1. Introduction
The infection process of plant RNA viruses is highly dynamic. These viruses employ various strategies to establish themselves within plant cells. This includes processes like frame-shifting, overlapping genome structures, and the production of subgenomic RNAs (sgRNAs). Additionally, during pathogenesis, many plant RNA viruses generate defective RNAs (D-RNAs) and satellite RNAs (satRNAs) to modulate the infection process. These D-RNAs and satRNAs are formed through a recombination and rearrangement of the wild-type virus genome during faulty replication, rendering them incapable of self-replication [
1,
2]. Instead, they rely on the parental wild-type virus acting as a helper virus for replication, when both are present in the same cell. These defective RNAs typically retain the necessary cis-regulatory elements for their replication [
3] and encapsidation [
4], which may facilitate their local or systemic movement within the plant as progeny particles.
Even though these defective RNAs represent only a portion of the viral genome, understanding their functionality is crucial for developing gene-delivery vehicles. Such vehicles can be used for expressing specific genes of interest, either for functional genomics studies or therapeutic purposes. For the expression of foreign genes, two main strategies are commonly employed: the “full” virus vector strategy and the “deconstructed” virus strategy [
5]. In the full virus genome-based strategy, the gene of interest is inserted within the coding frame of the full virus genome, allowing foreign genes to be expressed along with the functional virus as fused mRNA or protein. However, this approach often faces challenges such as low stability, limited expression levels, and restricted cargo capacity, making it less suitable for industrial-scale applications [
6]. To overcome these limitations, deconstructed virus genome-based vectors have gained popularity. In this approach, the virus genome is redesigned to retain only the essential regulatory elements for replication and translation, while eliminating other gene elements necessary for various viral functions such as symptoms, transmission, and movement. Consequently, deconstructed virus vectors rely on a “parental” helper virus for genome functions like replication and translation. This strategy offers a stable and efficient expression of large and multiple foreign proteins, is easy to engineer, and does not pose biosafety concerns associated with the loss of virus function. Several deconstructed virus vectors have been developed using the genomes of popular viruses, with each designed for specific host systems.
In this context, the cucumber green mottle mosaic virus (CGMMV), a member of the Tobamovirus genus with a single-stranded, monopartite RNA genome, is an attractive candidate. CGMMV infects certain cucurbits and
Nicotiana benthamiana, a model plant, causing mild symptoms in a large number of hosts [
7]. Therefore, it holds potential as a biological toolkit for cucurbitaceous crops. The CGMMV genome consists of a single-stranded, positive-sense RNA (approximately 6.4 kb) with four overlapping open reading frames (ORFs) encoding four proteins: two replication proteins (129 kDa and 186 kDa), one 30 kDa movement protein (MP), and a 17–18 kDa coat protein (CP) [
8]. The 5′ terminus of the CGMMV genome plays a crucial role in expressing replicase enzymes, while two 3′ co-terminal subgenomic RNAs (sgRNAs) express the MP and CP [
9]. These sgRNAs are naturally generated due to erroneous replication mechanisms and are shorter than their cognate genomic RNAs, often including 5′ regulatory sequences. The production of sg mRNAs is a strategy employed by eukaryotic RNA viruses for the quantitative and temporal expression of proteins [
10]. This strategy can also be valuable for the heterologous expression of other proteins, using a CGMMV-based expression vector.
However, limited efforts have been made to utilise CGMMV as an expression vector. Initial attempts focused on expressing smaller foreign genes (approximately 100 to 150 nt long) using the full-length CGMMV genome [
11]. Later, Zheng et al. [
12] attempted to express GFP (~800 nt) but got a weak expression. Subsequent efforts, like the one conducted in our laboratory, aimed to express larger genes such as GFP (approximately 800 nt). However, achieving a strong expression was challenging, especially when the foreign gene was inserted at the end of the CP gene. More promising results were obtained when the foreign gene was inserted at a specific location within the CP gene [
13]. Nevertheless, instability in expression beyond 5–10 days post-inoculation (DPI) remained a challenge, highlighting the limitations of full-length viral genome-based vectors. Additionally, the lack of suitable restriction sites and undefined regulatory elements within the CGMMV genome, along with difficulties in loading multiple foreign genes, further restrict its utility.
To address these limitations, our goal is to design a new expression system using CGMMV. Our hypothesis is twofold: (1) deconstructing the CGMMV genome will lead to the development of shorter replicons that can replicate with the help of the main virus, and (2) these short replicons can serve as the basis for a new expression system capable of accommodating large and multiple foreign genes. However, before utilising them as potential expression vectors, it is essential to artificially create deconstructed genomic RNAs of CGMMV. These deconstructed RNAs (dRNAs) should retain minimal regulatory sequences, while preserving their biological properties, including in planta replication, movement, and persistence. This study will pave the way for a more versatile and efficient expression system using CGMMV.
