

Comprehensive Transcriptome and Proteome Analyses Reveal the Drought Responsive Gene Network in Potato Roots


 ,
,
Abstract
:1. Introduction
2. Results
2.1. Results of the Sequencing of the Entire Transcriptome and Proteome
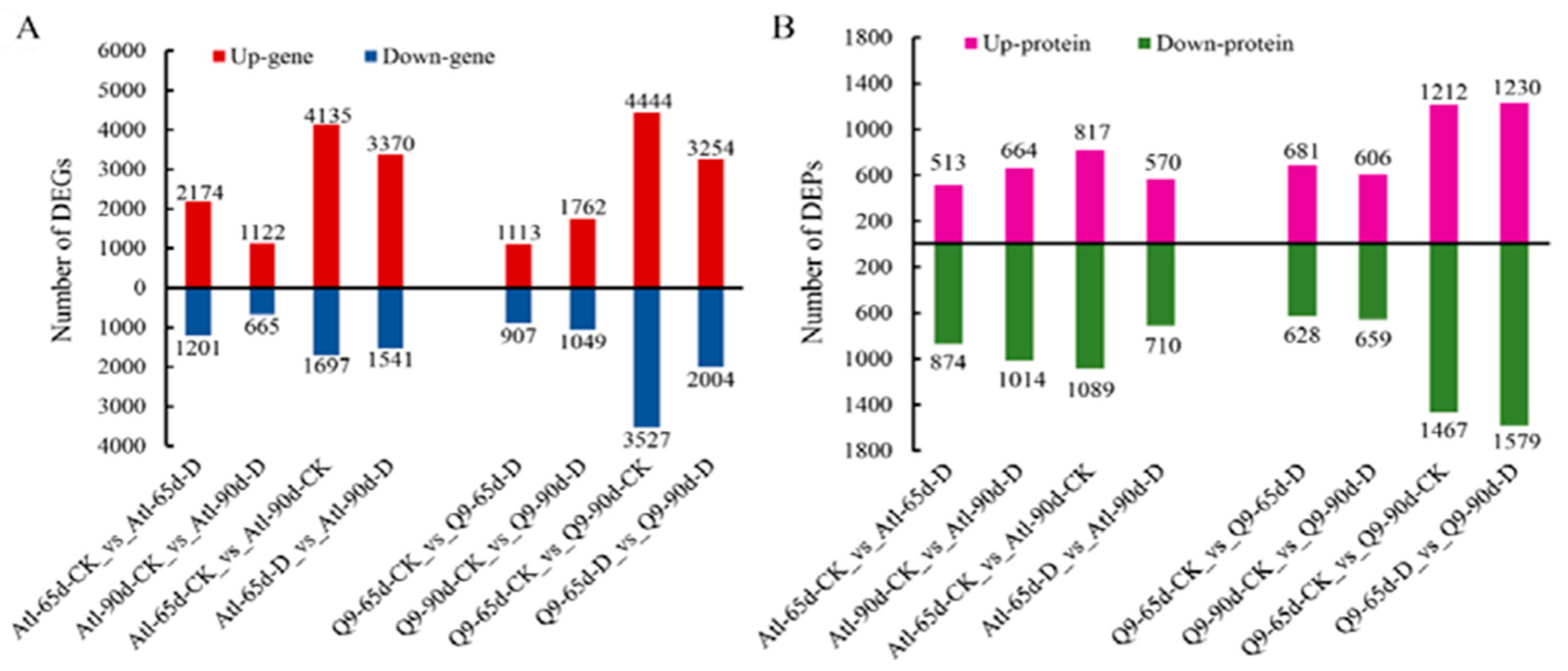
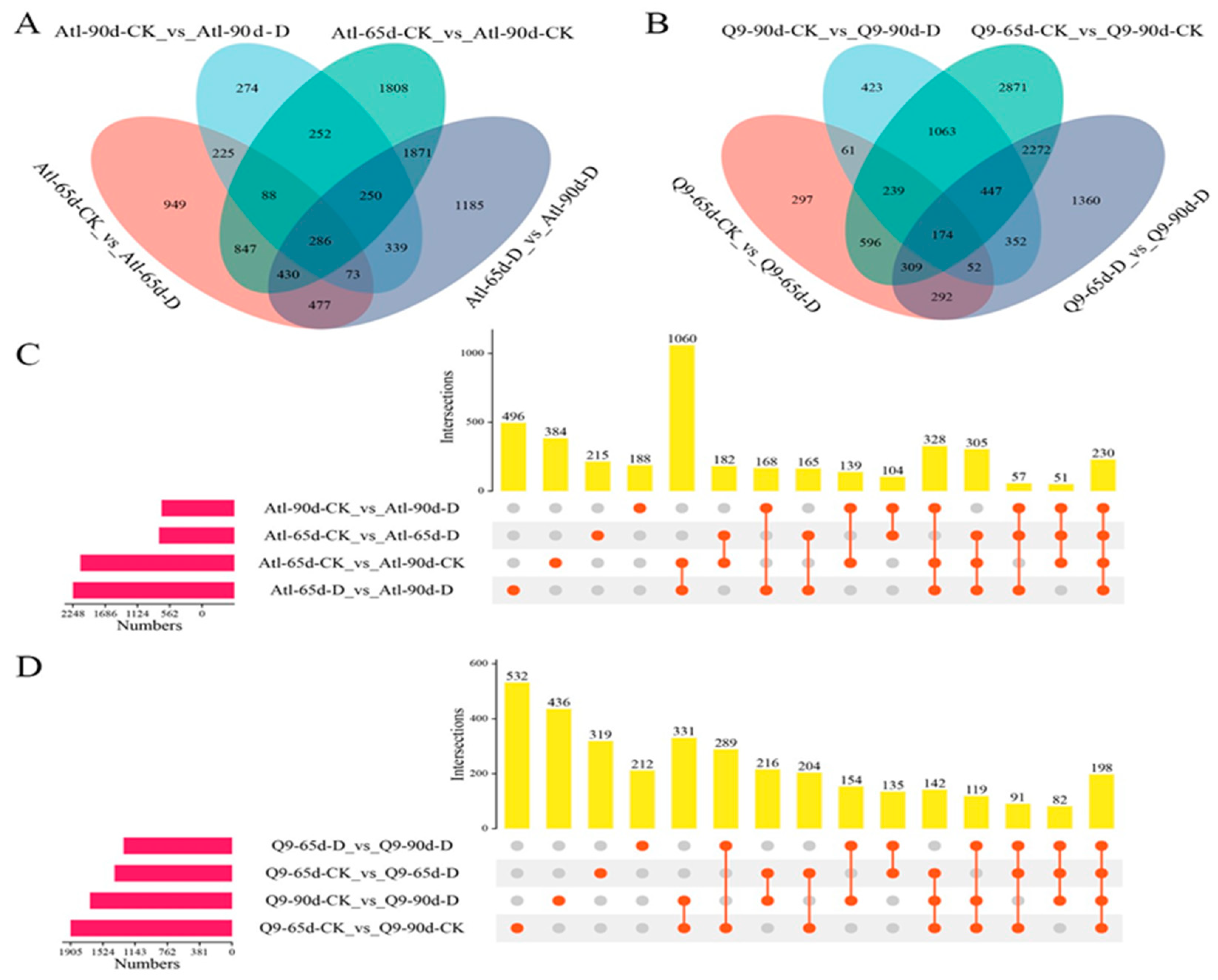
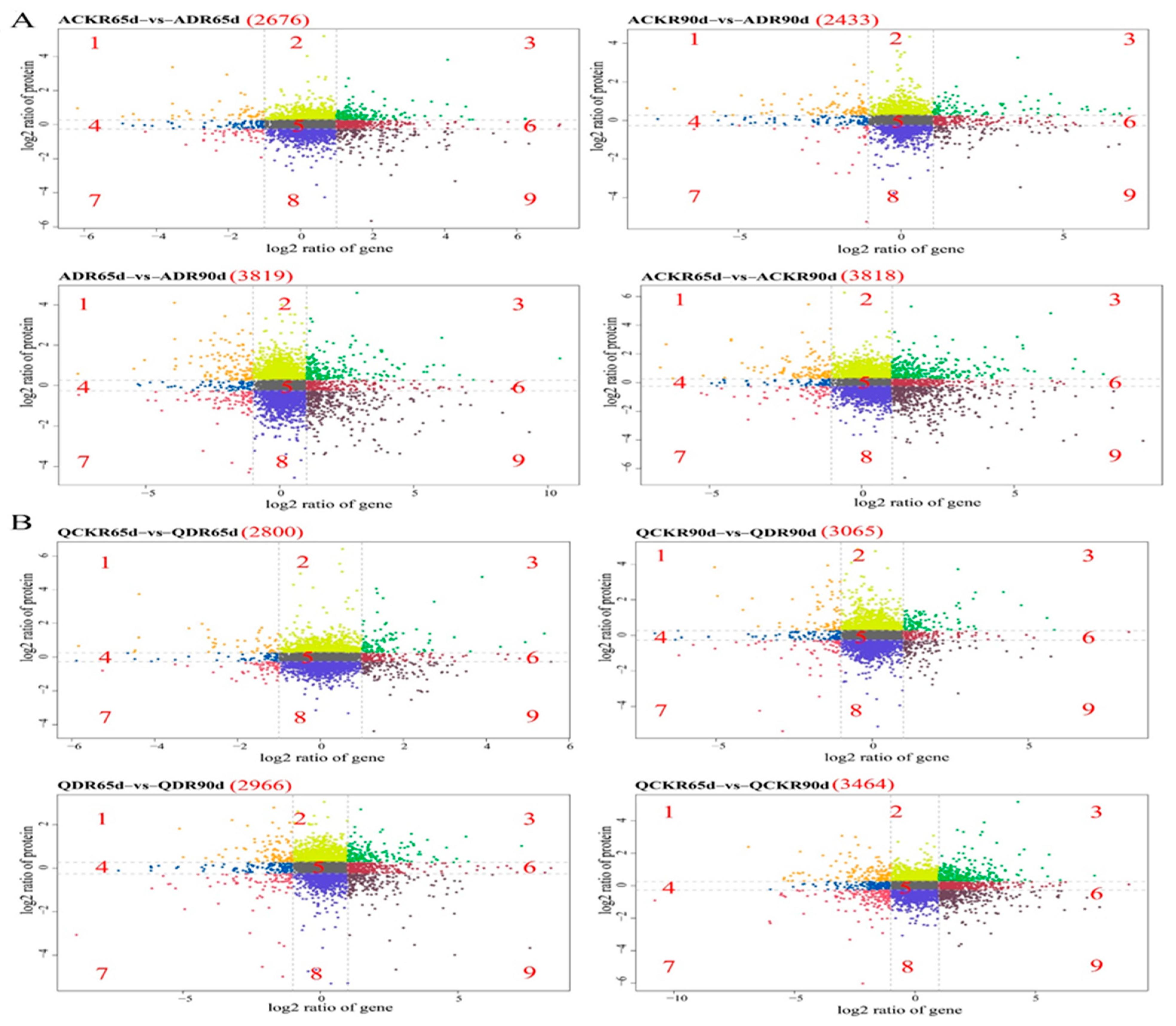
