
Integrative Physiological, Transcriptome, and Proteome Analyses Provide Insights into the Photosynthetic Changes in Maize in a Maize–Peanut Intercropping System

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
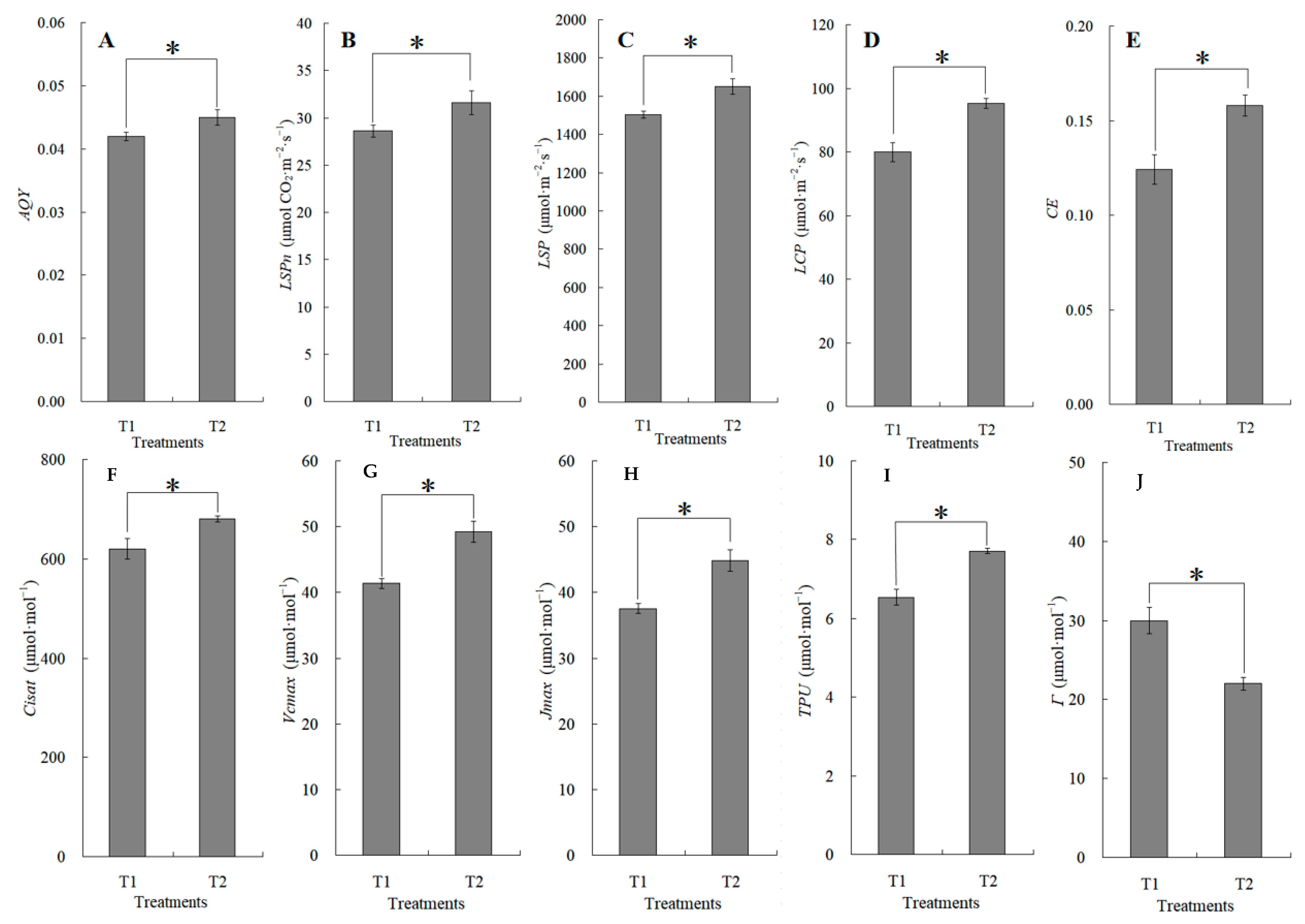
2.1. Photosynthetic Characteristic Changes in Response to Maize–Peanut Intercropping
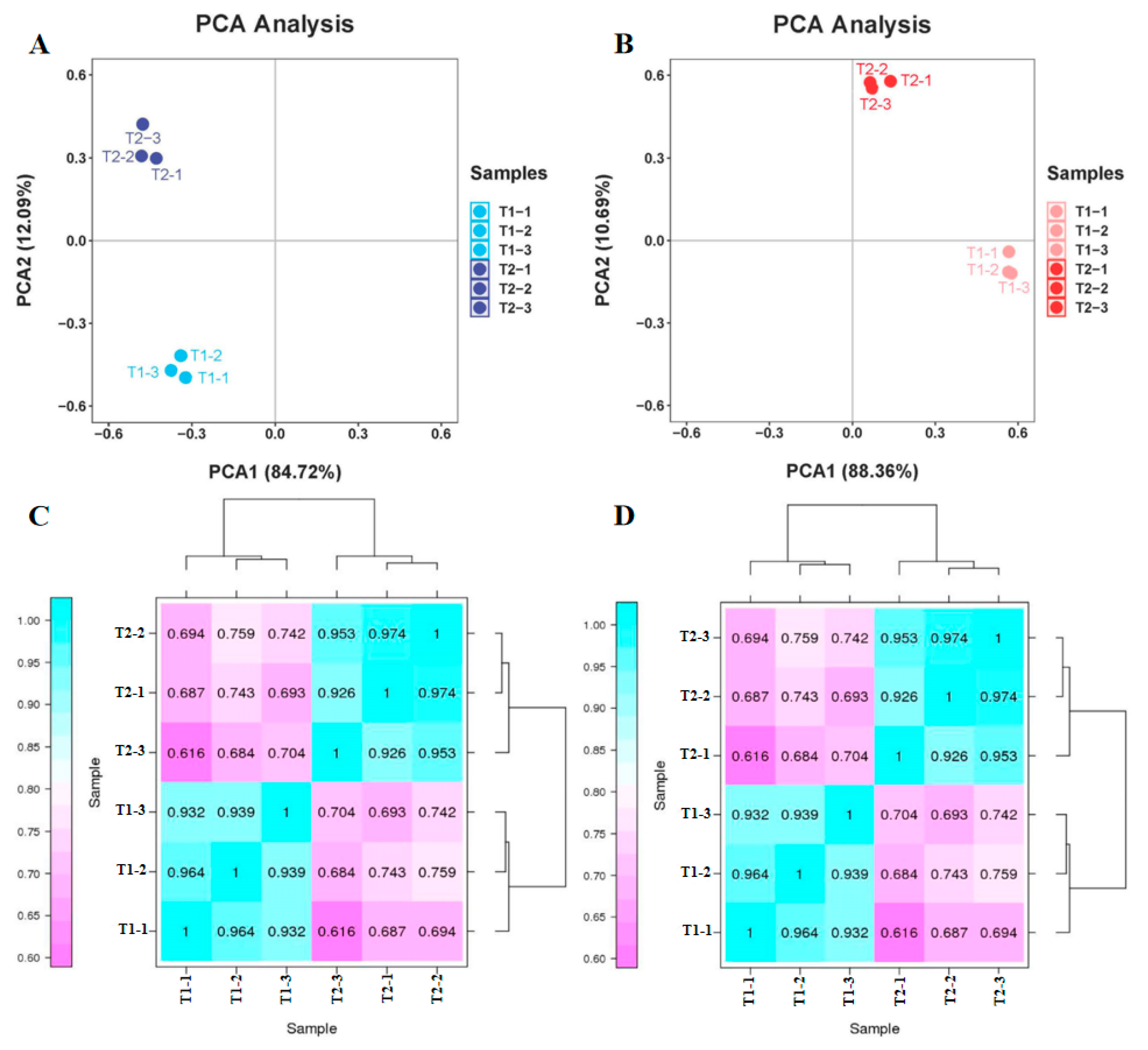
2.2. Primary Transcriptome and Proteome Data Analyses
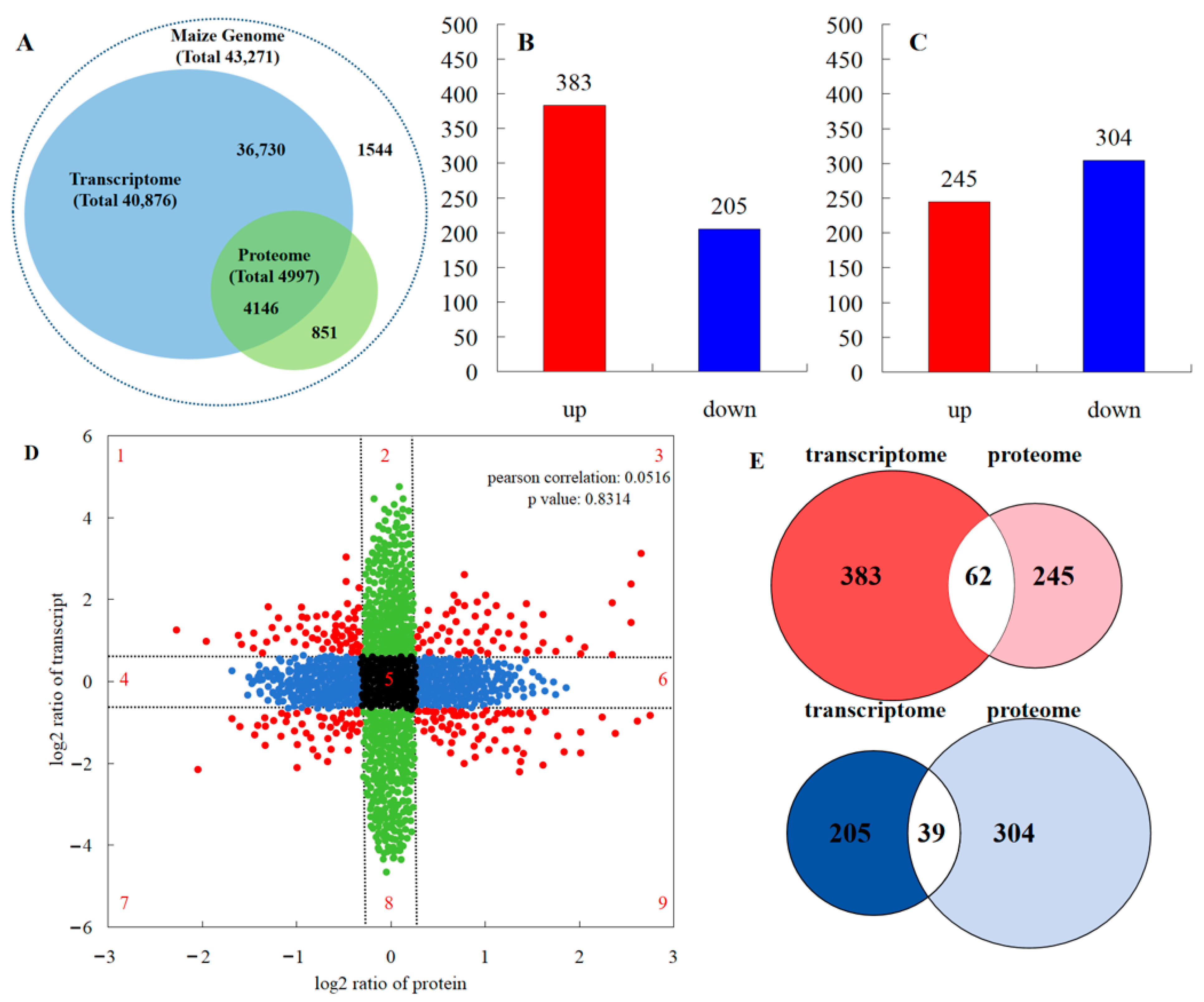
2.3. The mRNA and Protein Expression Changes in the Genome under Maize–Peanut Intercropping
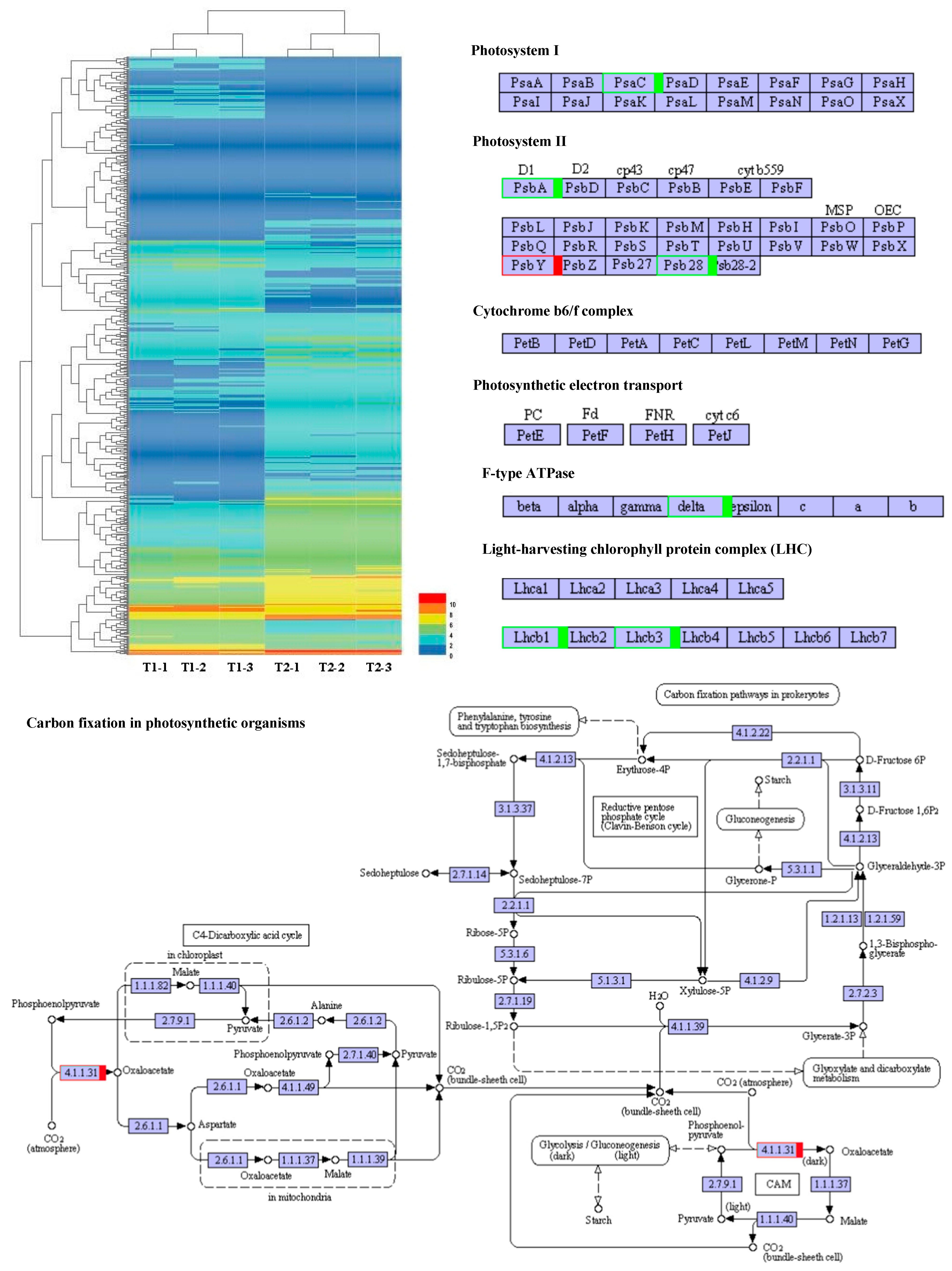
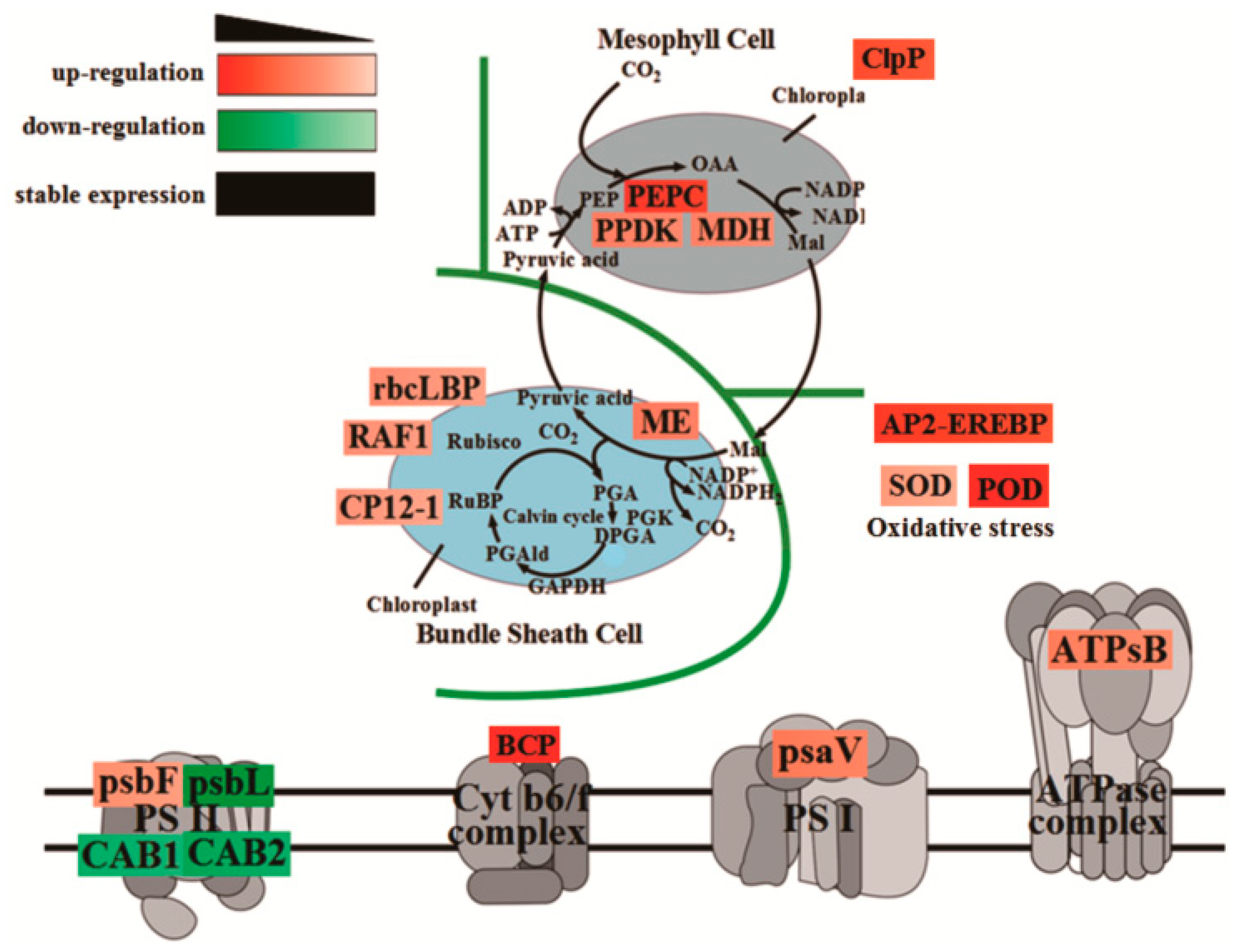
2.4. Photosynthetic Response Transcriptome Differences under Maize–Peanut Intercropping
2.5. Photosynthetic Response Proteome Differences under Maize–Peanut Intercropping
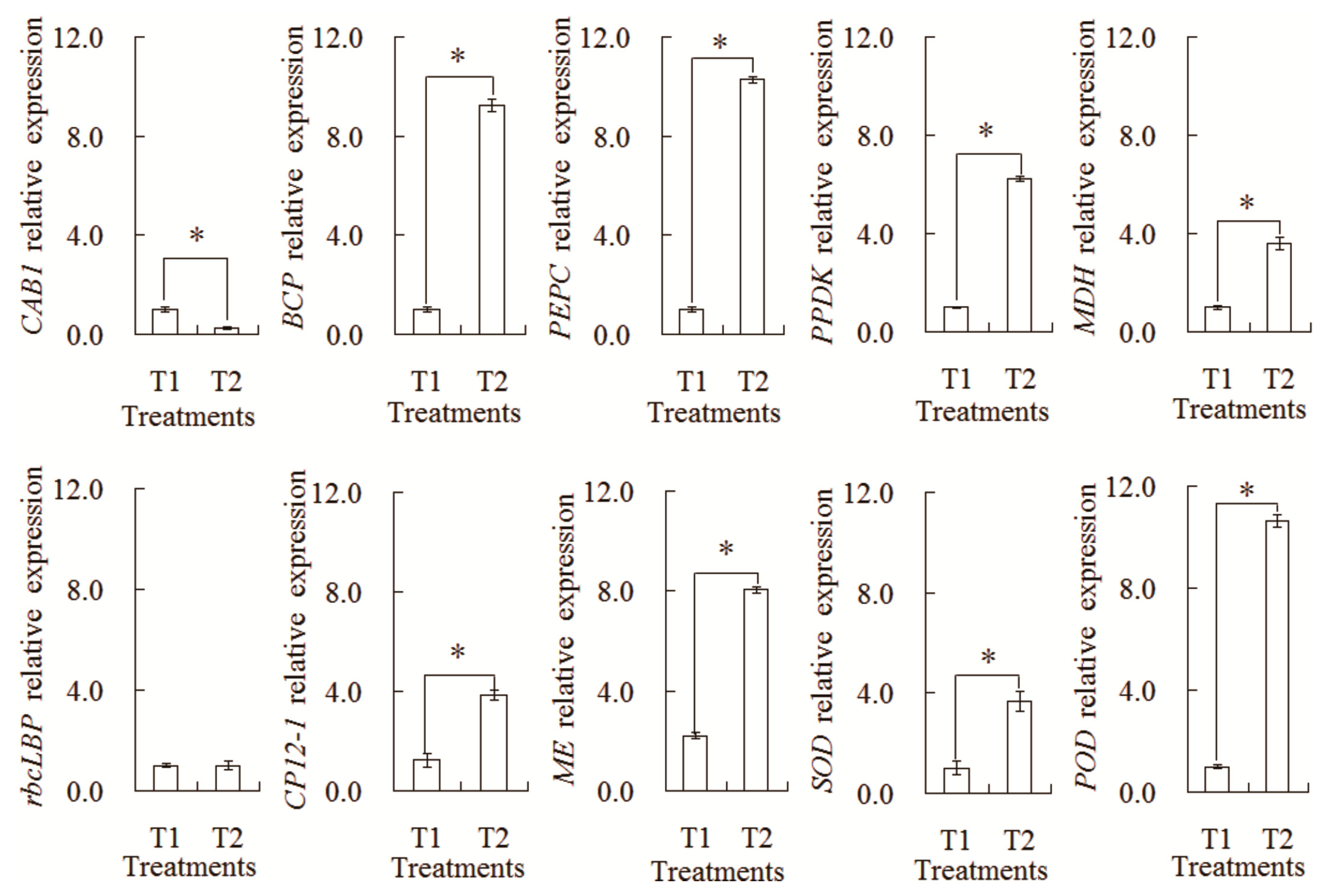
2.6. Transcriptional Expression Analysis by qRT-PCR
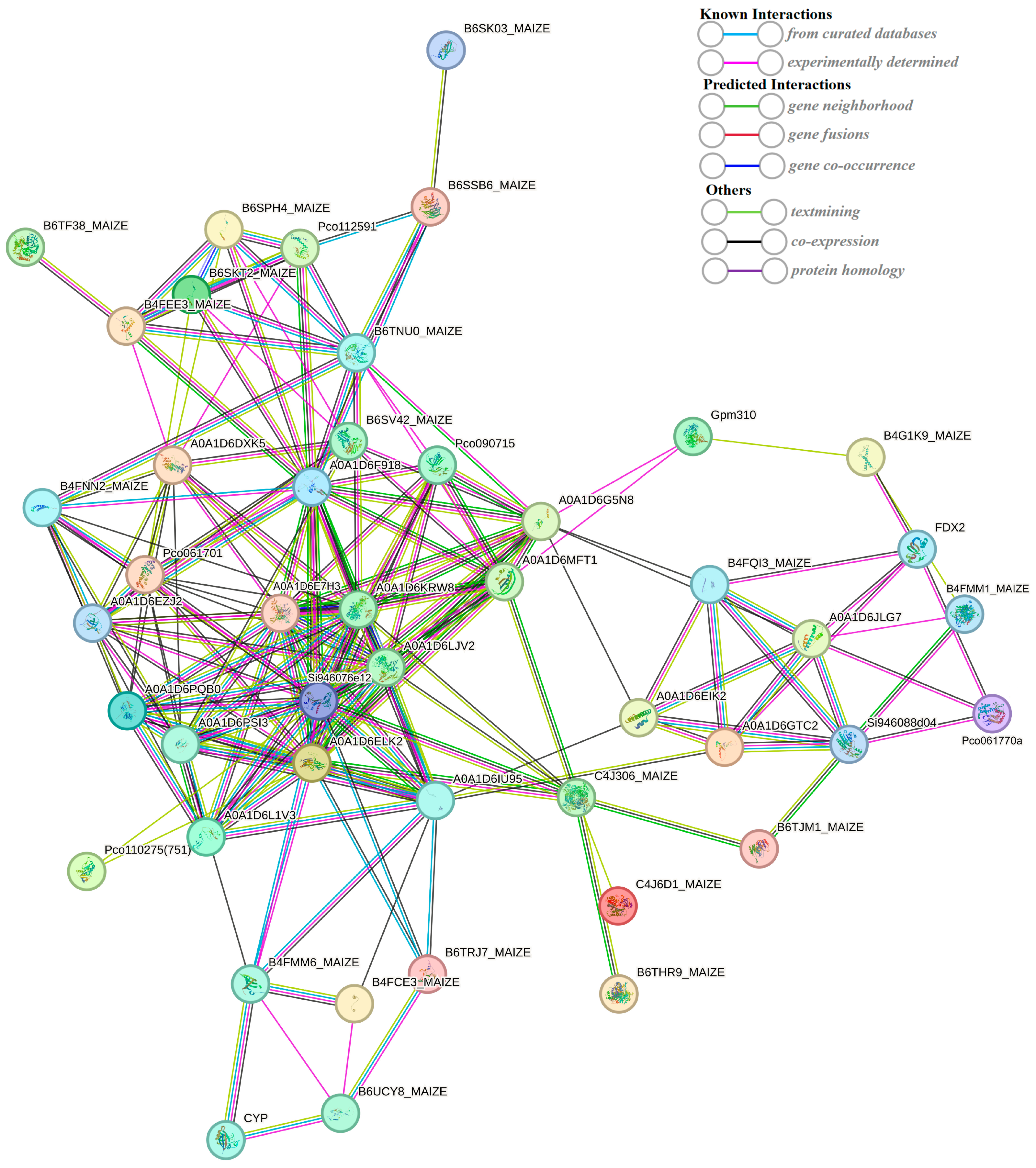
2.7. String-Based Protein–Protein Interaction (PPI) Analysis
3. Discussion
4. Materials and Methods
4.1. Plant Growth Condition, Treatments, and Sampling
4.2. RNA Extraction, Library Construction, and Sequencing
4.3. Protein Extraction, Concentration Determination, Enzymatic Digestion, and iTRAQ Reagent Labeling
4.4. Chromatography, LC–MS/MS Analysis, and Raw Data Analysis
4.5. Total RNA Extraction and qPCR
4.6. Assay of Photo Flux Density and Photosynthetic Characteristics
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Jiao, N.Y.; Wang, F.; Ma, C.; Zhang, F.S.; Jensen, E.S. Interspecific interactions of iron and nitrogen use in peanut (Arachis hypogaea L.)-maize (Zea mays L.) intercropping on a calcareous soil. Eur. J. Agron. 2021, 128, 126303. [Google Scholar] [CrossRef]
- Ren, B.; Li, L.; Dong, S.; Liu, P.; Zhao, B.; Zhang, J. Photosynthetic characteristics of summer maize hybrids with different plant heights. Agron. J. 2017, 109, 1454–1462. [Google Scholar] [CrossRef]