2. Materials and Methodology
2.1. Designing of Deconstructed Virus Genomes and Analysing Their RNA Secondary Structures
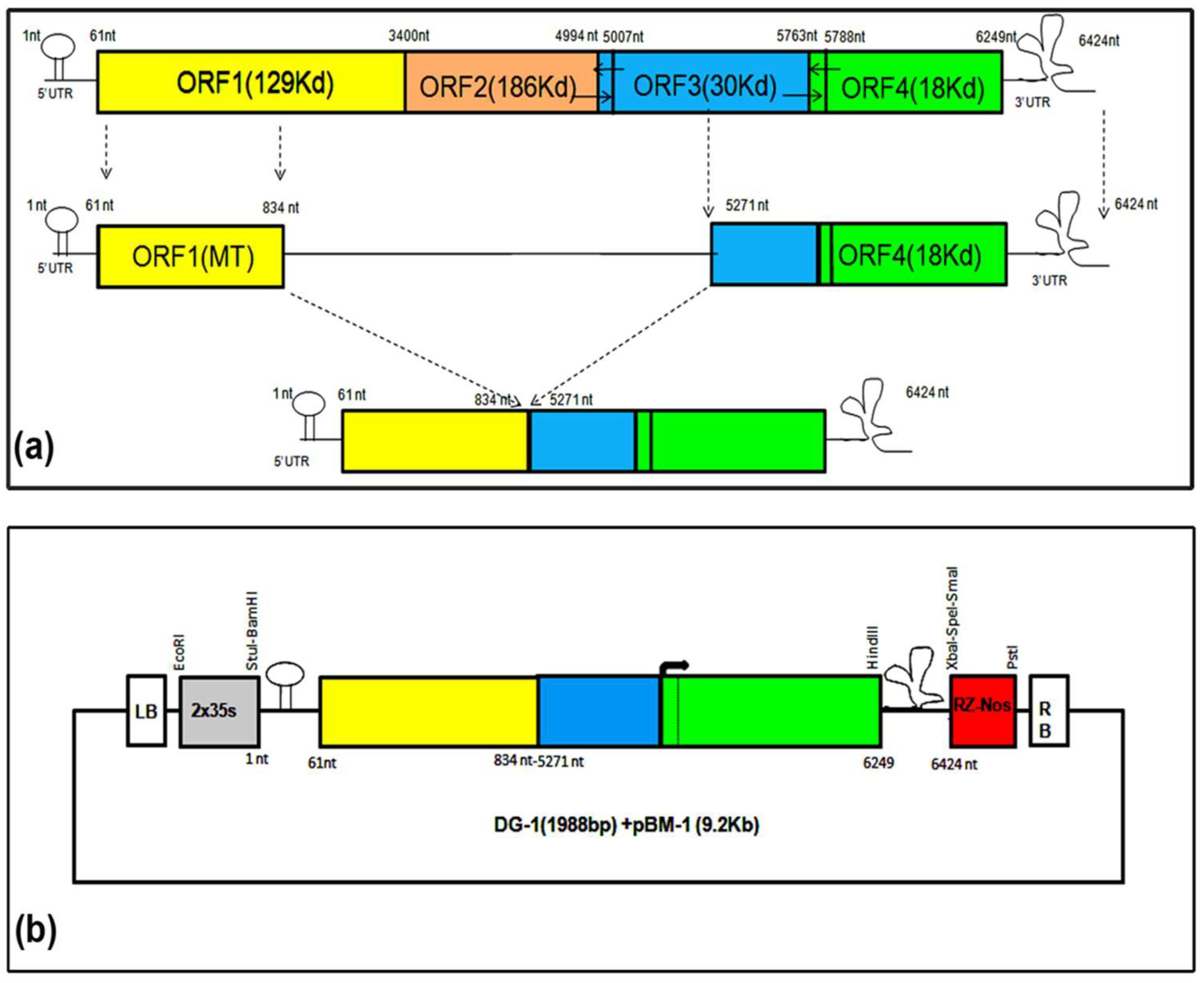
Initially, we designed a deconstructed RNA genome (DG-1) from the CGMMV genome, based on the consensus sequence alignment of the previously reported dRNA genome structure of TMV, which included nucleotides 1–841 from the 5′ terminal and 5182–6424 from the 3′ terminal of the genome [
14]. We used the Mfold Web server (
http://mfold.rna.albany.edu/) to predict the RNA secondary structure of both the wild-type CGMMV and its deconstructed genome [
15]. The input files included the complete RNA genome sequence and the dRNA genome of CGMMV. The secondary structure of dRNA was predicted at 37 °C using Mfold version 2.3 [
15,
16]. We selected potential stable structures with minimal Gibbs free energy from the output file and conducted a comparative analysis to predict RNA pseudoknots and the putative regulatory regions located at the 5′ and 3′ terminals of the genome. This analysis helped us to determine the possibility of trans-replication of the deconstructed genome.
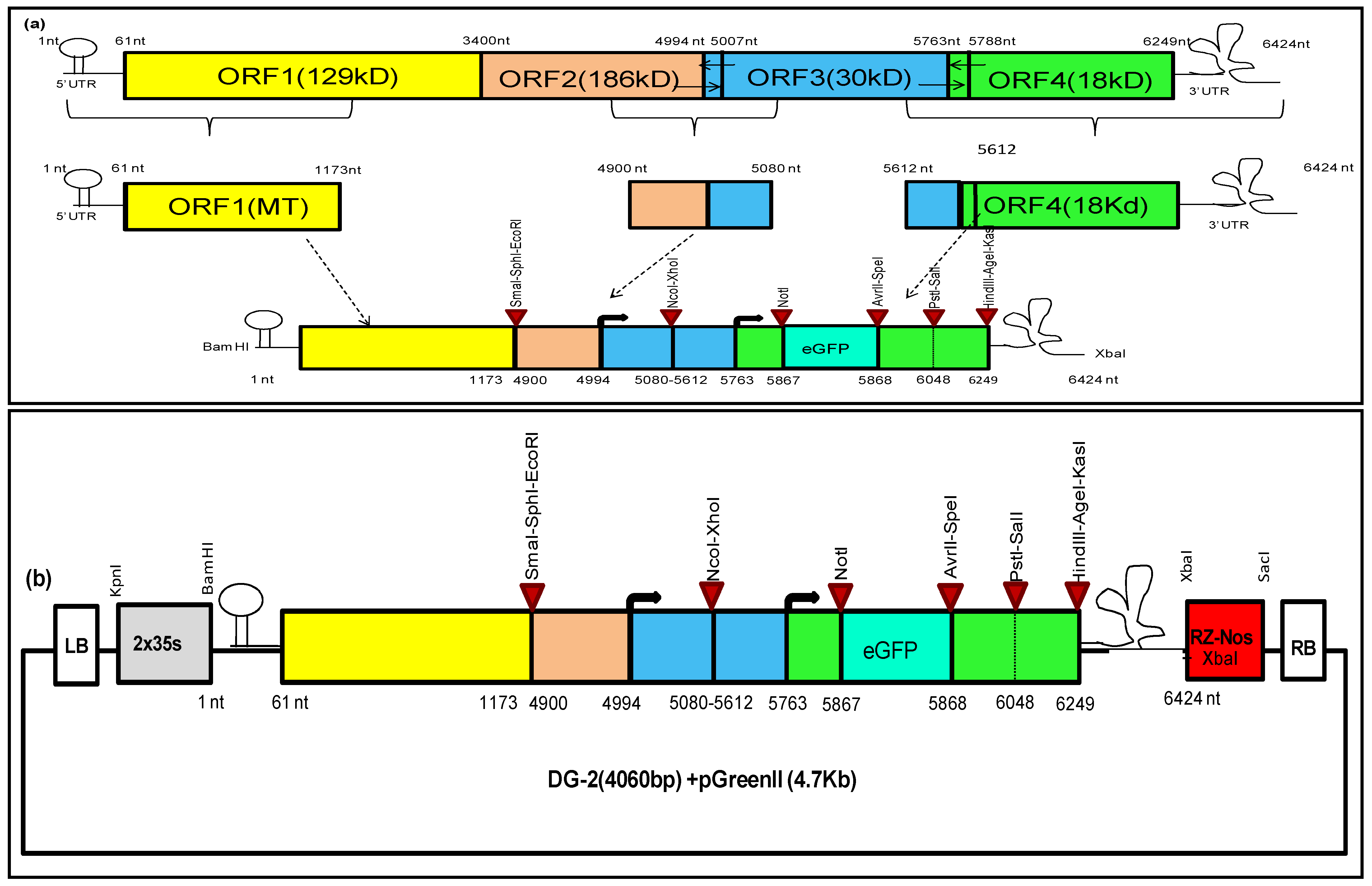
Subsequently, we designed another deconstructed genome (DG-2) of CGMMV based on the in silico prediction of cis-regulatory elements within the CGMMV genome. Our in silico prediction identified the specific cis-regulatory elements crucial for replication and translation. These elements were primarily located within the 5′ and 3′ untranslated regions (UTRs) of the genome and the subgenomic promoter region of the MP and CP [
17]. We merged these sequences to create a complete replicative deconstructed genome, DG-2. To facilitate the insertion of multiple foreign genes, we incorporated some restriction sites at various locations within the DG-2 genome. We used the Mfold web server (
http://mfold.rna.albany.edu/) to predict the RNA secondary structure of DG-2 and compared it with that of the wild-type CGMMV. Similar to DG-1, we used the complete RNA genome sequence and the dRNA genome of CGMMV as input files, selecting potentially stable structures with minimal Gibbs free energy from the output file. This comparative analysis aimed to identify the conservation of regulatory elements located at the 5′ and 3′ terminals of the genome.
