1. Introduction
Arnica montana has been used for several hundred years for the treatment of various ailments, including contusion, wounds, rheumatism and inflammation [
1]. Numerous studies have shown the great medicinal value of Arnica flowers (Arnicae flos), most often applied as tincture or ointment on affected skin areas [
1,
2,
3,
4,
5,
6].
A. montana tincture is mainly produced from Arnicae flos by ethanolic extraction. The tincture is desiccated by evaporation, and the extract (3–30%) is incorporated into a variety of herbal preparations or pharmaceutical products [
1].
A. montana contains a high number of biologically active substances, including sesquiterpene lactones, flavonoids and phenolic acids [
1,
5,
6,
7]. While sesquiterpene lactones mediate the anti-inflammatory properties of
A. montana, flavonoids and phenolic acids exert significant antioxidant and antimicrobial activities [
1,
2,
3,
5,
8,
9]. The content of bioactive substances varies in different parts of the plant, but also with geographic location, climate, altitude and maturity of flower head (from bud to withered flower) [
1,
6,
7]. Flower heads are particularly rich in sesquiterpene lactones, such as helenalin (up to 1% dry weight) [
1,
2], while roots contain high levels of thymol, known for its antioxidant, antimicrobial and anti-inflammatory properties [
1,
7,
10,
11,
12,
13,
14,
15].
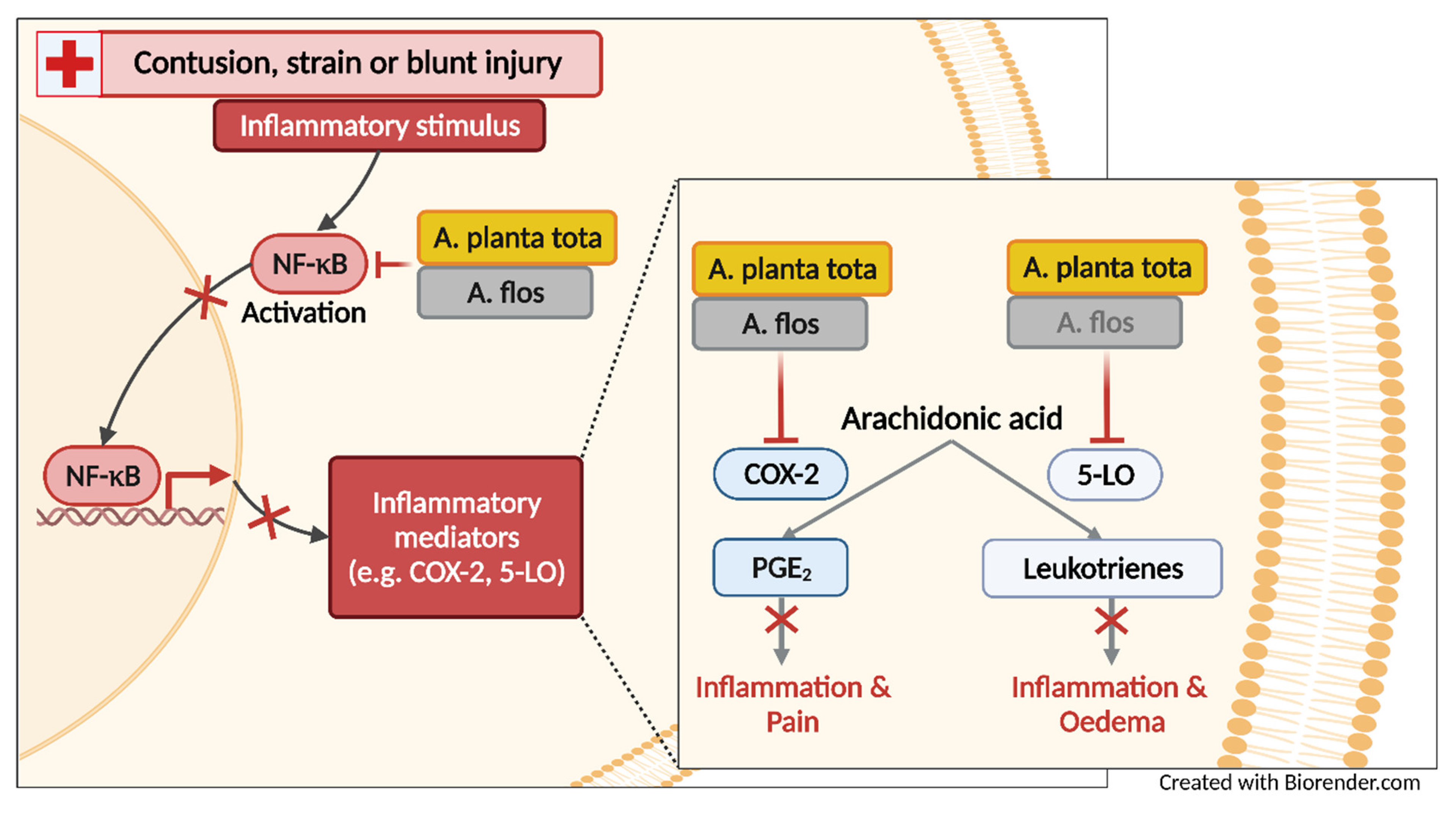
The anti-inflammatory activity of Arnicae flos, notably its ability to inhibit NF-κB signaling and its downstream 5-lipoxygenase (5-LO)- and cyclooxygenase-2 (COX-2)-mediated pathways, is well documented [
1,
2,
8,
9,
16]. NF-κB is an important mediator of immune response and inflammation [
17]. It regulates the transcription of proinflammatory genes such as arachidonate 5-lipoxygenase (ALOX5) and prostaglandin-endoperoxide synthase 2 (PTGS2). ALOX5 and PTGS2 encode 5-LO and COX-2, which are key enzymes in the biosynthesis of eicosanoids, namely leukotrienes and prostaglandins, respectively, thereby promoting inflammation, pain and fever [
16,
18]. Both leukotrienes and prostaglandins are directly involved in the processes leading to the early signs of inflammation, including redness, swelling (oedema) and pain [
16,
18,
19,
20,
21,
22,
23,
24]. While most studies have investigated the anti-inflammatory activities of Arnicae flos, very little is known of the anti-inflammatory properties of Arnicae planta tota [
1,
25,
26]. We hypothesized that, due to an increased and broader content of bioactive substances, Arnicae planta tota extracts might exert greater anti-inflammatory activities than Arnicae flos extracts.
In this study, we compared the ability of Arnicae planta tota and Arnicae flos to inhibit the NF-κB signaling pathway and its messengers (5-LO, COX-2), using in vitro enzymatic assays, cell-based assays and an in vivo mouse model of inflammation. We showed that Arnicae planta tota presented superior anti-inflammatory activity in these assays, compared to Arnicae flos. Our data suggest that products containing Arnicae planta tota might be more effective in alleviating inflammation symptoms than those based on Arnicae flos only.
3. Discussion
While the anti-inflammatory activity of Arnicae flos is well described in the literature [
1,
2,
8,
9,
16], data on Arnicae planta tota are scarce [
25,
26]. This study compared the anti-inflammatory properties of Arnicae planta tota to those of Arnicae flos, using a variety of
in vitro and
in vivo assays.