- Huang, M.; Wang, Z.H.; Luo, L.C.; Wang, S.; Hui, X.L.; He, G.; Cao, H.B.; Ma, X.L.; Huang, T.M.; Zhao, Y.; et al. Soil testing at harvest to enhance productivity and reduce nitrate residues in dryland wheat production. Field Crop Res. 2017, 212, 153–164. [Google Scholar] [CrossRef]
- Li, C.X.; Li, Y.Y.; Li, Y.J.; Fu, G.Z. Cultivation techniques and nutrient management strategies to improve productivity of rain-fed maize in semi-arid regions. Agric. Water Manag. 2018, 210, 149–157. [Google Scholar] [CrossRef]
- Xia, H.Y.; Wang, L.; Jiao, N.Y.; Mei, P.P.; Wang, Z.G.; Lan, Y.F.; Chen, L.; Ding, H.B.; Yin, Y.L.; Kong, W.L.; et al. Luxury absorption of phosphorus exists in maize when intercropping with legumes or oilseed rape-covering different locations and years. Agronomy 2019, 9, 314. [Google Scholar] [CrossRef]
- Jiao, N.Y.; Wang, J.T.; Ma, C.; Zhang, C.C.; Guo, D.Y.; Zhang, F.S.; Jensen, E.S. The importance of aboveground and belowground interspecific interactions in determining crop growth and advantages of peanut/maize intercropping. Crop J. 2021, 9, 1460–1469. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, Z.H.; Fu, Z.Y.; Liu, Z.H.; Hu, Y.M. Comparative QTL analysis of maize seed artificial aging between an immortalized F2 population and its corresponding RILs. Crop J. 2016, 4, 30–39. [Google Scholar] [CrossRef]
- Zhang, L.H.; Yu, F.Y.; Shi, W.M.; Li, Y.J.; Miao, Y.F. Physiological characteristics of selenite uptake by maize roots in response to different pH levels. J. Plant Nutr. Soil. Sci. 2010, 173, 417–422. [Google Scholar] [CrossRef]
- Zhang, G.Q.; Shen, D.P.; Ming, B.; Xie, R.Z. Using irrigation intervals to optimize water-use efficiency and maize yield in Xinjiang, northwest China. Crop J. 2019, 7, 322–334. [Google Scholar] [CrossRef]
- Yang, F.; Liao, D.; Wu, X.; Gao, R.; Fan, Y.; AliRaza, M.; Wang, X.; Yong, T.; Liu, W.; Liu, J.; et al. Effect of aboveground and belowground interactions on the intercrop yields in maize-soybean relay intercropping systems. Field Crop Res. 2017, 203, 16–23. [Google Scholar] [CrossRef]