2.2. Development of Deconstructed Virus Genomes
The deconstructed plasmid (pDG1) was derived from the infectious clone (pBP4) of CGMMV-BgDel isolate (CGMMV-BP4) [
13,
18] by deleting the original plasmid construct (pBP4), using an LC-PCR-based cloning strategy [
19,
20]. This mechanism involved a one-step PCR, using two long primers (P5R and P6F) designed with a high GC content and sharing some degree of homology at their 5′ end (
Table S1). Another extra primer (P7) was used for self-circularisation. The PCR reactions were performed using a 50 µL master mix containing 1× PCR buffer, 0.8 mM dNTPs, 1 U hi-fidelity Phusion Taq DNA polymerase, 100 ng of the vector (plasmid), and 1 mM of each primer. The thermal cycling included 1 cycle at 98 °C for 5 min; 20 cycles at 98 °C for 20 s and at 65–70 °C for 8 min (@ 1 kb/30 s); and a final hold at 4 °C. After the PCR, the products were treated directly with the Dpn1 enzyme at a rate of 1U per 50 µL for 1 h 30 min without further purification. The resulting products were then transformed into highly efficient competent
E. coli DH5α cells (10
8 to 10
9 cfu/µg). A colony PCR was used to screen for colonies containing the desired plasmid constructs, and positive colonies were further verified through restriction digestion of the plasmids. The PCR-derived deletion of the constructs was confirmed via sequencing, to ensure the presence of only the desired modifications.
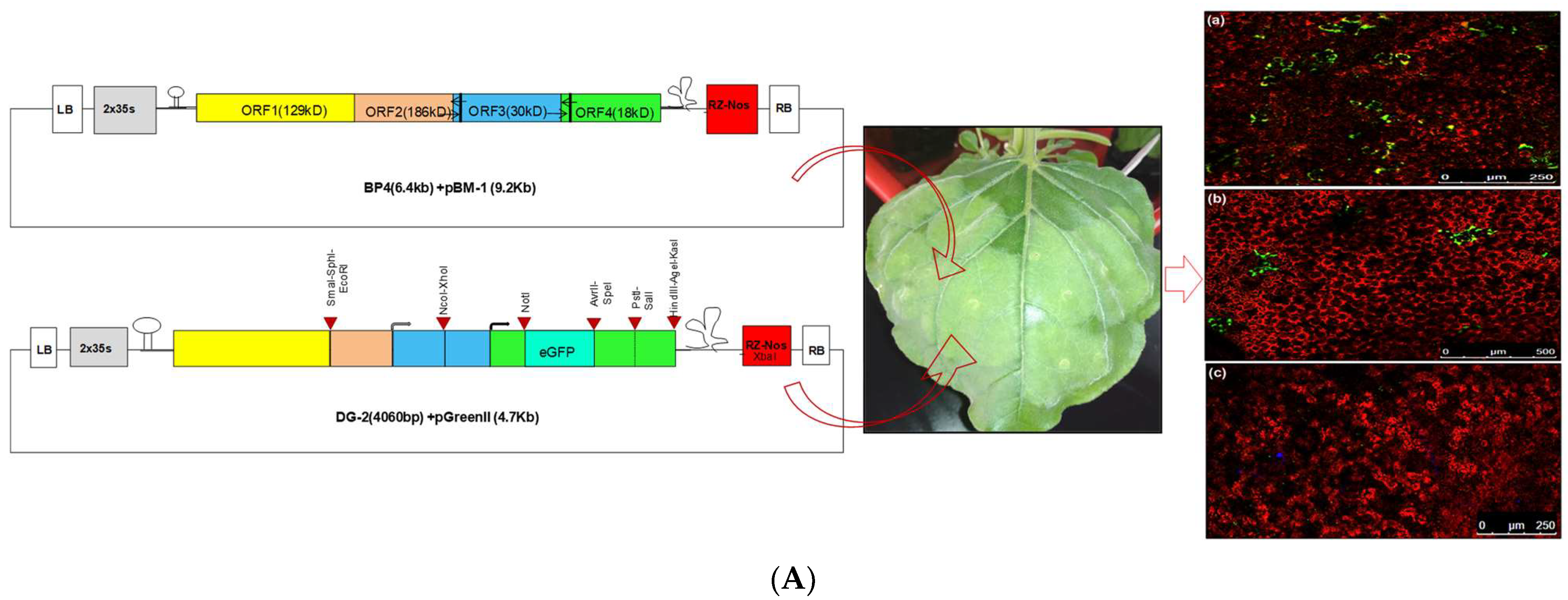
The second deconstructed genome, DG-2, was designed by incorporating the partial genome sequence of CGMMV within the 2 × 35 s promoter and ribozyme (Rz) site with a Nos terminator. After synthesis, the entire cassette (approximately 4062 nt) was incorporated into the pUC57 vector backbone (2.7 Kb) using the KpnI and SacI restriction sites. Subsequently, the cassette was directly cloned into pGreen II (0029) using the same restriction sites. This process removed the entire multiple cloning sites (MCS) from the pGreenII (0029) backbone (4632 bp), allowing for the utilisation of these sites for further DG-2 genome modifications. Positive clones were verified through restriction digestion using the BamH1 and HindIII restriction enzymes, releasing desirable 2.78 kb fragments only from the vector backbone (4.7 Kb). The confirmed plasmid constructs were transferred into Agrobacterium GV-3101, along with the pSoup helper plasmid, via electroporation. The co-transformation of pGreen and pSoup was conducted at equal concentrations of both constructs, with only Kanamycin (not tetracycline) and Rifampicin for the selective screening of the transformed Agrobacterium colonies. The transformed Agrobacterium colonies were confirmed via a colony PCR, and positive colonies were maintained for further use.