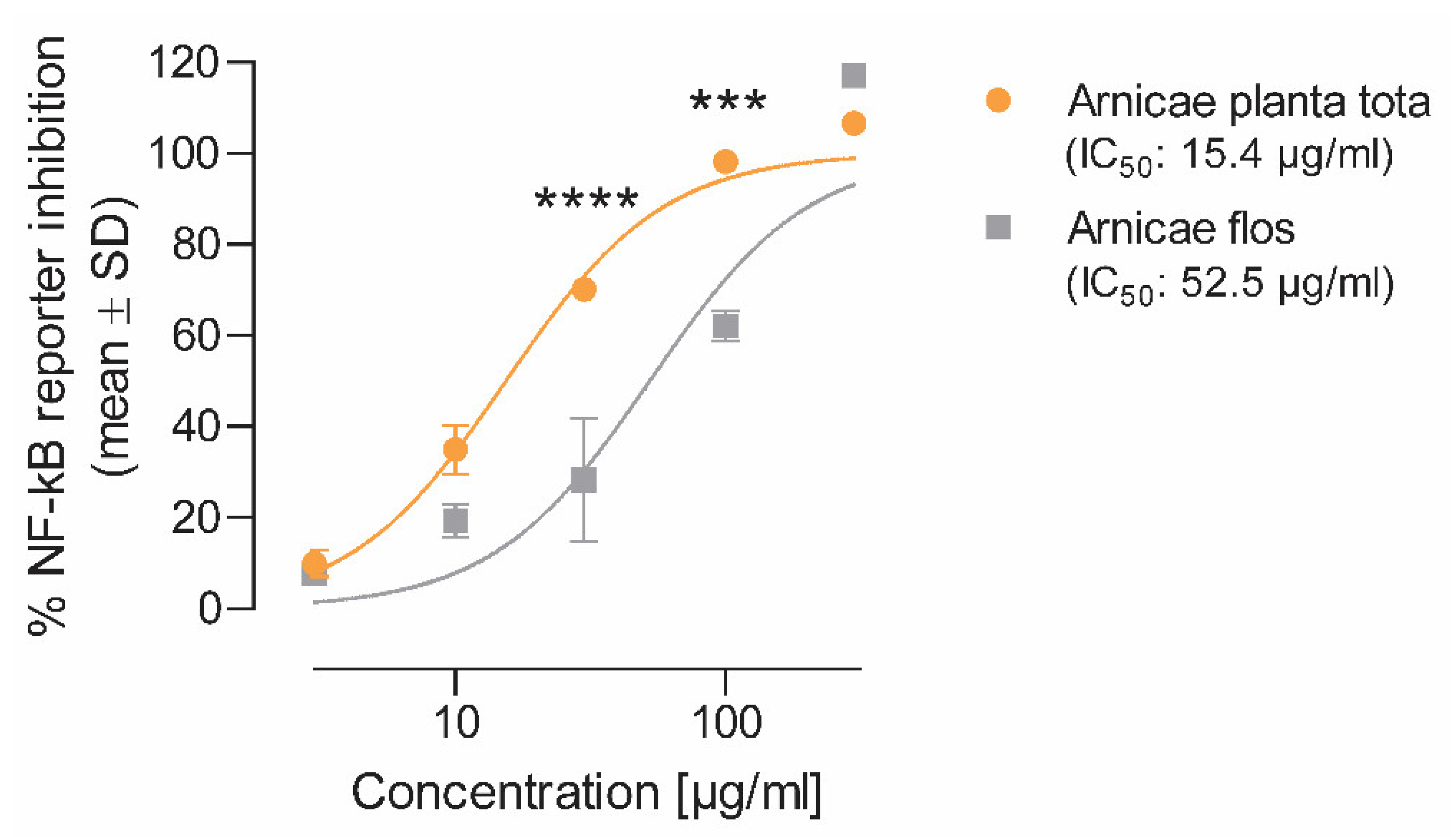
We showed that in most models of inflammation investigated, both Arnicae planta tota and Arnicae flos exerted inhibitory functions toward tested pro-inflammatory targets, and that Arnicae planta tota was a stronger inhibitor compared to Arnicae flos (lower IC
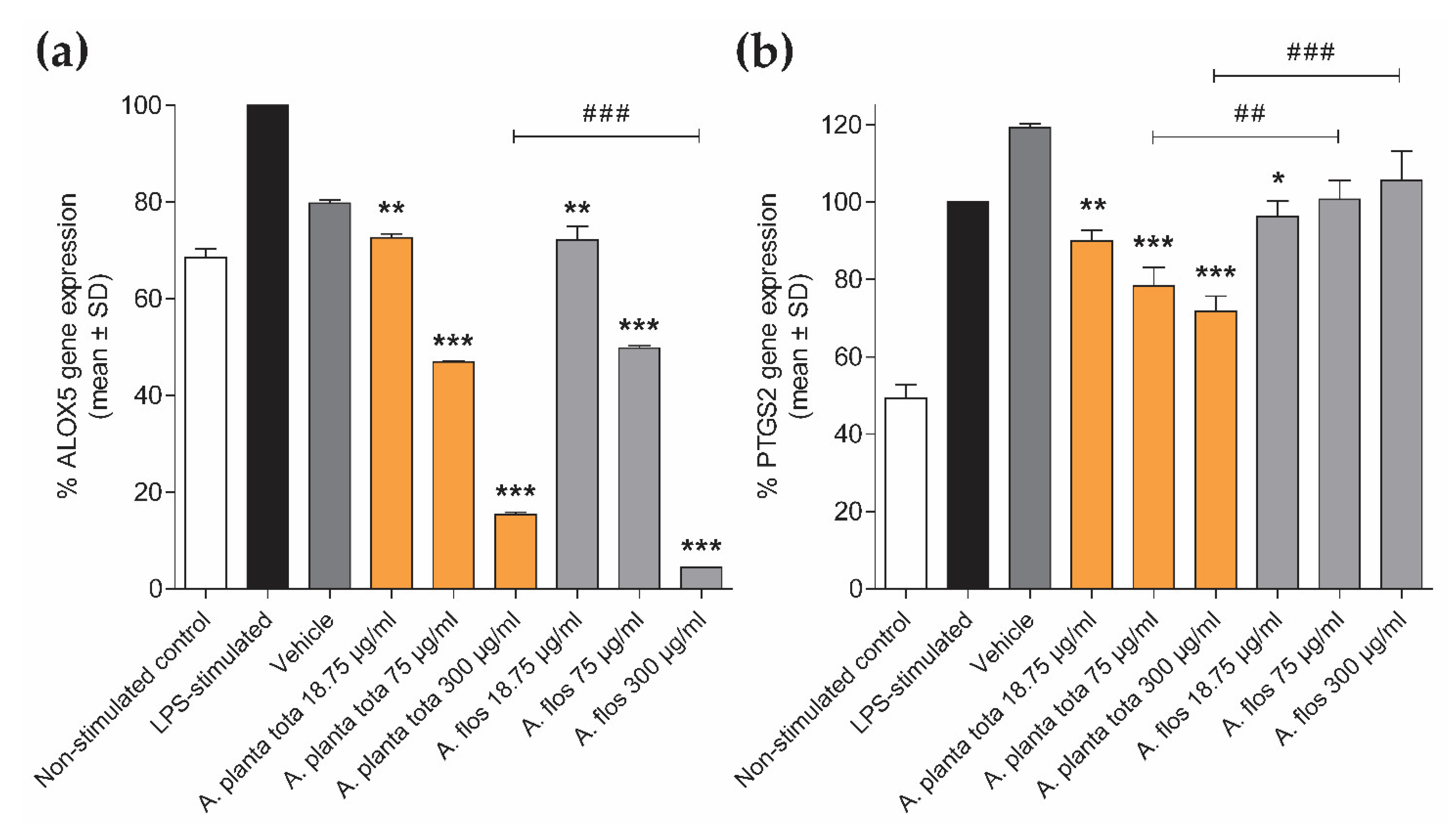
50). Arnicae planta tota inhibited NF-κB activation in a reporter assay and the LPS-induced expression of the NF-κB target genes ALOX5 and PTGS2 in differentiated THP-1 cells (
Figure 7). The enzymatic activity of the proteins encoded by ALOX5 and PTGS2 (5-LO and COX-2, respectively,) was also directly inhibited by Arnicae planta tota in a cell-free assay, demonstrating a second level of inhibition downstream of NF-κB activation (
Figure 7). Inhibition of 5-LO and COX-2 enzymatic activity was confirmed in human primary cells, as evidenced by the reduced release of the proinflammatory arachidonic acid metabolites leukotrienes and prostaglandins, in cells pretreated with Arnicae planta tota (
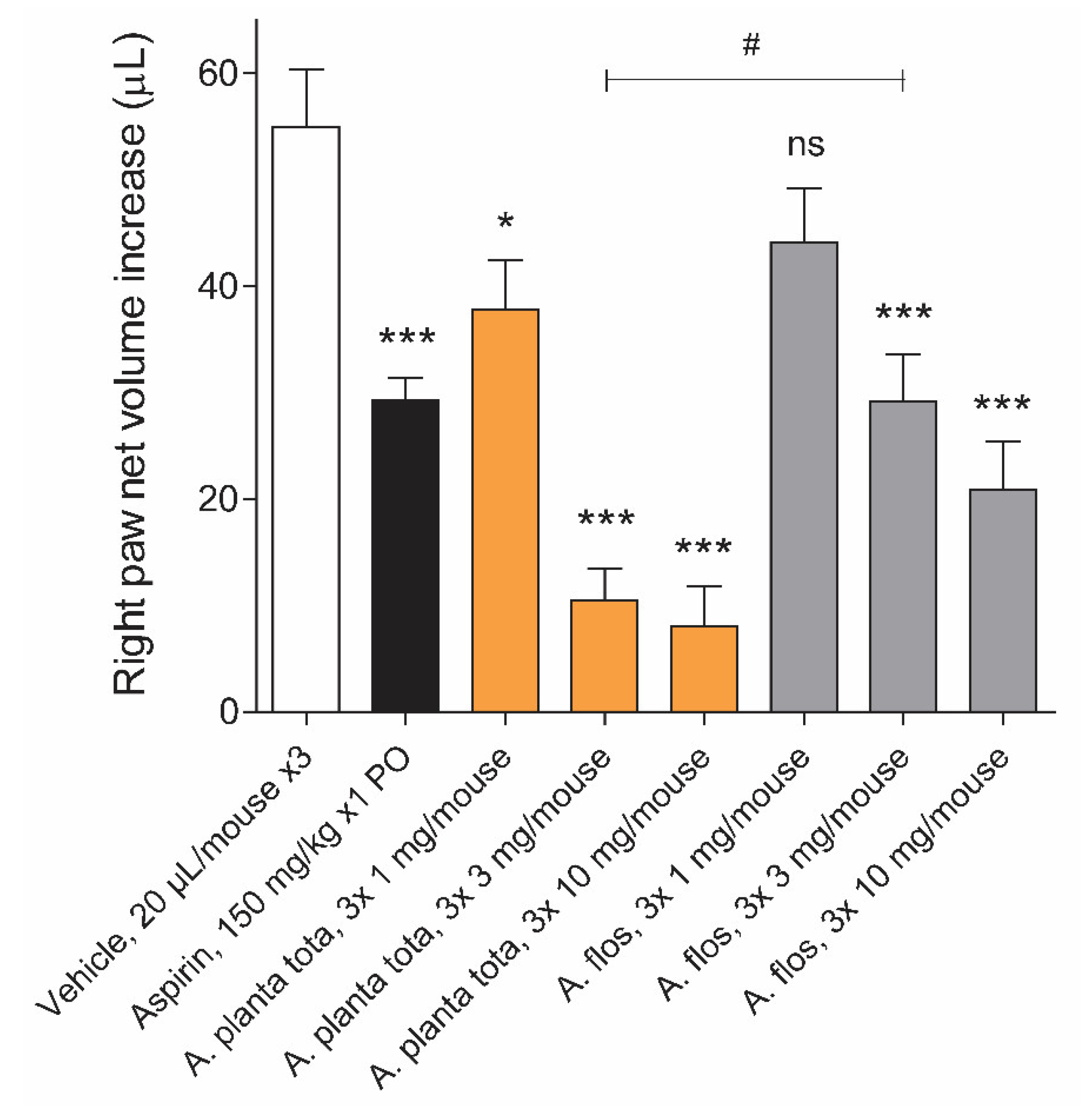
Figure 7). Finally, topical application of Arnicae planta tota (and Arnicae flos) partly prevented foot swelling in a mouse model of carrageenan-induced paw oedema (
Figure 7). The anti-oedema activity of Arnica is in line with its inhibitory effect on the production of leukotrienes and prostaglandins, which are both directly involved in oedema formation [
16,
18,
19,
20,
23]. Our results therefore confirm the anti-inflammatory activities attributed to Arnicae flos [
