseng" Plants 12, no. 17: 3070. https://doi.org/10.3390/plants12173070