- Gao, Y.; Wu, P.; Zhao, X.N.; Wang, Z.K. Growth, yield, and nitrogen use in the wheat/maize intercropping system in an arid region of northwestern China. Field Crop Res. 2014, 167, 19–30. [Google Scholar] [CrossRef]
- Li, Q.; Chen, J.; Wu, L.; Luo, X.; Li, N.; Arafat, Y.; Lin, S.; Lin, W. Belowground interactions impact the soil bacterial community, soil fertility, and crop yield in Maize/Peanut intercropping systems. Int. J. Mol. Sci. 2018, 19, 622. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Zhang, A.; Wang, F.; Han, X.; Wang, D.; Li, S. Arbuscular mycorrhizal fungi and rhizobium facilitate nitrogen uptake and transfer in soybean/maize intercropping system. Front. Plant Sci. 2015, 6, 339. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.W.; Lu, D.K.; Wang, H.Z.; Li, Y.J. Morphological and physiological traits of rice roots and their relationships to yield and nitrogen utilization as influenced by irrigation regime and nitrogen rate. Agric. Water Manag. 2018, 203, 385–394. [Google Scholar] [CrossRef]
- Wang, Y.L.; Tang, J.W.; Zhang, H.L.; Gao, Z.Q.; Kou, T.J. Aggregate-associated organic carbon and nitrogen impacted by the long-term application of fertilizers, rice straw, and pig manure. Soil. Sci. 2014, 179, 522–528. [Google Scholar] [CrossRef]
- Liu, X.; Rahman, T.; Song, C.; Su, B.; Yang, F.; Yong, T.; Wu, Y.; Zhang, C.; Yang, W. Changes in light environment, morphology, growth and yield of soybean in maize-soybean intercropping systems. Field Crop Res. 2017, 200, 38–46. [Google Scholar] [CrossRef]
- Nozoye, T.; Aung, M.S.; Masuda, H.; Nakanishi, H.; Nishizawa, N.K. Bioenergy grass [Erianthus ravennae (L.) Beauv.] secretes two members of mugineic acid family phytosiderophores which involved in their tolerance to Fe deficiency. Soil Sci. Plant Nutr. 2017, 63, 543–552. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, C.X.; Wang, Y.C.; Zheng, L.L. Cloning and functional validation of RtVAMP2-2 gene of Reaumuria trigyna. Pratacultural Sci. 2021, 38, 2363–2371. [Google Scholar]