1,
2,
8,
9], while demonstrating—to the best of our knowledge, for the first time—superior anti-inflammatory activities for Arnicae planta tota due to interference with the NF-κB—eicosanoid inflammation pathway. The superior anti-inflammatory properties of Arnicae planta tota are likely due to the increased and broader content of biologically active substances in the whole Arnica plant, compared to the flowers alone [
1,
2,
3,
4,
5,
6,
7,
8,
9]. The stronger inhibitory effect of Arnicae planta tota on leukotriene release in PMNL, and its stronger anti-oedema effect in the carrageenan-induced inflammation mouse model, suggest that leukotriene-mediated inflammation might be a major target of the anti-inflammatory activity of Arnicae planta tota, compared to Arnicae flos.
In addition to anti-inflammatory activities, we also unveiled an unexpected inhibitory effect of Arnicae planta tota on the basal expression of ALOX5 in differentiated THP-1 cells. This effect was also observed upon treatment with Arnicae flos. The transcription factors responsible for basal ALOX5 expression in differentiated THP-1 cells that are targeted for inhibition by
A. montana extracts are yet to be identified. Previous studies indicated that tandem consensus-binding motifs for the Egr-1/Sp1 transcription factors within ALOX5 core promoter are essential for its basal activity [
29,
37,
38]. Additional studies would be necessary to determine whether Egr-1/Sp1 transcription factors are inhibited by