2.2. Analysis of Differential Genes and Proteins
2.2.1. Heatmap of the Association between Modules and Traits in Gene Co-Expression Networks
2.2.2. Characterized Gene Module Eigengenes in Gene Co-Expression Networks
2.2.3. Gene Ontology Enrichment Analysis of Genes in Candidate Modules in Gene Co-Expression Networks
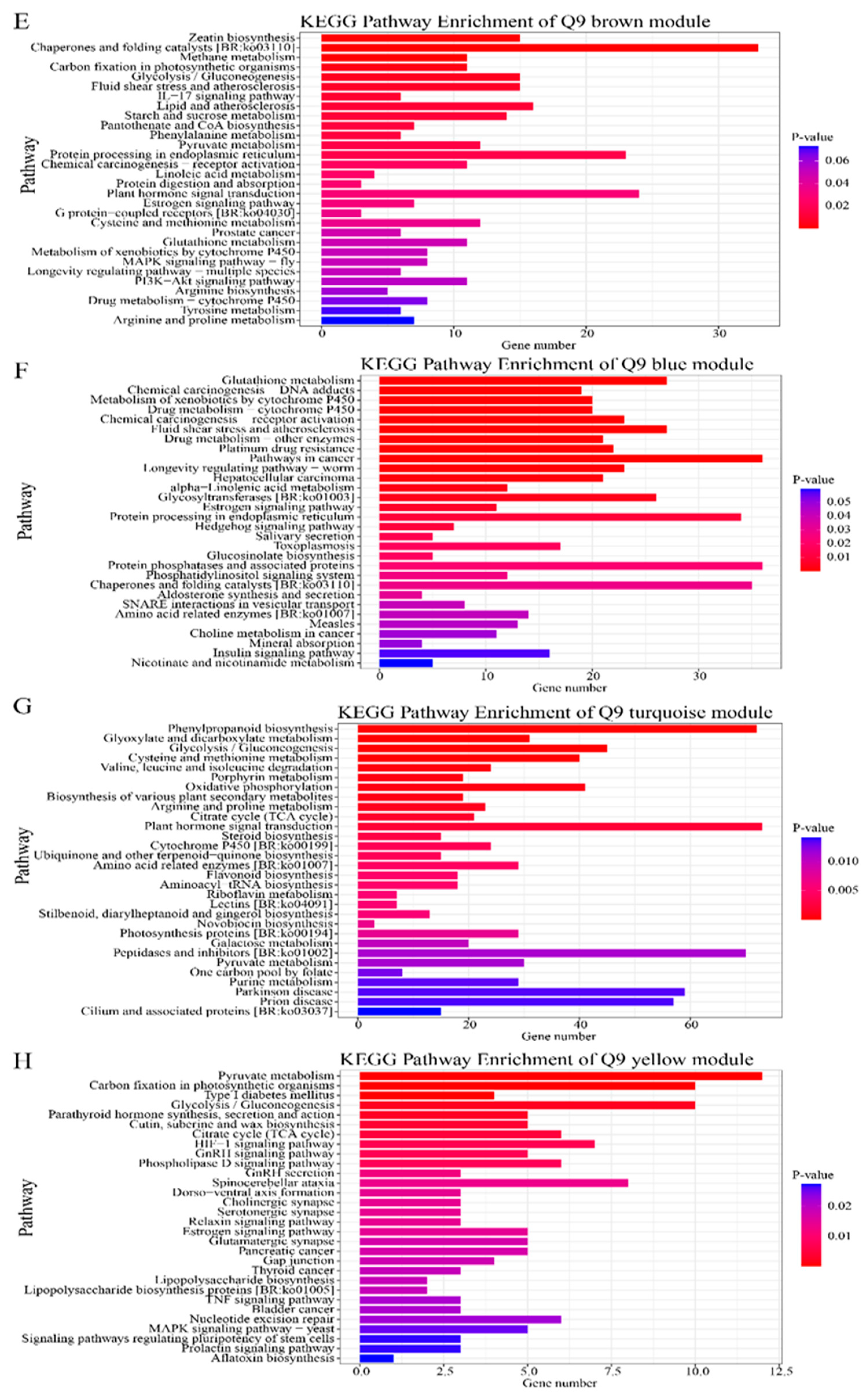
2.2.4. Kyoto Encyclopedia of Genes and Genomes Enrichment Analysis of Genes in Candidate Modules in Gene Co-Expression Networks
2.2.5. Visualization of the Core Genes in Candidate Modules in Gene Co-Expression Networks
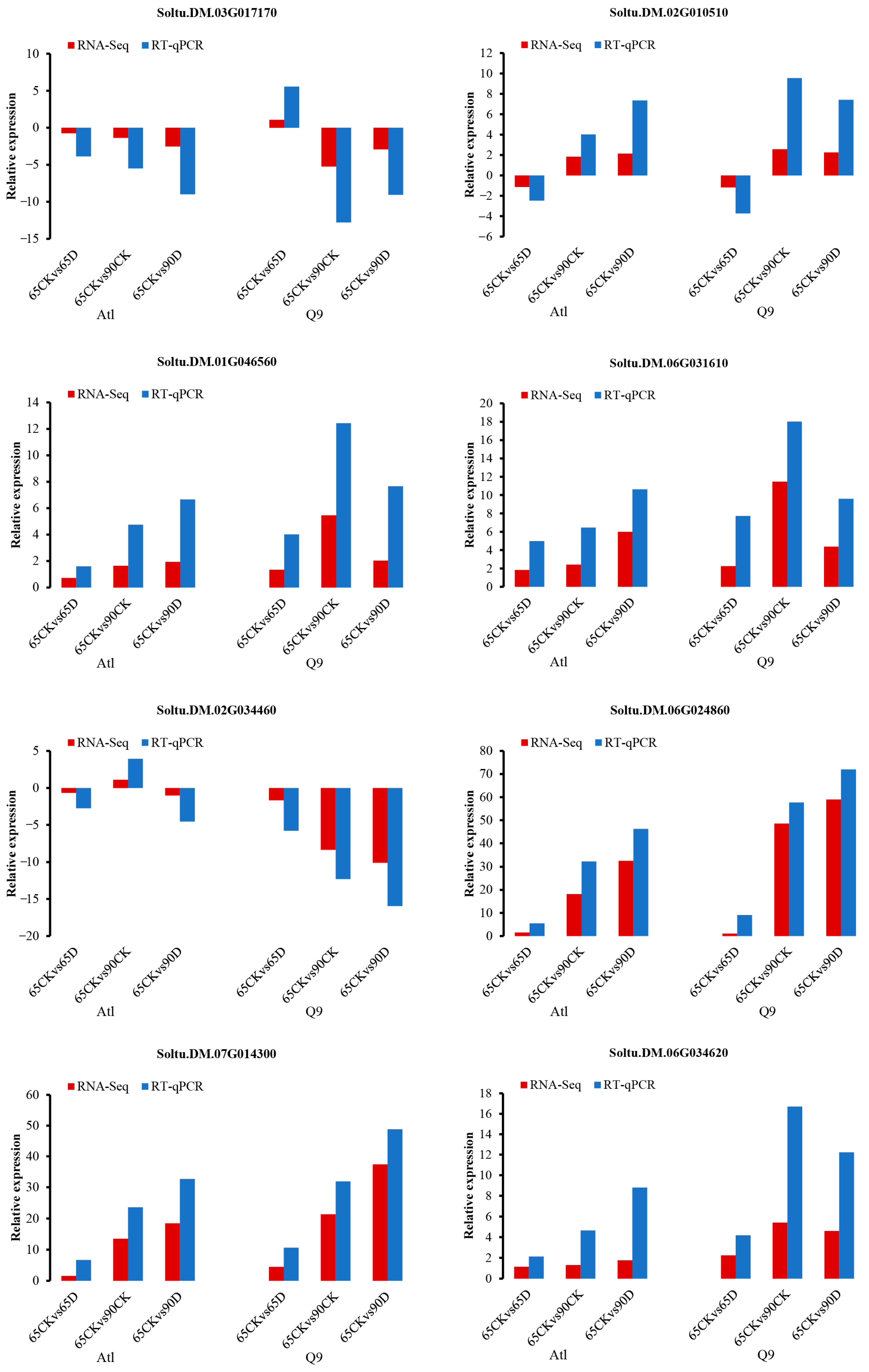