- Liu, H.; Chong, P.; Yan, S.; Liu, Z.; Bao, X.; Tan, B. Transcriptome and proteome association analysis to screen candidate genes related to salt tolerance in Reaumuria soongorica leaves under salt stress. Plants 2023, 12, 3542. [Google Scholar] [CrossRef]
- Chai, L.; Li, H.; Zhao, X.; Cui, C.; Zheng, B.; Zhang, K.; Jiang, J.; Zhang, J.; Jiang, L. Analysis of altered flowering related genes in a multi-silique rapeseed (Brassica napus L.) line zws-ms based on combination of genome, transcriptome and proteome Data. Plants 2023, 12, 2429. [Google Scholar] [CrossRef]
- Jia, Y.; Liu, H.L.; Qu, Z.J.; Wang, J.; Wang, X.P.; Wang, Z.Q.; Yang, L.; Zhang, D.; Zou, D.T.; Zhao, H.W. Transcriptome sequencing and iTRAQ of different rice cultivars provide insight into molecular mechanisms of cold-tolerance response in Japonica rice. Rice 2020, 13, 43. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, J.; Wen, W.; Sun, J.; Shu, S.; Guo, S. Transcriptome and proteome analysis identifies salt stress response genes in bottle gourd rootstock-grafted watermelon seedlings. Agronomy 2023, 13, 618. [Google Scholar] [CrossRef]
- Schmollinger, S.; Muhlhaus, T.; Boyle, N.R.; Blaby, I.K.; Casero, D.; Mettler, T.; Moseley, J.L.; Kropat, J.; Sommer, F.; Strenkert, D.; et al. Nitrogen-sparing mechanisms in Chlamydomonas affect the transcriptome, the proteome, and photosynthetic Metabolism. Plant Cell 2014, 26, 1410–1435. [Google Scholar] [CrossRef] [PubMed]
- Yong, T.W.; Chen, P.; Dong, Q.; Du, Q.; Yang, F.; Wang, X.; Liu, W.; Yang, W. Optimized nitrogen application methods to improve nitrogen use efficiency and nodule nitrogen fixation in a maize-soybean relay intercropping system. J. Inter. Agric. 2018, 17, 60345–60347. [Google Scholar] [CrossRef]
- Li, L.; Tilman, D.; Lambers, H.; Zhang, F.S. Plant diversity and overyielding: Insights from belowground facilitation of intercropping in agriculture. New Phytol. 2014, 203, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Dong, N.; Tang, M.; Zhang, W.; Bao, X.; Wang, Y.; Christie, P.; Li, L. Temporal differentiation of crop growth as one of the drivers of intercropping yield advantage. Sci. Rep. 2018, 8, 3110. [Google Scholar] [CrossRef]