A. montana extracts.
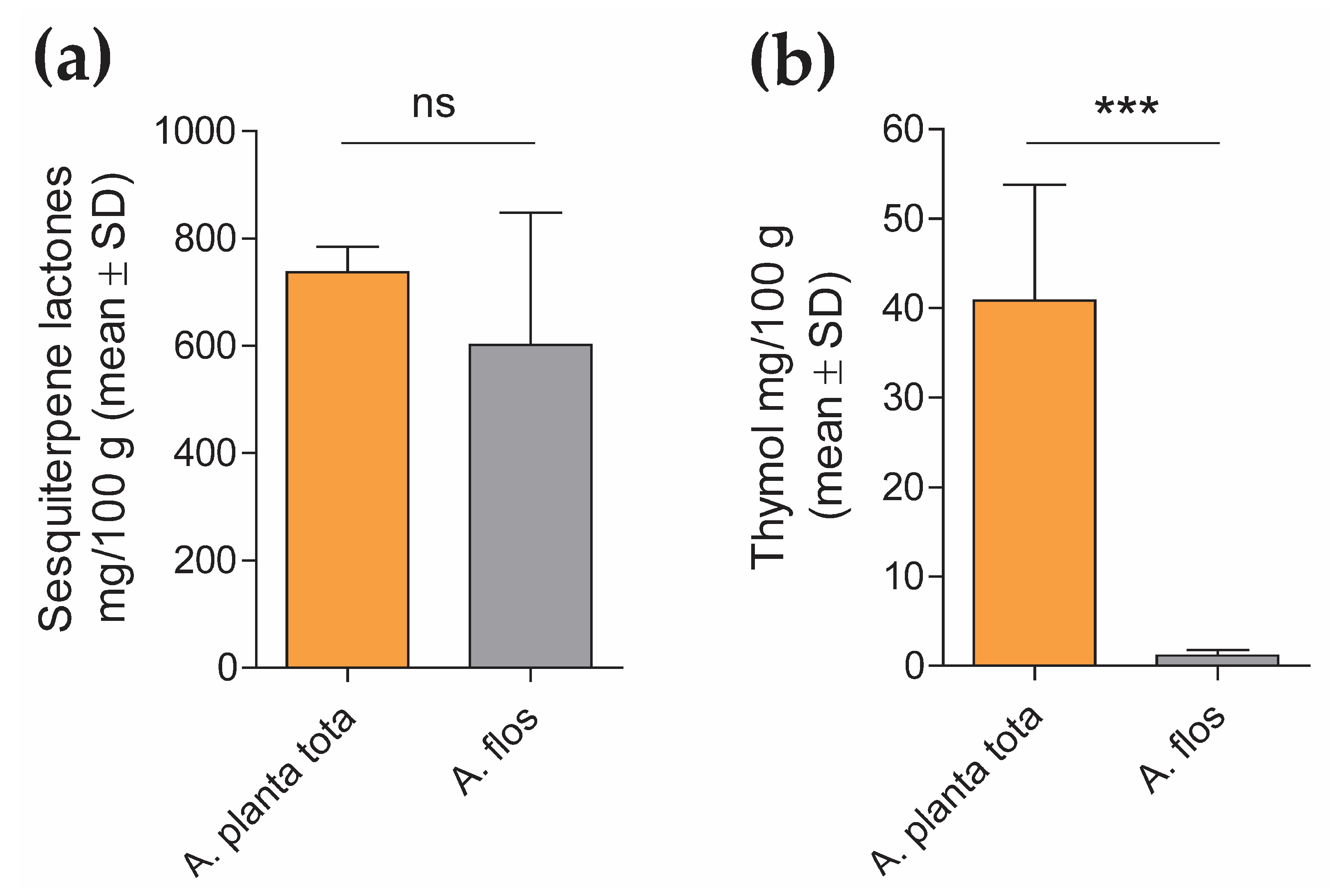
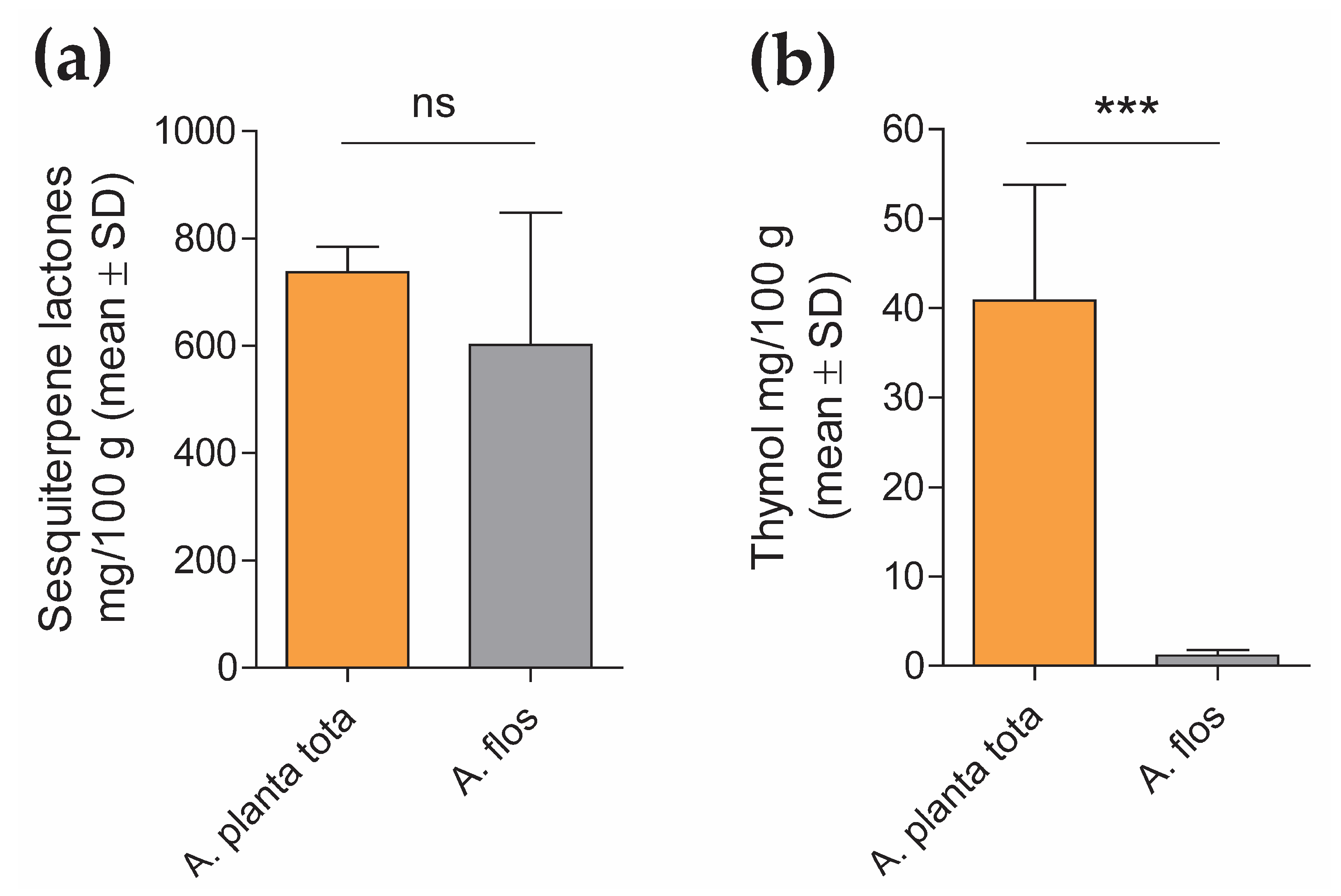
Interestingly, while LPS-induced expression of PTGS2 in differentiated THP-1 was inhibited by Arnicae planta tota in a concentration-dependent manner, it remained unaffected by pre-treatment with Arnicae flos. This suggests that some bioactive substances contained in Arnicae planta tota but not in Arnicae flos were responsible for this inhibition. Indeed, bioactive substance content varies in different parts of the plant [
1,
6,
7], as confirmed by our preliminary HPLC experiments. Whether it is thymol, which was detected in a higher amount in Arnicae planta tota compared to Arnicae flos, and is known for its anti-inflammatory, antioxidant, and anti-microbial properties [
1,
7,
10,
11,
12,
13,
14,
15], or other substances within Arnicae planta tota which are responsible for the inhibition of LPS-induced PTGS2 in differentiated THP-1 cells, remains to be uncovered.
It is worth noting that, although ALOX5 and PTGS2 have been described as NF-κB targets [
16,
18], we have not demonstrated that LPS-induced expression of these genes in differentiated THP-1 cells is actually mediated by NF-κB. In fact, a recent study showed that LPS-induced expression of ALOX5 in differentiated THP-1 cells does not involve NF-κB but an as-yet-uncharacterized factor [
29]. If LPS-induced expression of PTGS2 in these cells is also independent of NF-κB, it would explain the absence of inhibition of LPS-induced expression of PTGS2 by Arnicae flos. Whatever the mechanism of induction of these genes in response to LPS is, our results emphasize the difference in the inhibitory effect of Arnicae planta tota and Arnicae flos.