2.3. Plant Inoculation of Deconstructed Virus Genomes along with Wild-Type Virus Strains
The cDNA plasmid constructs of the deletion mutant, deconstructed genome-1 (pDG1), and the synthetic genome construct (pDG2), along with the wild-type CGMMV infectious clones, viz., symptomatic constructs (pBP4) [
20] as well as asymptomatic constructs (pBP7) were introduced into highly efficient competent cells of the Agrobacterium tumefaciens strain GV3101, using the freeze–thaw method [
21]. Plasmid constructs (1–2 µg) were mixed with 200 µL of Agro-competent cells and incubated on ice for 20 min. The mixture was then subjected to freezing in liquid nitrogen for 20 s, followed by thawing at 37 °C for 5 min, with subsequent incubation on ice (0 °C) for 20 min. Afterwards, 5 mL of LB medium was added, and the culture was incubated at 28 °C for 3 h with continuous shaking (220 rpm). Transformed Agro cells were plated on appropriate antibiotic-containing plates (kanamycin and rifamycin) and incubated at 28 °C. The presence of recombinant clones in the Agro-culture was confirmed via a colony PCR, using gene-specific primers. The confirmed colonies were picked and individually grown in 5 mL of LB broth containing 50 μg/mL kanamycin and 15 μg/mL rifampicin at 28 °C for 18–24 h, with shaking at 200 rpm. The overnight Agro-culture was centrifuged at 5000 rpm for 10 min and resuspended in 50 mL of agroinfiltration buffer containing 10 mM 2-(N-morpholino) ethane sulfonic acid (MES, pH 5.7), 10 mM MgCl
2, and 150 µM Acetosyringone, to reach a final OD of 0.6 for infiltration. The resuspended Agro-culture was further incubated at 28 °C for 1–3 h, with agitation at 50 rpm. Subsequently, the Agro-culture containing the pBP4 construct was infiltrated into 3-week-old
Nicotiana benthamiana plants, using a sterile syringe on the abaxial leaf surface. To confirm the trans-replication of the deconstructed genomes, the Agro-cultures of the pDG1 (cDNA clone) and pDG2 (synthetic) constructs of CGMMV were either co-infiltrated (mixed at a 1:1 ratio) simultaneously with the wild-type CGMMV (pBP4/pBP7) or post-infiltrated (after 7 days post-inoculation of the wild-type CGMMV) into CGMMV-infected host plants. The infiltrated plants were maintained in a greenhouse with 16 h of daylight and 8 h of darkness at 25 °C, and observations were recorded. Each experiment was repeated three times for data replication and robustness.
2.4. Infectivity Assay of Wild-Type CGMMV
Leaf samples from pBP4-infected plants were harvested at 7 days post-inoculation (DPI) to confirm virus infection. The total plant RNA was isolated from symptomatic leaves, using a plant total RNA isolation kit (Promega, Madison, WI, USA) following the manufacturer’s protocol. For the cDNA synthesis, the total RNA was used as a template for a reverse transcriptase reaction with Superscript III (Invitrogen, Waltham, MA, USA), using a CGMMV-specific 3′ terminal primer (BM489R) according to the manufacturer’s protocol. The infectivity of the wild-type virus was detected through RT-PCR using CP gene-specific primers (BM-1178F and BM-1179R). After wild-type CGMMV infection, the deletion mutant was infiltrated in the case of post-infiltration.