- Yao, X.; Zhou, H.; Zhu, Q.; Li, C.; Zhang, H.; Wu, J.J.; Xie, F. Photosynthetic response of soybean leaf to wide light-fluctuation in Maize-Soybean intercropping system. Front. Plant Sci. 2017, 8, 1695. [Google Scholar] [CrossRef]
- Cai, Z.; Liu, G.; Zhang, J.; Li, Y. Development of an activity-directed selection system enabled significant improvement of the carboxylation efficiency of Rubisco. Protein Cell 2014, 5, 552–562. [Google Scholar] [CrossRef]
- Sharwood, R.E.; Ghannoum, O.; Whitney, S.M. Prospects for improving CO2 fixation in C3-crops through understanding C4-Rubisco biogenesis and catalytic diversity. Curr. Opin. Plant Bio. 2016, 31, 135–142. [Google Scholar] [CrossRef]
- Wang, F.S.; Dong, S.R.; Zhang, H.Y.; Wang, S.Y. Putative model based on iTRAQ proteomics for Spirulina morphogenesis mechanisms. J. Proteom. 2018, 171, 73–80. [Google Scholar] [CrossRef]
- Carstensen, A.; Herdean, A.; Schmidt, S.B.; Sharma, A.; Spetea, C.; Pribil, M.; Husted, S. The impacts of phosphorus deficiency on the photosynthetic electron transport chain. Plant Physiol. 2018, 177, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Ma, J.; Su, X.; Cao, P.; Chang, W.; Liu, Z.; Zhang, X.; Li, M. Structure of the maize photosystem I supercomplex with light-harvesting complexes I and II. Science 2018, 6393, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.E.; Haldrup, A.; Zhang, S.; Scheller, H.V. The PSI-O subunit of plant photosystem I is involved in balancing the excitation pressure between the two photosystems. J. Biol. Chem. 2004, 297, 24212–24217. [Google Scholar] [CrossRef] [PubMed]