A strength of our study is the use of different
in vitro and
in vivo experimental systems directly comparing planta tota and flos extracts prepared from the same source of
A. montana and employing the same extraction procedure. The data obtained in the mouse model of acute inflammation using a mode of application comparable to that typically used in humans (topical application) are very appealing, and should now be reproduced in humans. It is worth noting that the dose of extract applied locally in the mouse model was in a range comparable to that recommended for existing Arnica ointments (e.g., Arnicae planta tota 30% ointment, available in Germany). Considering a recommended average application of 2 mg/cm
2, three to five times a day, an amount of 1.8 to 3 mg Arnicae planta tota would be applied locally, using Arnica 30% ointment (compared to the 3 to 30 mg Arnicae planta tota applied in the mouse model of acute inflammation, described in this study). In addition to validating its use in preventing local inflammation (i.e., following a pre-treatment protocol, as described in this study), it would also be interesting to evaluate the potential benefit of the topical application of Arnicae planta tota to treat local inflammation (i.e., following induced inflammation), as shown for other natural products in the paw oedema mouse model [
36].
4. Materials and Methods
4.1. Arnica Montana Planta Tota and Flos Extracts
Preparation of dry extracts from Arnica montana L., planta tota and Arnica montana L., flos, organically cultivated in Germany on fields dedicated to medicinal-plant cultivation, was conducted using liquid extracts from Arnicae planta tota fresh plant or Arnicae flos fresh flowers (Arnicae planta tota recens, ethanolic extract 1:1.1, ethanol 30% [m/m] and Arnicae flos recens, ethanolic extract 1:1.1, ethanol 30% [m/m]). The dry herbal extracts were dissolved using 50% ethanol/50% purified water (m/m), resulting in a 30 mg/mL stock solution. The suspension was vortexed for 10 min and sonicated for 30 min with occasional vortexing. Samples were centrifuged for 10 min at 3000 x g. The supernatant was carefully harvested and used for experiments. If not stated otherwise, 50% ethanol/50% purified water (m/m), diluted to a final concentration of 0.5% ethanol, served as vehicle control in the respective assays.
4.2. Characterisation of A. Montana Extracts by HPLC
4.2.1. Composition and Semi-Quantitative Analysis of A. Montana Extracts
The composition of Arnicae planta tota and Arnicae flos extracts was evaluated using ultra-high-performance liquid chromatography with quadrupole time-of-flight tandem mass spectrometry (UHPLC-UV-hr-qTOF/MS). Each sample was measured in the form of technical triplicates. A total of 100 mg of each dry extract was dissolved in 10 mL, 1:1, v:v, acetonitrile:water containing 1% formic acid, vortexed for 2 min and ultrasonicated for 15 min. Finally, 1:50 v:v dilution was performed for each extract in 1:1, v:v, acetonitrile:water containing 1% formic acid, and subjected to instrumental analysis.
Chromatographic separation was performed on a Thermo Scientific Dionex UltiMate 3000 (ThermoFisher Scientific, Waltham, MA, USA), using an RRHD Zorbax C18 (2.1 × 100 mm, 1.8 μm) column (Agilent Technologies, Santa Clara, CA, USA). The mobile phase consisted of acetonitrile (B) and 0.1% formic acid in water (A). The flow rate was 0.4 mL/min, and the injection volume was 5 μL. The column oven was set at 45 °C. The following gradient was used: 0.0 min 3% B, 15.0 min 50% B, 18.0 min 80% B, 19 min 100% B, 21.0 min 100% B, 21.5 min 3 % B, and 23 min 3 % B.
The eluate from the liquid chromatography was directly introduced into the maXis Impact Ultra High Resolution TOF-MS mass spectrometer (Bruker Daltonics, Bremen, Germany), with mass scanning from 50–800 m/z and spectra rate 4 Hz, using electrospray ionization (ESI) in positive mode. The mass accuracy before each run was verified by comparison with sodium formate adducts. The mass accuracies were rounded to 1 mDa, and the corresponding retention times to 0.05 min. The UV spectra were recorded at 280 nm. Interpretation of the mass signals was conducted using Compass Data Analysis 4.2 (Bruker, Billerica, MA, USA). Identification of the detected sesquiterpene lactones and thymol derivatives was performed, based on the reported literature [
39,
40,
41], and their characteristic transition and relative abundance was determined according to the respective peak area.
4.2.2. Quantification of Sesquiterpene Lactones and Thymol Content in A. Montana Extracts
The content of sesquiterpene lactones and thymol in Arnicae planta tota and Arnicae flos extracts was quantified by high-performance liquid chromatography (HPLC), using five independent batches of extract preparation for each. Each HPLC measurement was carried out in duplicate.
For sesquiterpene lactones quantification, a modified HPLC method adapted from the monograph “Arnica tincture” of the European Pharmacopoeia (Ph. Eur.) 10.0/1809 was used, as follows. Arnica dry extracts (0.1 g) were dissolved in 5 mL ethanol R2 30% (w/w) and mixed with 1 mL internal standard solution (1 mg/mL santonin (chemical reference substance, European Directorate for the Quality of Medicines & HealthCare (EDQM), Council of Europe, Strasbourg, France), 2 mg/mL butyl 4-hydroxybenzoate R in methanol R2) and 3 g anhydrous aluminum oxide R. The mixture was shaken for 120 s, filtered through filter paper and evaporated to dryness under reduced pressure in a water bath. The desiccated residue was dissolved in 2 mL 50% methanol R2/50% chromatography-grade water R, and filtered through a 0.45 μm PVDF membrane filter. HPLC analysis was performed on an Ultimate 3000 RSLC 02 HPLC system equipped with a UV detector (ThermoFisher Scientific, Waltham, MA, USA), using a Symmetry C18 Column (150 mm length × 3.9 mm inner diameter, 5 μm particle size; Waters). The injection volume was 20 μL, the flow rate was 1.0 mL/min, the column temperature was maintained at 22 °C, and the detection wavelength of quantification was set at 225 nm. The mobile phase for chromatography was distilled water R (A) and methanol R2 (B). Following column equilibration in 40% methanol for 5 min, separation was obtained by gradient elution through increasing the methanol from 40% to 45% linearly over 17 min and holding for 10 min, followed by a second gradient, from 45% to 55% methanol for 25 min. The column was then washed by increasing the methanol to 90% for 1 min, and held for another 1 min, before re-equilibration at starting mobile-phase conditions (40% methanol in chromatography-grade water R). Retention time was about 10 min for santonin and about 49 min for butyl 4-hydroxybenzoate. Data were interpreted based on peak area determination (the sum of the peaks of helenalin and dihydrohelenalin derivatives between the reference substances santonin and butyl 4-hydroxybenzoate) and the santonin internal standard, using the Chromeleon 7.2 Chromatography Data System software (ThermoFisher Scientific, Waltham, MA, USA). Sesquiterpene lactone content was calculated according to the santonin and dihydrohelenalin tiglate peak area, using the conversion factor 1.187, as recommended by the Ph. Eur. 10.0/1809. Data (mean ± SD of five analyte batch quantification, each expressed as mean of duplicate measurement) were expressed in mg sesquiterpene lactones/100 g Arnica extract.
For thymol quantification, Arnica dry extracts (0.05 g) were dissolved in 50% methanol R2, mixed with 0.8 mL internal standard solution (1 mg/mL carvacrol [primary reference standard, Sigma-Aldrich/Merck, 04270590, Darmstadt, Germany] in methanol R2), and diluted to 100 mL with 50% methanol R2. The mixture was filtered through a 0.45 μm PVDF membrane. A solution containing 0.2 μg/mL thymol (primary reference standard, PhytoLab, 89287, Vestenbergsgreuth, Germany) in 50% methanol R2 was also measured as the reference standard. HPLC analysis was performed on an Ultimate 3000 RSLC 02 HPLC system equipped with an RF-20A fluorescence detector (ThermoFisher Scientific, Waltham, MA, USA), using a SunFire C18 Column (150 mm length × 3.0 mm inner diameter, 3.5 μm particle size; Waters). The injection volume was 10 μL, the flow rate was 1.0 mL/min, the column temperature was maintained at 30 °C, and detection was performed at 280 nm/310 nm. The mobile phase for chromatography was 0.1% trifluoroacetic acid R (A) and chromatography-grade acetonitrile R2 (B). Following equilibration in 40% acetonitrile (B) / 60% 0.1% trifluoroacetic acid (A) for 2 min, separation was obtained by gradient elution through increasing the acetonitrile from 40% to 65% linearly for 8 min. The column was then washed by increasing the acetonitrile to 95% for 2 min, and held for another 3 min, before re-equilibration at starting mobile-phase conditions. Retention time was about 9.0 min for carvacrol and about 9.6 min for thymol. Data were interpreted based on peak height using internal standard correction, with the Chromeleon 7.2 Chromatography Data System software (ThermoFisher Scientific, Waltham, MA, USA). Thymol content in the Arnica extracts was calculated according to the thymol external reference standard. Data (mean ± SD of the quantification of the five analyte batches) were expressed in mg thymol/100g Arnica extract.