2.5. Trans-Replication Assay of Deconstructed Genomes of CGMMV in Different Hosts
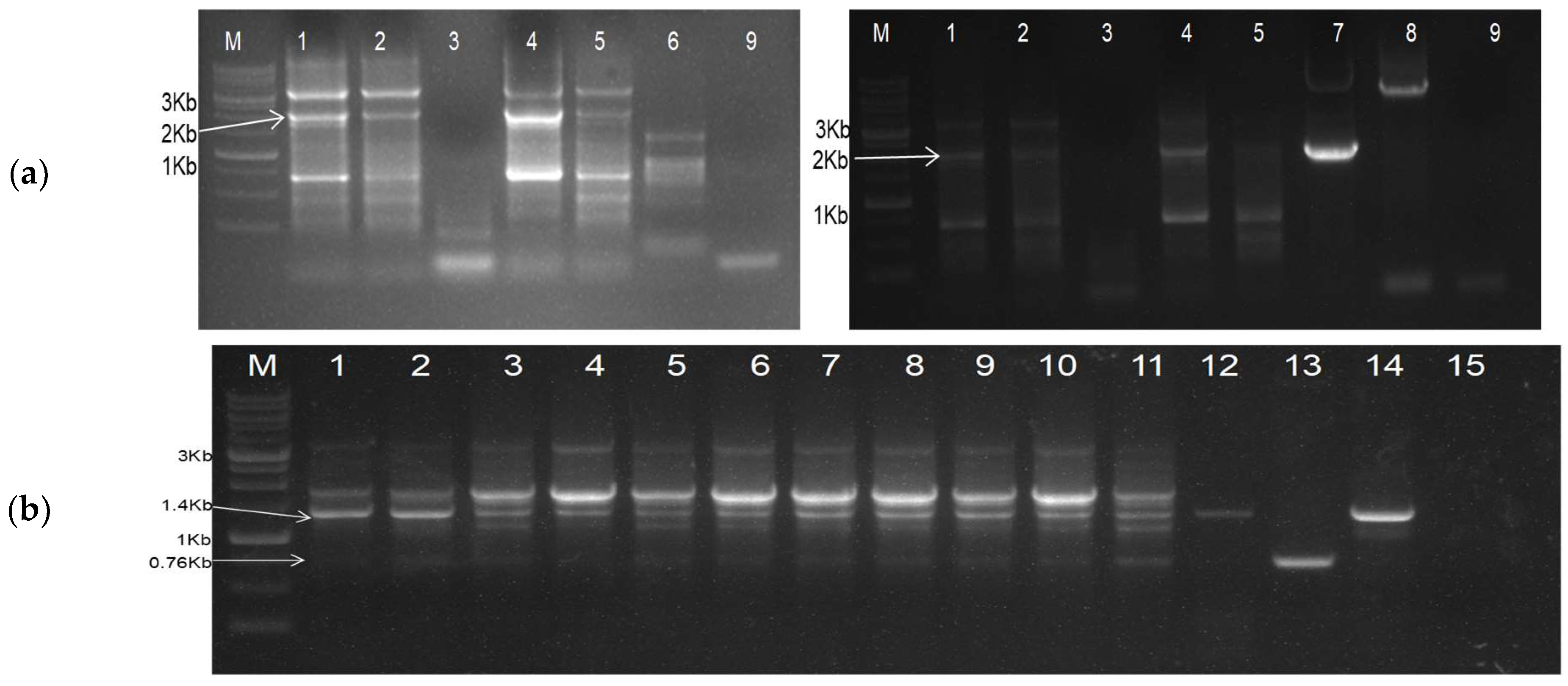
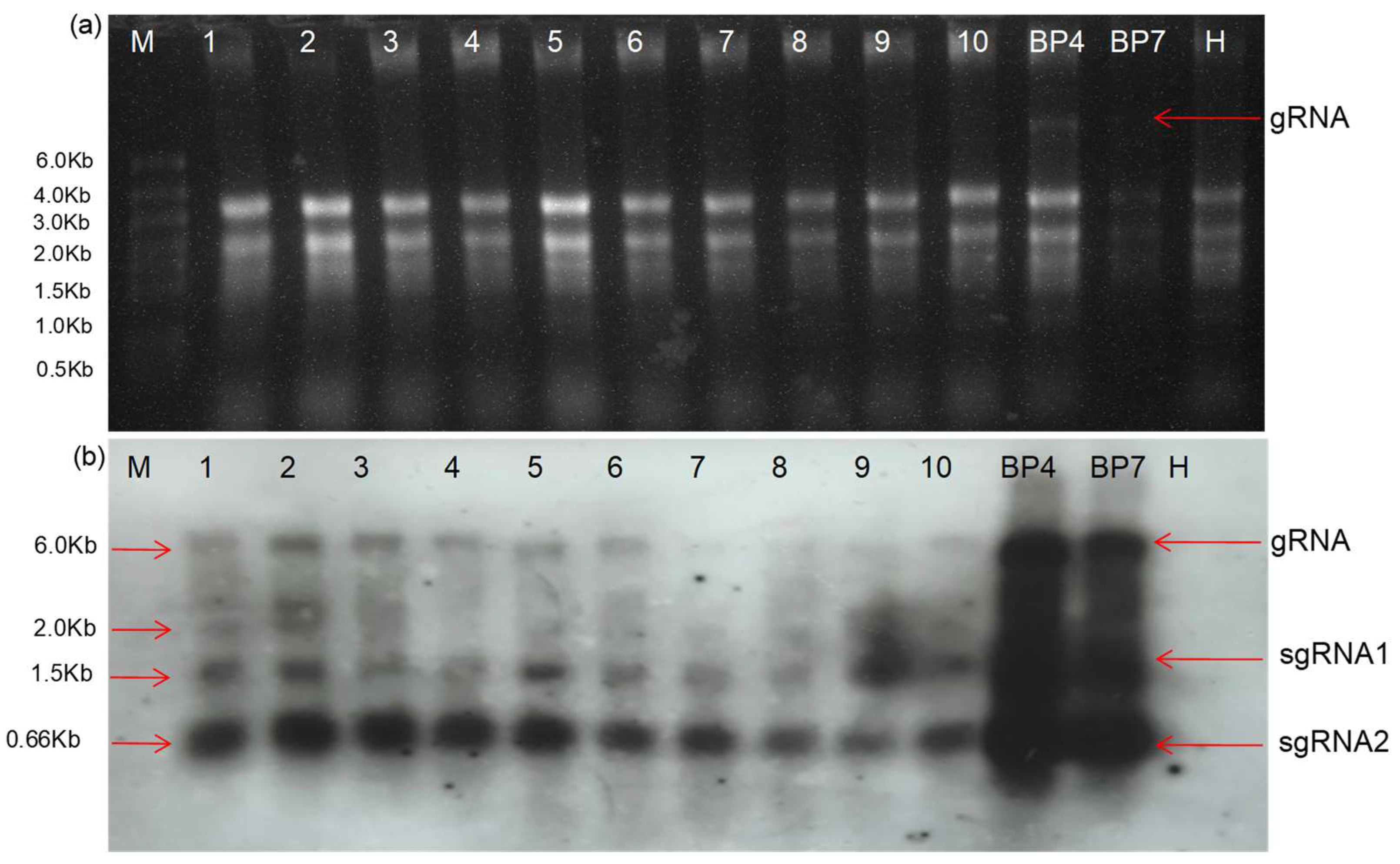
To detect the trans-replication of DG-1, RNA was extracted from infiltrated leaves and the systemic leaves of different host plants at different time intervals (7 DPI, 14 DPI), followed by a cDNA synthesis using the above-described protocol. The trans-replication of DG-1 was detected via RT-PCR, using virus-specific terminal primers (BM-486F and BM-489R). For additional confirmation, another duplex RT-PCR-based detection approach was employed, using virus-specific internal primer pairs (BM-1172F and BM-1173R), where the forward primer (BM-1172F) was designed from the flanking sequences (802–827 of DG-1 and BP4) located just prior to the junction/deleted site (835 nt) of DG-1, and the reverse primer (BM-1173R) was derived from the conserved sequence at 5981–6005 nt of BP4 and DG-1 (i.e., CP gene of CGMMV). On the other hand, the trans-replication and movement of DG-2 were detected using specific primers, BM-1265F and BM-1266R. RNA extracted from infiltrated and systemic tissues was used for testing. A simultaneous detection of helper CGMMV was carried out using primers 96F and 1268R. The PCR protocol was standardised, with denaturation at 94 °C for 3 min, followed by 35 cycles of denaturation at 94 °C for 30 s, annealing at 60 °C for 30 s, extension at 72 °C for 30 s, and a final extension at 72 °C for 10 min. Confirmation was made based on gel electrophoresis.
2.6. Movement Assay of Deconstructed Genomes of CGMMV
The in planta systemic movement of CGMMV’s dRNA was traced in different temporal and spatial scales. A total of ten CGMMV-infected plants were agroinfiltrated with DG-1, and leaf samples were harvested at various time intervals (from 1 DPI at 24 h intervals) and spatial scales (from infiltrated leaves to upper systemic leaves). RNA was extracted, and cDNA was prepared as per the manufacturer’s protocol. An RT-PCR was employed to detect DG-1 using terminal primers BM-486F and BM-489R. Systemic movement across different leaves was assessed at 14 DPI, to determine its distribution within the plant. The experiment was repeated three times for reliability.
2.7. Vector Function Assay of Deconstructed Genomes within CGMMV-Infected N. benthamiana
2.7.1. Gene-Silencing Assay of Deconstructed Genome-1-Expressing NbPDS Gene Sequence
Functional Validation of pDG(PDS)-1 Constructs
The phenotypic expression of the gene silencing was monitored during the 15–60 DPI period, starting from the appearance of photobleaching symptoms. The efficiency of the gene-silencing vector was estimated based on the percentage of plants showing photobleaching symptoms [
22]. The infiltrated plants were tested for confirmation of the replication and movement of the pDG(PDS)-1 construct using an RT-PCR. A duplex RT-PCR was established using two pairs of primers: one set containing BM 1180R and BM 1269R for the specific detection of pDG(PDS)-1, while the other set, containing BM-96F and BM-1268R, was used for the specific detection of helper CGMMV simultaneously in the same PCR reaction.