- Steppuhn, J.; Hermans, J.; Nechushtai, R.; Ljungberg, U.; Thümmler, F.; Lottspeich, F.; Herrmann, R.G. Nucleotide sequence of cDNA clones encoding the entire precursor polypeptides for subunits IV and V of the photosystem I reaction center from spinach. FEBS Lett. 1988, 237, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Rahoutei, J.; García-Luque, I.; Barón, M. Inhibition of photosynthesis by viral infection: Effect on PSII structure and function. Physiol. Plantarum. 2001, 110, 286–292. [Google Scholar] [CrossRef]
- Iida, S.; Kobiyama, A.; Ogata, T.; Murakami, A. The D1 and D2 proteins of dinoflagellates: Unusually accumulated mutations which influence on PSII photoreaction. Photosynth. Res. 2008, 98, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Otani, T.; Kato, Y.; Shikanai, T. Specific substitutions of light-harvesting complex I proteins associated with photosystem I are required for supercomplex formation. Plant J. 2018, 94, 122–130. [Google Scholar] [CrossRef]
- Yu, X.; Tan, P.; Shen, G.Z.; Zhao, J.D. Recombinant PsbF from Synechococcus sp. PCC 7002 forms β: β homodimeric cytochrome b559. Chin. Sci. Bull. 2003, 48, 563–569. [Google Scholar] [CrossRef]
- Qin, J.; Zhang, J.N.; Liu, D.; Yin, C.C.; Wang, F.M.; Chen, P.Y.; Chen, H.; Ma, J.B.; Zhang, B.; Xu, J.; et al. iTRAQ-based analysis of developmental dynamics in the soybean leaf proteome reveals pathways associated with leaf photosynthetic rate. Mol. Genet. Genom. 2016, 291, 1595–1605. [Google Scholar] [CrossRef]
- Kretchmer, J.S.; Boekelheide, N.; Warren, J.J.; Winkler, J.R.; Gray, H.B.; Miller, T.F. Fluctuating hydrogen-bond networks govern anomalous electron transfer kinetics in a blue copper protein. Proc. Natl. Acad. Sci. USA 2018, 115, 6129–6134. [Google Scholar] [CrossRef]
- Peng, C.; Xu, W.; Hu, L.; Li, Y.; Qi, X.; Wang, H.; Hua, X.; Zhao, M. Effects of the maize C4 phosphoenolpyruvate carboxylase (ZmPEPC) gene on nitrogen assimilation in transgenic wheat. Plant Growth Regul. 2018, 84, 191–205. [Google Scholar] [CrossRef]
- Slewinskia, T.L.; Anderson, A.A.; Price, S.; Withee, J.R.; Gallagher, K.; Turgeona, R. Short-root1 plays a role in the development of vascular tissue and Kranz anatomy in maize leaves. Mol. Plant 2014, 7, 1388–1392. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Zhang, B.J.; Chen, Q.Z. Promotive effect of ATP on photosynthesis and photooxidation in transgenic rice expressing PEPC, PPDK and ME enzyme genes. Nanosci. Nanotech. Lett. 2017, 9, 2133–2138. [Google Scholar] [CrossRef]