4.3. NF-κB Reporter Assay
The inhibitory effect of Arnicae planta tota and Arnicae flos on NF-κB activity was measured using an NF-κB reporter assay (Eurofins Panlabs #361000-1, New Taipei City, Taiwan). Briefly, human T lymphocytic Jurkat cells (2 × 106 cells/mL) stably transfected with a construct comprising a NF-κB response element controlling the transcription of the beta-galactosidase reporter gene (Eurofins Panlabs Taiwan #C13900, New Taipei City, Taiwan) were preincubated for 20 min at increasing concentrations (3–300 μg/mL in 0.5% ethanol final concentration) of either Arnicae planta tota or Arnicae flos extract, or vehicle (0.5% ethanol) in RPMI-1640 buffer pH 7.4 at 37 °C (two wells per condition). Increasing concentrations (3–300 nM) of cyclosporin A (Sigma-Aldrich/Merck C3662, Darmstadt, Germany), serving as positive control for NF-κB inhibition, were tested in parallel. Cells were then stimulated for 4 h with 0.5 μM Calcium Ionophore A23187 and 50 ng/mL phorbol 12-myristat 13-acetat (PMA), and harvested to quantify reporter-gene activity. Beta-galactosidase reporter-gene activity was determined by the conversion of fluorescein di-β-D-galactopyranoside (FDG; ThermoFisher Scientific, F1179, Waltham, MA, USA) to fluorescein, and the fluorescence intensity was measured on a SpectroFluor Plus plate reader. A decrease of ≥50% in fluorescence intensity, relative to 10 μM cyclosporin A, indicates significant inhibitory activity. Data (mean ± SD of duplicate cell treatment) were expressed as the percentage of reporter inhibition relative to vehicle, and the half-maximal inhibitory concentration (IC50) for Arnicae planta tota and Arnicae flos was calculated.
Cytotoxicity assays were conducted in triplicate under the same experimental conditions, as a control for specific NF-κB inhibition. Staurosporine (Sigma-Aldrich/Merck S4400, Darmstadt, Germany; 0.001–100 μg/mL) was tested in parallel (in duplicate) as positive control for cytotoxicity. Following cell stimulation, CellTiter-Glo reagent (CellTiter-Glo® Luminescent Cell Viability Assay, Promega, Madison, WI, USA) was added to each well, and luminescence was measured on a Safire2 Microplate Reader (Tecan, Männedorf, Switzerland). CellTiter-Glo reagent measures the amount of ATP present in the cell-culture well. ATP is an indicator of metabolically active cells, and its amount is directly proportional to the number of living cells. The percentage of cell viability was calculated relative to the vehicle control.
4.4. ALOX5 and PTGS2 Gene Expression Analysis Using Quantitative RT-PCR
4.4.1. THP-1 Cell Differentiation and Stimulation
Human monocytic leukemia THP-1 cells (American Type Culture Collection (ATCC) #TIB-202, Manassas, VA, USA) were seeded in a 48-well plate and incubated in culture medium (RPMI 1640 supplemented with 10% heat-inactivated fetal calf serum, 1% L-glutamine, and 1% penicillin-streptomycin) for 4 h. Cells were differentiated into macrophages by the addition of 200 nM PMA in culture medium for 72 h. PMA-containing medium was replaced by fresh medium, and differentiated cells were pretreated for 4 h with the indicated concentrations of Arnicae planta tota or Arnicae flos, or vehicle (0.5% ethanol) as control. Cells (three wells per condition) were further stimulated for 24 h with 100 ng/mL lipopolysaccharide (LPS).
4.4.2. Cytotoxicity Control Assay
Cytotoxicity assays were conducted in triplicate under the same experimental conditions. Following stimulation, cells were incubated with WST-8 (CCK-8 #311KTA1020, Tebubio, Le Perray, France) and optical density was measured at 450 nm using a VERSAmax spectrophotometer (Molecular Devices, San Jose, CA, USA). WST-8 is a tetrazolium salt that is reduced in a yellow-colored formazan dye by mitochondrial succinate dehydrogenase (an indicator of metabolically active cells). Formazan product formation is directly proportional to the number of living cells and their metabolic activity. The percentage of cell viability was calculated relative to the LPS-stimulated condition.
4.4.3. RNA Isolation and Quantitative RT-PCR
Following LPS stimulation, cells were harvested and stored at −80 °C until RNA isolation. Total RNA was isolated using TriPure Isolation Reagent® (Sigma-Aldrich/Merck, Darmstadt, Germany) according to the supplier’s instructions. RNA quality was verified using capillary electrophoresis (Bioanalyzer 2100, Agilent Technologies, Santa Clara, CA, USA) and RNA concentration was measured using spectrophotometry (Synergy H1, BioTek Instruments/Agilent Technologies, Santa Clara, CA, USA). Complementary DNA (cDNA) was synthetized by reverse transcription of total RNA in the presence of oligo(dT) using Transcriptor Reverse Transcriptase (Roche, Basel, Switzerland). Quantitative PCR was performed using ONEGreen® FAST qPCR Premix (Ozyme, Saint-Cyr-l’Ecole, France) according to the manufacturer’s instructions, and triplicate reactions were run on a LightCycler® 480 System (Roche Molecular System Inc., Pleasanton, CA, USA), using a two-step protocol (95 °C 5 s, 60 °C 34 s; 45 cycles). The forward and reverse primers used for amplification of ALOX5 (NM_000698), PTGS2 (NM_000963) and the normalizer gene GAPDH (NM_002046) were designed using Primer3 and NCBI Blast, and were ordered from Sigma-Aldrich/Merck (Darmstadt, Germany). The respective sequences (5′–3′) of the forward and reverse primers were as follows: ALOX5, TCATCGTGGACTTTGAGCTG and CAGAAGGTGGGTGATGGTCT; PTGS2, TGAGCATCTACGGTTTGCTG and TGCTTGTCTGGAACAACTGC; GAPDH, GGCTCTCCAGAACATCATCCCTGC and GGGTGTCGCTGTTGAAGTCAGAGG. Cycle threshold (Ct) values were expressed in arbitrary units (AU) according to the formula: AU = (1/2Ct) × 106. AU of the genes of interest ALOX5 and PTGS2 (AUGOI) were normalized to that of the housekeeping gene GAPDH (AUGAPDH), according to the formula AUGOI/AUGAPDH, and ALOX5 and PTGS2 expression data were expressed as a percentage of gene expression relative to that in the LPS-stimulated control.