2.2.6. RT-qPCR (Real-Time Quantitative PCR) Validation of Core Genes in Candidate Modules in Gene Co-Expression Networks
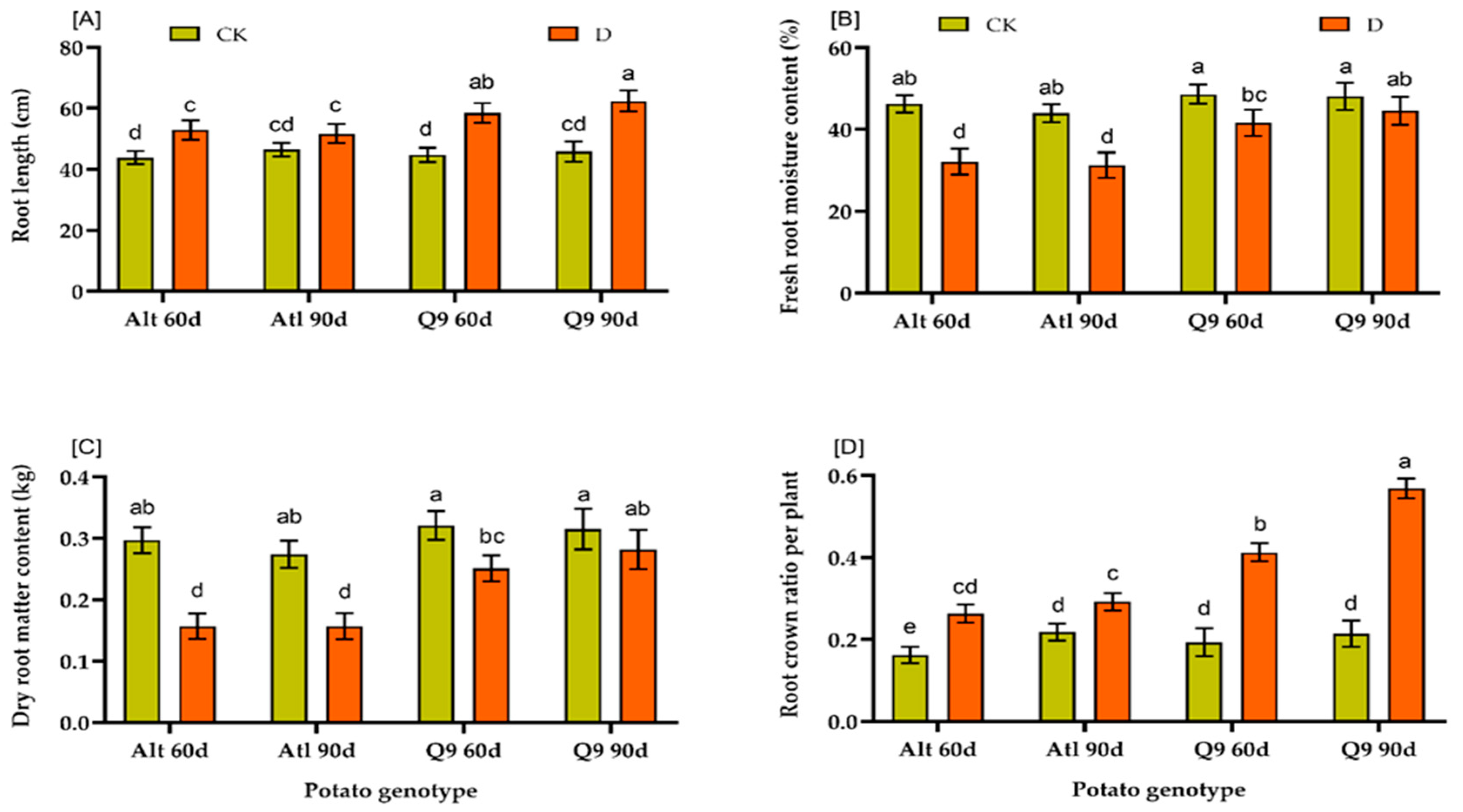
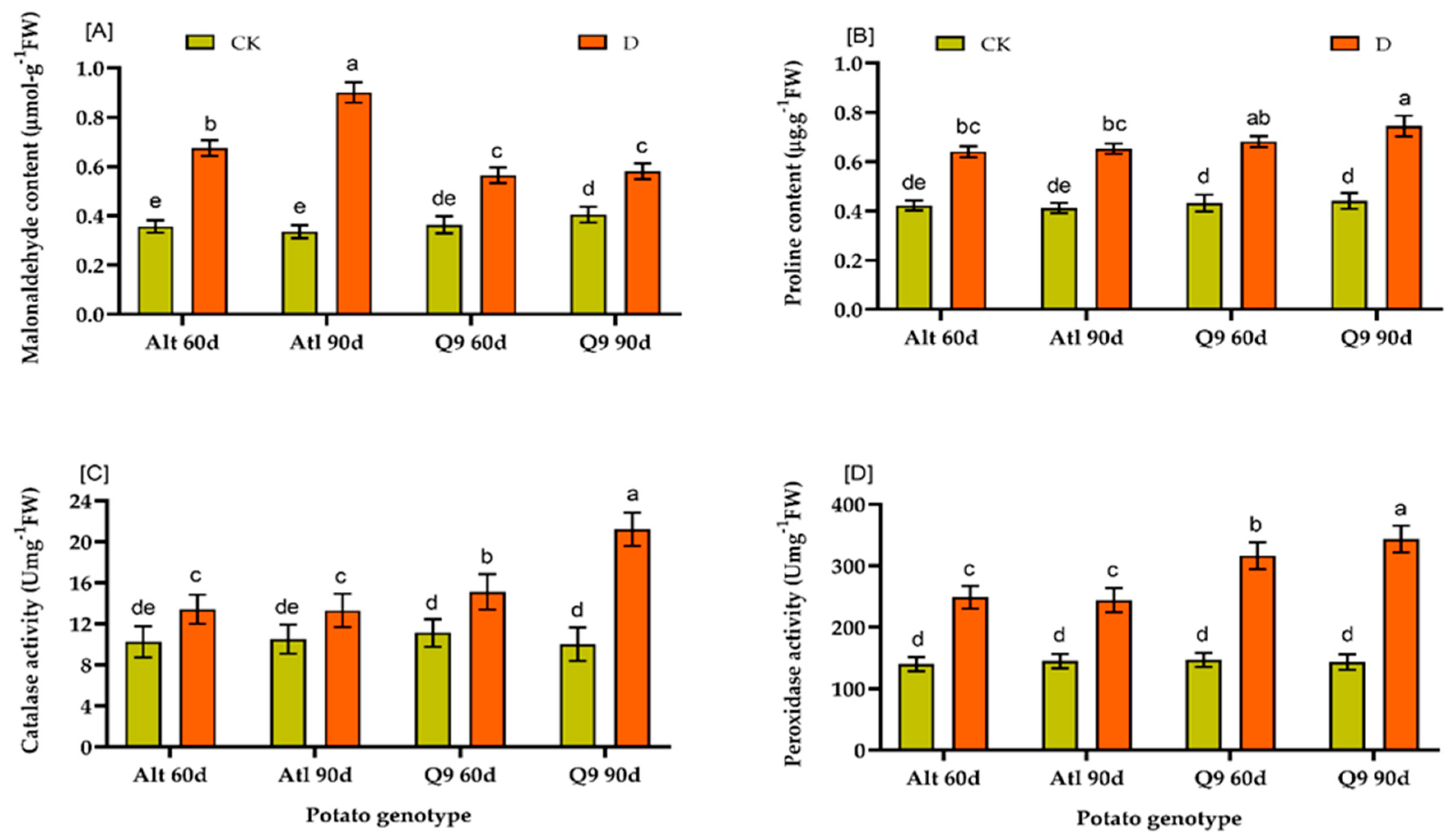
2.3. Effects of Drought on Physiological and Biochemical Indices of Two Potato Genotypes
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. Experimental Design
4.3. Extraction and Detection of Total RNA and Proteins from the Roots
4.4. Construction and Sequencing of Whole Transcriptome Libraries and Mass Spectrometry of Proteome
4.5. Quality Assessment of Sequencing Data
4.6. Comparative Analysis of the Reference Sequences
4.7. Analysis of Differential Expression
4.8. Weighted Gene Correlation Network Analysis of Differential Genes in Atl and Q9
4.8.1. Checking for Outlier Samples
4.8.2. Determination of Soft Thresholds in Gene Co-Expression Networks
4.8.3. Number of Gene Clusters and Module Slices for Gene Co-Expression Networks
4.9. GO Enrichment Analysis of DEGs and Proteins
4.10. KEGG Enrichment Analysis of DEGs and Proteins
4.11. Determination of Morpho–Physiological Indices and Antioxidant Enzyme Activities, Proline, and MDA Content in the Roots
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Food and Agriculture Organization of the United Nations (FAO). FAOSTAT. 2022. Available online: http://faostat.fao.org/ (accessed on 14 May 2024).