- Sjögren, L.L.; Stanne, T.M.; Zheng, B.; Sutinen, S.; Clarke, A.K. Structural and functional insights into the chloroplast ATP-dependent Clp protease in Arabidopsis. Plant Cell 2006, 18, 2635–2649. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Li, X.F.; Zhou, Z.J.; Feng, X.P.; Yang, W.J.; Jiang, D.A. Two rubisco activase isoforms may play different roles in photosynthetic heat acclimation in the rice plant. Physiol. Plant. 2010, 139, 55–67. [Google Scholar] [CrossRef] [PubMed]
- López-Calcagno, P.E.; Abuzaid, A.O.; Lawson, T.; Raines, C.A. Arabidopsis CP12 mutants have reduced levels of phosphoribulokinase and impaired function of the Calvin-Benson cycle. J. Exp. Bot. 2017, 68, 2285–2298. [Google Scholar] [CrossRef] [PubMed]
- Feiz, L.; Williams-Carrier, R.; Wostrikoff, K.; Belcher, S.; Barkan, A.; Stern, D.B. Ribulose-1,5-bis-Phosphate carboxylase/oxygenase accumulation factor1 is required for holoenzyme assembly in maize. Plant Cell 2012, 24, 3435–3446. [Google Scholar] [CrossRef]
- Foyer, C.H. Reactive oxygen species, oxidative signaling and the regulation of photosynthesis. Environ. Exp. Bot. 2018, 154, 134–142. [Google Scholar] [CrossRef]
- Moschen, S.; Bengoa, L.S.; Di, R.J.A.; Caro, M.P.; Tohge, T.; Watanabe, M.; Hollmann, J.; González, S.; Rivarola, M.; García-García, F.; et al. Integrating transcriptomic and metabolomic analysis to understand natural leaf senescence in sunflower. Plant Biotechnol. J. 2016, 14, 719–734. [Google Scholar] [CrossRef]