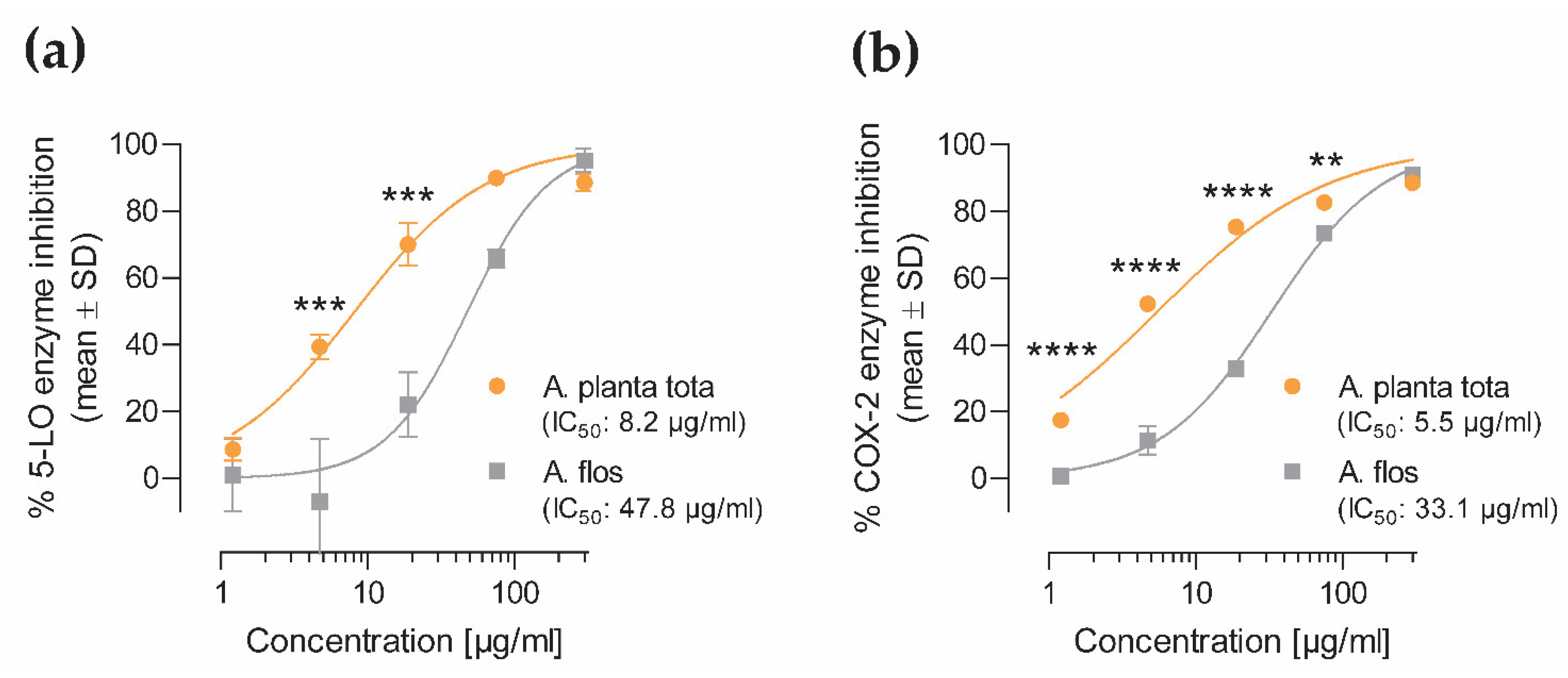
4.5. 5-LO and COX-2 In Vitro Enzymatic Assays
The inhibitory effect of Arnicae planta tota and Arnicae flos on the enzymatic activity of human 5-lipoxygenase (5-LO) and cyclooxygenase-2 (COX-2) was assessed using cell-free enzymatic assays (Eurofins Cerep #772, Celle-Lévescault, France, and Eurofins Panlabs #118030, New Taipei City, Taiwan, respectively).
For the 5-LO enzymatic assay, 2.4 U/mL purified human recombinant 5-LO protein was preincubated in duplicate with increasing concentrations (1.2–300 μg/mL) of either Arnicae planta tota or Arnicae flos extract, or vehicle (0.5% ethanol), for 15 min at 25 °C in Tris buffer pH 7.4. Nordihydroguaiaretic acid (NDGA; Cayman Chemical 70,300-1, Ann Arbor, MI, USA; 0.01–30 µM) served as control inhibitor. The 5-LO enzymatic reaction was performed in the presence of 25 µM arachidonic acid (Biomol Cay90010-100, Hamburg, Germany), as substrate, and non-fluorescent dihydrorhodamine 123 for 5 min at 25 °C. The 5-LO enzymatic activity was measured using fluorescence spectroscopy at 500 nm/590 nm of rhodamine 123, generated by 5-LO-mediated conversion of dihydrorhodamine 123.
For the COX-2 enzymatic assay, 34 U/mL purified human recombinant COX-2 protein was preincubated in duplicate with increasing concentrations (1.2–300 μg/mL) of either Arnicae planta tota or Arnicae flos extract, or vehicle (0.5% ethanol), for 15 min at 25 °C in modified Tris-HCl buffer pH 8.0. Rofecoxib (Glentham Life Sciences GP0159, Corsham, UK; 0.03–3 µM) served as control inhibitor. The COX-2 enzymatic reaction was initiated by adding 3 µM arachidonic acid substrate and 100 µM Amplifu Red (10-Acetyl-3,7-dihydroxyphenoxazine), and further incubated for 3 min at 25 °C. The COX-2 enzymatic activity was analysed using fluorescence spectroscopy at 535 nm/590 nm of resorufin, produced by COX-2-catalysed oxidation of Amplifu Red.
Measurements were performed in duplicate, and data were expressed as the percentage of enzyme inhibition relative to vehicle. IC50 values were calculated for Arnicae planta tota and Arnicae flos, as well as for the reference inhibitors nordihydroguaiaretic acid (5-LO assay) and rofecoxib (COX-2 assay).
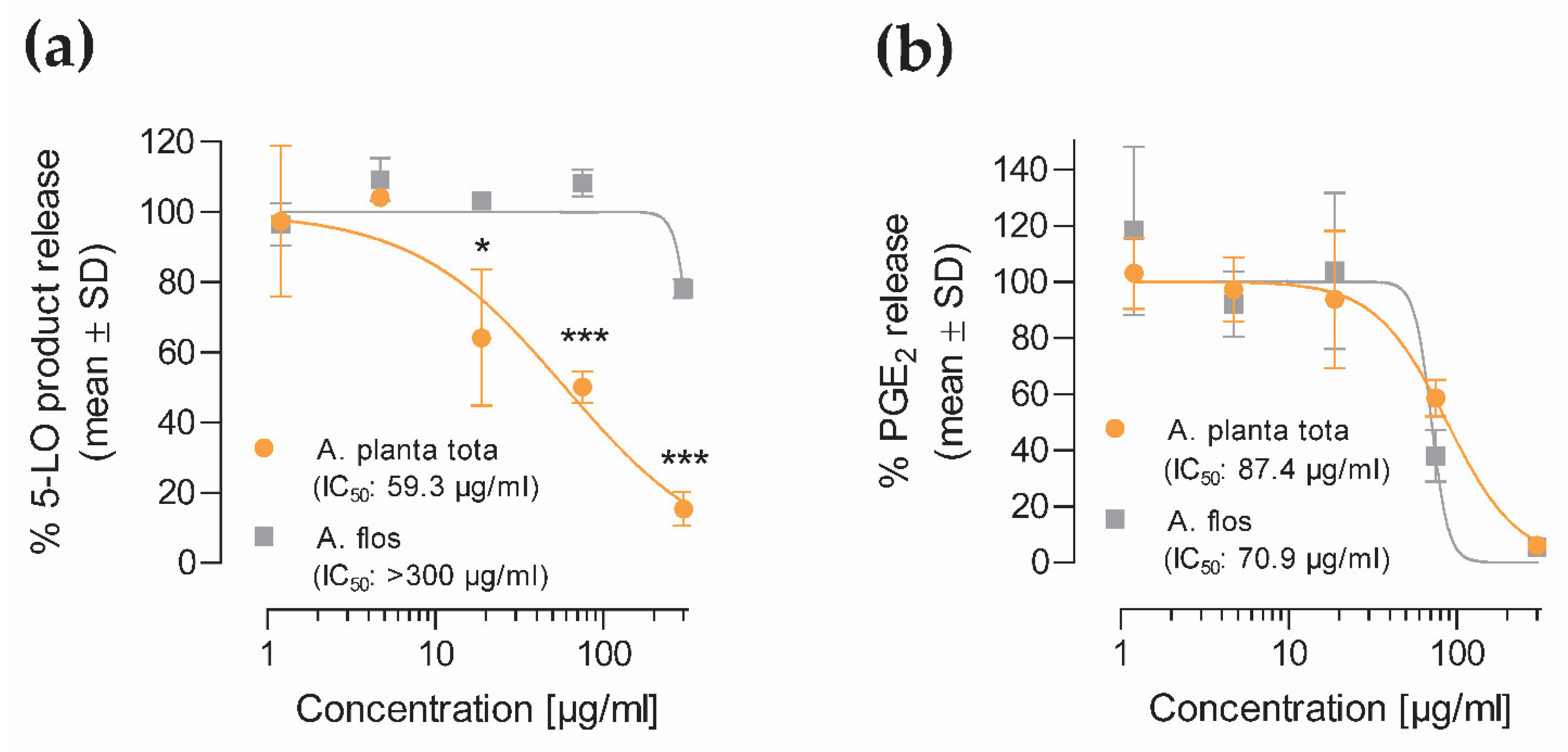
4.6. 5-LO and COX-2 Product Release from Human Peripheral Blood Cells
The inhibitory effect of Arnicae planta tota and Arnicae flos on 5-LO and COX-2 product formation was investigated in cellular assays.