Northern Blot Analysis for the Confirmation of the Deletion RNA Replicon
RNA extracted from different experimental samples was used for the Northern blot analysis. The study was conducted using the DIG-High Prime DNA Labelling and Detection Starter Kit II (Roche Applied Science, Penzberg, Germany). A digoxigenin-labelled positive-strand RNA genome-specific DNA probe was generated with DIG-High Prime and tagged with the 662 nt from the 3′ terminal of the CGMMV genome. This DIG-labelled probe was used for hybridisation with membrane-blotted RNA, which had been previously separated in an agarose gel using the standard methodology. The hybridised probes were immuno-detected with anti-digoxigenin-AP and Fab fragments, and then visualised with the chemiluminescence substrate CSPD, following the manufacturer’s protocol. The enzymatic dephosphorylation of the CSPD by alkaline phosphatase led to light emission at a maximum wavelength of 477 nm, which was recorded on X-ray films. Film exposure times ranged from 5 to 30 min. An in vitro RNA transcript was used as the positive control.
2.7.2. In Planta Expression of Foreign Protein Using Deconstructed Genome-2
Detection of GFP Fluorescence through Confocal Microscopy
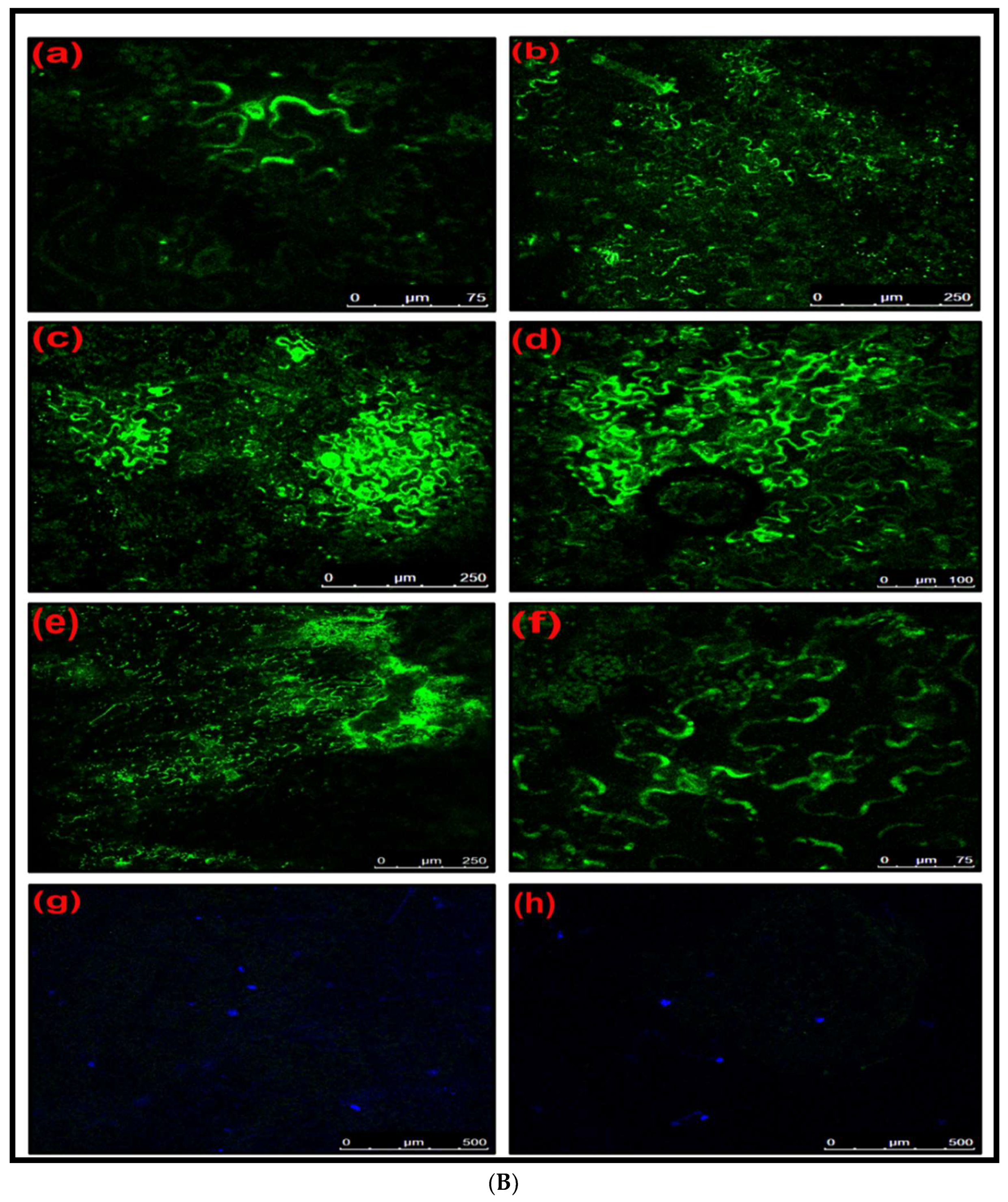
Initially, CGMMV-infected plants were selected for the agroinfiltration of the DG-2 construct. Starting from 2 DPI, plants were periodically analysed for GFP expression through confocal microscopy. Periodic observations of GFP expression were taken at 2-day intervals. pDG-2-infiltrated plants exhibited GFP expression in the inoculated area. CGMMV-infected plants, pDG-2-infiltrated plants, and PBS buffer-inoculated (mock) plants were chosen as control groups. GFP expression was confirmed by confocal microscopy, with a wavelength range of 490–510 nm.
Western Blot Assay for the Confirmation of GFP Expression
Protein extraction was performed using leaf tissue (0.8 g) from inoculated and systemic leaves of
N. benthamiana. The tissue was initially ground to a fine powder in liquid nitrogen, using an autoclaved, pre-chilled mortar and pestle (−70 °C). The powder was then resuspended by inversion mixing in 300 µL of protein extraction buffer (0.1 M KH
2PO
4, pH 6.5, 0.5 mM PMSF, and 10 mM β-mercaptoethanol). Extracts were centrifuged for 20 min at 15,000 rpm at 4 °C, and the supernatant was collected and stored at −20 °C. Protein samples were prepared by mixing the crude protein with an equal volume of sample buffer and boiling it for 5 min in water. A Standard Sodium Dodecyl Sulphate Polyacrylamide Gel Electrophoresis (SDS-PAGE) was performed as per the protocol by Laemmli [
23] to separate the proteins. The proteins were then transferred to a nitrocellulose membrane (NCM). The NCM was treated with a GFP-specific primary antibody, followed by a secondary antibody, goat anti-rabbit IgG conjugated with alkaline phosphatase (AP). After two washes with TTBS and one wash with TBS, the reaction was detected by adding the substrate (BCIP/NBT).