- Wobbe, L.; Bassi, R.; Kruse, O. Multi-level light capture control in plants and green algae. Trends Plant Sci. 2016, 21, 55–68. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Wang, S.; Tian, L.; Wu, L.; Li, M.; Zhang, J.; Li, P.; Zhang, W.; Chen, Y. Comparative proteomic analysis of the maize responses to early leaf senescence induced by preventing pollination. J. Proteom. 2018, 177, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.Z.; Huang, J.J.; Ding, Y.; Wu, J.M.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L.P. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, 316–322. [Google Scholar] [CrossRef]
- Ma, C.; Zhang, J.; Yuan, J.L.; Guo, J.R.; Xiong, Y.; Feng, Y.L. Differential expression of the micrornas are responsive to drought stress and exogenous methyl jasmonate in wheat (Triticum aestivum). Int. J. Agric. Biol. 2019, 22, 475–486. [Google Scholar]
- Long, S.P.; Bernacchi, C.J. Gas exchange measurements, what can they tell us about the underlying limitations to photosynthesis? Procedures and sources of error. J. Exp. Bot. 2003, 54, 2393–2401. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, C.; Feng, Y.; Wang, J.; Zheng, B.; Wang, X.; Jiao, N. Integrative Physiological, Transcriptome, and Proteome Analyses Provide Insights into the Photosynthetic Changes in Maize in a Maize–Peanut Intercropping System. Plants 2024, 13, 65. https://doi.org/10.3390/plants13010065
Ma C, Feng Y, Wang J, Zheng B, Wang X, Jiao N. Integrative Physiological, Transcriptome, and Proteome Analyses Provide Insights into the Photosynthetic Changes in Maize in a Maize–Peanut Intercropping System. Plants. 2024; 13(1):65. https://doi.org/10.3390/plants13010065
Chicago/Turabian StyleMa, Chao, Yalan Feng, Jiangtao Wang, Bin Zheng, Xiaoxiao Wang, and Nianyuan Jiao. 2024. "Integrative Physiological, Transcriptome, and Proteome Analyses Provide Insights into the Photosynthetic Changes in Maize in a Maize–Peanut Intercropping System" Plants 13, no. 1: 65. https://doi.org/10.3390/plants13010065
APA StyleMa, C., Feng, Y., Wang, J., Zheng, B., Wang, X., & Jiao, N. (2024). Integrative Physiological, Transcriptome, and Proteome Analyses Provide Insights into the Photosynthetic Changes in Maize in a Maize–Peanut Intercropping System. Plants, 13(1), 65. https://doi.org/10.3390/plants13010065