4.6.1. 5-LO Product Formation
For 5-LO product formation analysis, polymorphonuclear leukocytes (PMNL) were freshly isolated from human peripheral blood of two consenting healthy donors (Institute of Transfusion Medicine, University Hospital Jena, Germany) who had declared that they had not taken any anti-inflammatory drugs within 10 days prior to blood withdrawal, as previously described [
42]. The experimental protocol was approved by the ethical committee of the Jena University Hospital, and all methods were performed in accordance with the relevant guidelines and regulations. PMNL (5 × 10
6/
mL) were preincubated with increasing concentrations (1.2–300 μg/mL) of either Arnicae planta tota or Arnicae flos extract, or vehicle (0.5% ethanol), for 15 min at 37 °C, and stimulated with 2.5 µM Ca
2+-Ionophore A23187 (Biomol Cay11016-10, Hamburg, Germany) in the presence of 20 µM arachidonic acid (Biomol Cay90010-100, Hamburg, Germany), as 5-LO substrate, for 10 min at 37 °C. The 5-LO- specific inhibitor zileuton (Biosynth Ltd. FZ28760, Staad, Switzerland; 15 µM) was used as a reference compound. The 5-LO products LTB
4, its trans-isomers, and 5-HETE released in the supernatant were analysed using reverse-phase high-performance liquid chromatography (RP-HPLC), as reported [
42], their sum defining 5-LO products. Data (mean ± SD of two donors) were expressed as the percentage of released 5-LO products relative to vehicle control.
4.6.2. COX-2 Product Formation (PGE2)
For COX-2 product formation analysis, monocytes were freshly isolated from human peripheral blood of three consenting healthy donors (Institute of Transfusion Medicine, University Hospital Jena, Germany) who had declared that they had not taken any anti-inflammatory drugs within 10 days prior to blood withdrawal, as previously described [
42]. The experimental protocol was approved by the ethical committee of the Jena University Hospital and all methods were performed in accordance with the relevant guidelines and regulations. Cells (1 × 10
6/
mL) were preincubated with increasing concentrations (1.2–300 μg/mL) of either Arnicae planta tota or Arnicae flos extract, or vehicle (0.5% ethanol), for 15 min at 37 °C, and then stimulated with 100 ng/mL LPS for 6 h. Indomethacin (Sigma-Aldrich/Merck I7378, Darmstadt, Germany; 10 µM) was used as reference inhibitor. Cell culture supernatants of the three donors were collected, and the released COX-2 product PGE
2 was analysed using ultra-performance liquid chromatography tandem mass spectrometry (UPLC-MS/MS), as previously reported [
43]. Data (mean ± SD of three donors) were expressed as the percentage of released PGE
2, relative to vehicle control.
To verify that the effect of Arnica extracts was not due to cytotoxicity during the 6-h incubation time, mitochondrial dehydrogenase activity was measured under comparable experimental conditions using an MTT-based assay. Briefly, monocytes were preincubated with increasing concentrations (1.2–300 μg/mL) of either Arnicae planta tota or Arnicae flos extract, 0.5% ethanol (vehicle), or 10 µM indomethacin (inhibition control) for 15 min at 37 °C, and stimulated with 100 ng/mL LPS for 6 h, as above. Following stimulation, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich/Merck, Darmstadt, Germany) was added to each well for 2 h. An additional LPS-stimulated well was treated with 0.1% Triton X-100, as a cytotoxicity control (100% dead cells), before the addition of 2 mmol/L MTT. Cells were lysed, purple-colored formazan crystals were solubilized with 10% SDS in 20 mmol/L HCl, and absorbance was measured at 570 nm, using a Multiskan Spectrum Microplate Reader (ThermoFisher Scientific, Waltham, MA, USA). Data (mean ± SD of three donors) were expressed as the percentage of cell viability relative to the stimulated vehicle control.
4.7. Carrageenan-Induced Inflammation Mouse Model
Male ICR mice (26 ± 2 g, 6 mice per group), purchased from BioLasco Taiwan (Taipei, Taiwan; under Charles River Laboratories’ License) were fasted overnight before the start of treatment and for the duration of the experiment. Arnicae planta tota, Arnicae flos, given at 1, 3 and 10 mg/paw, or vehicle (50% ethanol), each in a volume of 20 µL, was applied topically to the right hind paw around the site of the carrageenan injection three consecutive times at 20 min intervals, starting 1 h before the carrageenan injection. The positive control Aspirin was given orally at a dosage of 150 mg/kg 1 h before the carrageenan injection. The mice received an intraplantar injection of carrageenan (50 µL of 1% suspension) into the right hind paw. Paw oedema was recorded 4 h after the carrageenan challenge, using a plethysmometer. The uninjected left hind paw served as an internal negative control. The animals were prohibited from licking their paws by wearing neck ruffs after drug application and for the duration of the experiment. The study was performed by Pharmacology Discovery Services (New Taipei City, Taiwan) under the Animal Care and Use Protocol No. IN018-10222020 approved by the Institutional Animal Care and Use Committee (IACUC) on October 10, 2020, in a laboratory animal facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC), and in general accordance with the Guide for the Care and Use of Laboratory Animals: Eighth Edition (National Academies Press, Washington, D.C., 2011).
4.8. Statistical Analysis
Statistical analysis, curve fitting and IC50 value calculation were performed using GraphPad Prism version 9.2.0 (GraphPad Software Inc., San Diego, CA, USA). All values are presented as mean ± standard deviation (SD). Data are representative of at least two independent experiments analysed with duplicate or triplicate measurements (biological replicates). IC50 values were calculated by non-linear regression using the equation (Y = 100/(1 + 10^((LogIC50-X)*HillSlope)), with X = log of concentration, Y = normalised response (0–100%)). The statistical analysis of dose-response experiments was performed using two-way ANOVA with Bonferroni’s correction for multiple comparisons, to compare inhibition by Arnicae planta tota and Arnicae flos at the respective concentrations. Two-group-comparison testing was performed using an unpaired t-test. Data from gene expression analysis and animal experimentation were statistically analysed using one-way ANOVA followed by Dunnett’s multiple comparison test, to assess the effect of Arnica extracts relative to the vehicle control or to compare the effects of Arnicae planta tota and Arnicae flos, as indicated. A p-value ≤ 0.05 was considered statistically significant.




 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}