2.8. Quantitative Estimation of Replication Rate of Deconstructed Genomes
To quantify the replication level of the deletion mutants of CGMMV, a semi-quantitative experiment was conducted. DG-1 was agroinfiltrated into previously infected N. benthamiana plants. Ten plants were selected, based on their similar age and growth pattern. The 5th leaf of each infected plant was chosen for the agroinfiltration of DG-1. Small leaf discs (5 mm) were cut from the infiltrated part of all ten plants regularly at 24 h intervals, starting from the 1st DPI. These discs were combined for RNA extraction. The RNA was extracted following the previously described protocol, quantified, and treated with RNase-free on-column DNaseI to remove any influence from the infiltrated plasmid construct. The RNA samples were then used to synthesise first-strand cDNAs with an oligo(dT)20 primer using Superscripts III (Invitrogen, USA). An RT-PCR was performed to confirm the infectivity of CGMMV infectious clones in plants, using internal specific primers BM-1172F and BM-1173R. A quantitative RT-PCR was carried out to examine the RNA level of the DG-1 construct in the silenced, as well as healthy, plant leaves, using the primers BM 1180F and BM 1269R. The previously described PCR reaction mixture and program conditions were used.
Further, to quantify the DG-2 transcripts in inoculated plants, as well as control healthy transcripts, a qPCR was performed, targeting the GFP gene. The total RNA was extracted from 100 mg of plant tissue, using a plant RNA extraction kit (SV total RNA isolation system, Promega, USA). The qPCR was performed using specific primers to GFP (BM-1265F and BM-1266R) and actin1 (as an internal control) from N. benthamiana (BM-849F & BM-850R) with the SYBR green master mix from KAPA SYBR FAST qPCR Kits (KAPA Biosystem, Wilmington, MI, USA). The thermal cycling program for both their primers was 95 °C for 1 min, followed by 45 cycles at 95 °C for 20 s, 60 °C for 20 s, and 72 °C for 20 s. The reaction was performed in a qPCR machine Light Cycler 96 SW (Roche, Indianapolis, IN, USA).
4. Discussion
The emergence of defective genomes during virus pathogenesis is a natural phenomenon. These defective genomes are spontaneously generated during the replication of a virus, especially those with RNA genomes [
27], particularly when their concentration reaches a high titer [
26]. It is hypothesised that the premature detachment of RdRp from its negative-strand template during replication (the breakpoint), and the subsequent reattachment to the same template, lead to the synthesis of defective genomes [
28,
29,
30]. Although these genomes are defective in terms of self-replication and pathogenesis due to the absence of substantial portions of their parental viral genome, they still carry some of the key regulatory genomic elements essential for replication. Consequently, these truncated forms can only replicate with the assistance of wild viruses, which act as helpers by providing replicase enzymes. They may either interfere with the replication of the parental virus [
31] or not, hence they are referred to as defective viral genomes (DVGs) [
32,
33]. They have a significant functional impact on the pathogenesis and evolution of the wild-type virus. Naturally, their occurrence has been reported during late infection events of many human and animal viruses, such as dengue virus [
34], Ebola virus [
35], hepatitis C virus [
36], influenza A virus [
37,
38], mumps virus [
39], poliovirus [
40], respiratory syncytial virus [
41], Sendai virus [
42], Sindbis virus [
43], etc. Similar subviral agents are also evident [
44] in the case of many plant viruses like Bromovirus [
45], Carmovirus [
46], Flexivirus [
47], Tombusvirus [
48], etc., which are synthesised de novo [
49]. However, such a documentation of naturally occurring DVGs is lacking in the case of the majority of Tobamoviruses [
50]. This raises questions about the efficacy of CGMMV in supporting the trans-replication of subgenomic virus particles. Therefore, different artificial forms of dRNAs were generated from their genome to study their functional significance.
Previously, DVGs were artificially generated from the full-length RNA genome of TMV by deleting internal genome sequences [
24,
50,
51] that contain cis-regulatory elements for replication and movement. Building on this information from TMV, we attempted to develop a deconstructed genome called DG-1 derived from the CGMMV genome, and its functionality was tested. The DG-1 construct of CGMMV lost its ability for self-replication and in planta movement due to the large internal deletion of the genome, including the helicase, RNA-dependent RNA polymerase domain of the replicase enzyme, and a significant part of the movement protein. As a result, the DG-1 replicon was not detected when infiltrated alone, neither in the infiltrated leaves nor in the systemic leaves of
N. benthamiana, consistent with the previous findings of Ogawa et al. [
52], where large deletions of the genes encoding the methyltransferase, helicase, and RdRp domains of replicase generated replication-defective mutants in TMV. DG-1 can only replicate and move systemically within
N. benthamiana with the help of wild-type CGMMV. In this scenario, wild-type CGMMV provides the replicase enzyme to support the replication of DG-1, similar to the earlier experiments by Raffo and Dawson [
53] in TMV. They reported that artificially created subgenomic replicons—named KL, which retained 1–256 nt of the 5′ terminal and 5237–6395 nt of the 3′ terminal ends, and KLL, containing only 1–258 nt of the 5′ terminal ends and 4900–6395 nt of the 3’ terminal ends of the TMV genome—could replicate and spread systemically within tobacco plants once co-inoculated with wild-type TMV [
53,
54]. This supports our hypothesis that wild-type CGMMV can assist in the trans-replication of subviral particles. However, the DG-1 replicon (dRNA) was not detected in the infiltrated leaves of
N. benthamiana when co-infiltrated with wild-type CGMMV (either symptomatic BP4 or asymptomatic BP7). This differs from previous findings for TMV, where the trans-replication of dRNAs was reported when co-inoculated with parental TMV, either in tobacco protoplasts or in the leaves of
N. benthamiana plants. This difference might be related to the methods of delivery in the plant system. Previously, in vitro transcripts of dRNA genomes were used for the inoculation of protoplasts, and after 1–2 days of growth in protoplasts, the cell lysate containing the active viral culture was directly inoculated into the plant leaves, allowing for the rapid multiplication of dRNAs along with their helper virus. In contrast, during co-agroinfiltration of the cDNA construct of both DG-1 and ‘helper’ CGMMV, the wild-type virus does not sufficiently support the amplification of DG-1 dRNA transcripts, due to the lack of sufficient replicase enzymes in planta. In the initial stage of infection by CGMMV, it produces replicase to replicate its own genome for the establishment of infection and colonisation. It requires time to replicate its own genome up to a critical level at which it will support the replication of other dependent genomes, such as the replication of dRNA immediately after infection. Identifying this critical time is necessary for achieving a sufficient amplification of DG-1, which may require further refinement in the delivery system.
Naturally occurring dRNAs possess certain cis-acting elements that are necessary for efficient replication and movement with the help of the parental virus. This principle was applied to generate DG-1 based on previously reported information on TMV dRNAs. DG-1 contains 1–834 nt of the N-terminal part and 5271 to 6424 nt of the C-terminal part of the genome, based on the genome map of the deconstructed RNA (∆HINC151) of TMV [
24], which retained 1–841 nt (258 aa) of the N-terminal and 5182–6395 nt of the C-terminal of its genome. The evidence of in planta trans-replication and systemic movement of DG-1 with the help of wild-type CGMMV is in accordance with the ∆HINC151 of TMV. Interestingly, the ∆HINC151 of TMV is also reported to accumulate in the inoculated leaves of
N. benthamiana about 4–5 times more highly than its helper TMV [
24]. However, in our case, DG-1 is found to replicate at a maximum of 4.5 times over actin, which is about 7.7 times lower than wild-type CGMMV. After the cellular accumulation of DG-1 within infected tissue, DG-1 can spread systemically up to the tenth leaf or even more (as per our observation) in
N. benthamiana via vascular bundles, which is not evident in the case of the ∆HINC151 of TMV, which is reported to move within specialised non-vascular tissue, thus restricting its systemic movement [
24]. By contrast, the DG-1 replicon can systemically move long distances throughout the plant, but its uniform distribution in all local tissue is limited. This suggests that the 258 aa of the MT domain of CGMMV is necessary for the long-distance movement of dRNA but has a negative impact on its higher accumulation. This finding is consistent with the previous report of Knapp et al. [
24,
51] on TMV, where they elucidated the functional significance of the N-terminal MT domain containing (1–258 aa) in virus movement [
24]. Furthermore, this MT domain also plays a crucial role in the formation of a methylated cap that enhances the stability of viral genomic RNA. Interestingly, this 5′ RNA capping mechanism is conserved among diverse members of the Alphavirus-like superfamily, despite their limited amino acid identity [
53]. A comparative analysis of the MT domain of various plant-infecting viruses reveals some conserved amino acid residues, including histidine (H81), aspartate (D134), arginine (R137), and tyrosine (Y271), located at its N-terminal part, forming a catalytic tetrad that might have a significant role in cap formation [
55]. Disruption of any one of these residues greatly hampers the methyltransferase and AdoMet-dependent guanylyltransferase activity of this enzyme, affecting RNA capping [
56] and resulting in the subsequent instability of newly synthesised RNA, as observed in the Semliki Forest virus [
57] and bamboo mosaic virus [
58]. The existence of a partial N-terminal domain in the DG-1 of CGMMV may also impair RNA stability. Thus, despite its replication, the limited stability of the DG-1 replicon affects its higher accumulation.
To address the shortcomings of DG-1, DG-2 was created by incorporating 1173 nt (371 aa) of the MT domain, with the aim of achieving higher replication. Additionally, multiple cloning sites were incorporated under the control of subgenomic promoters, allowing for the simultaneous expression of multiple foreign genes. When the DG-2 construct was infiltrated into CGMMV-infected
N. benthamiana, DG-2 was trans-replicated with the help of wild-type CGMMV. Detection was carried out using an RT-PCR, but DG-2 was not detected within the systemic leaf tissue. This restricted movement of the deconstructed genome was previously reported for TMV [
51], where two categories of dRNAs were identified: ones that could not move but were replicated by the wild-type virus and others that could move but were not replicated by the wild-type virus. RT-PCR-based detection showed that the replication rate of DG-2 gradually increased but remained restricted within the infiltrated tissue. This indicates that the C’-terminal part of the MT domain (i.e., 258–371 aa) is involved in the higher accumulation of dRNAs, possibly due to the proper capping mechanism. Furthermore, this region may also be involved in the restricted movement of dRNAs. Similar evidence was reported by Knapp et al. [
24], demonstrating the higher accumulation but restricted movement of the deconstructed RNA genome, ∆Cla151, which possessed the 425 aa of the MT domain. Previously, the role of a non-conserved region located between the MT domain and helicase domain in cell-to-cell movement was reported, and the mutation of certain nucleotides in the 248 aa to 538 aa region hampered this function in TMV [
59]. From the confocal microscopic analysis of DG-2 in the infiltrated tissue, it was evident that the localisation of DG-2 was scattered within the lamina. Therefore, it could be predicted that the cell-to-cell movement of DG-2 might be restricted due to the extensive modification of the MT domain. Further, a qPCR assay of the DG-2 replicon indicated an increase in its replication rate over time (within 15 DPI), from 0.067 to 0.098 ng/g tissue. The quantification of GFP expression within the infiltrated tissue also indicates that trans-replication of DG-2 is assisted by wild-type CGMMV. Apart from its trans-replication, DG-2 also exhibits better translational ability. To demonstrate this, the GFP protein was inserted within the coding frame of the CP and was successfully expressed within infiltrated tissue. The expression of GFP was visualised over time in confocal microscopy, showing that GFP expression began on the 2nd DPI and reached a high level between the 6th and 10th DPI, gradually decreasing but remaining expressed up to the 15th DPI. This finding was also supported by Western blot assays, where the amount of GFP translation in the form of the CP-fused protein (48 kDa) was quantified using polyclonal anti-GFP antibodies. The pattern of GFP expression was consistent with the confocal visualisation, confirming that DG-2 can be used to express a foreign protein in plant tissues. Therefore, DG-2 can be utilised to establish a bipartite vector system using the CGMMV genome. A similar bipartite system based on TMV and its dRNA was developed by Knapp et al. [
51], which was later explored as a novel two-component vector system (dRT-V) for the high-level expression of multiple therapeutic proteins, including a human monoclonal antibody, in plants [
14]. This new design based on the deconstructed genome of TMV proved to be a successful alternative. In this system, a defective/deconstructed RNA (dRNA) of TMV was created by a major deletion of replicase and movement protein, and its functionality (trans-replication and movement) was demonstrated using a helper TMV construct. Importantly, two foreign genes were inserted, one in the helper construct and one in the deleted construct, enabling simultaneous expression in the same cell. This non-competitive bipartite module is named the defective RNA (dRNA)-based TMV vector (dRT-V), and it is used for the expression of mAbs IgG against the antigen of Bacillus anthracis in
N. benthamiana at 120 mg/kg FWT. Similarly, our bipartite system can also be exploited for the expression of multiple proteins in plants.
In conclusion, two novel two-component CGMMV-based vector systems have been developed (
Figure 10). The DG-1 replicon can replicate
in trans with the assistance of its helper CGMMV but exhibits lower stability, reducing its accumulation in high copy numbers. However, it can move cell to cell and cross the vascular bundle barrier to reach the upper leaves of
N. benthamiana. In this way, DG-1, along with helper CGMMV, represents a bipartite model system with functional similarities to the multipartite genome systems of other plant viruses. In planta functional validation of these bipartite systems in CGMMV demonstrates their utility as gene-silencing vectors for deciphering plant functional genomics. In the second system, DG-2 replicates at a much higher rate but remains localised within the infiltrated tissue. It also exhibits a translational ability for foreign proteins, as demonstrated through the expression of GFP within
N. benthamiana. Thus, the creation of a bipartite system using the CGMMV genome has been successfully achieved.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}