- Li, Q.; Zhang, S. Impacts of recent climate change on potato yields at a provincial scale in Northwest China. Agronomy 2020, 10, 426. [Google Scholar] [CrossRef]
- Li, Y.; Tang, J.; Wang, J.; Zhao, G.; Yu, Q.; Wang, Y.; Hu, Q.; Zhang, J.; Pan, Z.; Pan, X. Diverging water-saving potential across China’s potato planting regions. Eur. J. Agron. 2022, 134, 126450. [Google Scholar] [CrossRef]
- Qin, R.; Zhang, F.; Yu, C.; Zhang, Q.; Qi, J.; Li, F.-M. Contributions made by rain-fed potato with mulching to food security in China. Eur. J. Agron. 2022, 133, 126435. [Google Scholar] [CrossRef]
- Luo, X.-L.; Sun, Y.; Liu, L.-M.; Wang, L.-W.; Gao, X.-N. Response of Potato Growth and Yield to Drought Scenarios Based on APSIM-Potato in the Single-Cropping Region in Northern China. Potato Res. 2024, 1–22. [Google Scholar] [CrossRef]
- Nasir, M.W.; Toth, Z. Effect of drought stress on potato production: A review. Agronomy 2022, 12, 635. [Google Scholar] [CrossRef]
- Martínez, I.; Muñoz, M.; Acuña, I.; Uribe, M. Evaluating the drought tolerance of seven potato varieties on volcanic ash soils in a medium-term trial. Front. Plant Sci. 2021, 12, 693060. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Liang, W.; Yan, K.; Xu, D.; Qin, T.; Fiaz, S.; Kear, P.; Bi, Z.; Liu, Y.; Liu, Z. Expression of potato StDRO1 in Arabidopsis alters root architecture and drought tolerance. Front. Plant Sci. 2022, 13, 836063. [Google Scholar] [CrossRef] [PubMed]
- Chen, B. The Role of Arbuscular Mycorrhizal Symbiosis in Plant Adaptation to Drought Stress. Ph.D. Thesis, Tohoku University, Sendai, Japan, 2018. [Google Scholar]
- Guo, W.; Chen, L.; Herrera-Estrella, L.; Cao, D.; Tran, L.-S.P. Altering plant architecture to improve performance and resistance. Trends Plant Sci. 2020, 25, 1154–1170. [Google Scholar] [CrossRef] [PubMed]
- Ogura, T.; Goeschl, C.; Filiault, D.; Mirea, M.; Slovak, R.; Wolhrab, B.; Satbhai, S.B.; Busch, W. Root system depth in Arabidopsis is shaped by EXOCYST70A3 via the dynamic modulation of auxin transport. Cell 2019, 178, 400–412.e16. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Liang, W.; Sturrock, C.J.; Pandey, B.K.; Giri, J.; Mairhofer, S.; Wang, D.; Muller, L.; Tan, H.; York, L.M. Rice actin binding protein RMD controls crown root angle in response to external phosphate. Nat. Commun. 2018, 9, 2346. [Google Scholar] [CrossRef]
- Uga, Y.; Sugimoto, K.; Ogawa, S.; Rane, J.; Ishitani, M.; Hara, N.; Kitomi, Y.; Inukai, Y.; Ono, K.; Kanno, N. Control of root system architecture by DEEPER ROOTING 1 increases rice yield under drought conditions. Nat. Genet. 2013, 45, 1097–1102. [Google Scholar] [CrossRef]
- Chen, Q.; Hu, T.; Li, X.; Song, C.-P.; Zhu, J.-K.; Chen, L.; Zhao, Y. Phosphorylation of SWEET sucrose transporters regulates plant root: Shoot ratio under drought. Nat. Plants 2022, 8, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Orman-Ligeza, B.; Morris, E.C.; Parizot, B.; Lavigne, T.; Babé, A.; Ligeza, A.; Klein, S.; Sturrock, C.; Xuan, W.; Novák, O. The xerobranching response represses lateral root formation when roots are not in contact with water. Curr. Biol. 2018, 28, 3165–3173.e5. [Google Scholar] [CrossRef] [PubMed]
- Maurel, C.; Nacry, P. Root architecture and hydraulics converge for acclimation to changing water availability. Nat. Plants 2020, 6, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Andrés, F.; Coupland, G. The genetic basis of flowering responses to seasonal cues. Nat. Rev. Genet. 2012, 13, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Zia, R.; Nawaz, M.S.; Siddique, M.J.; Hakim, S.; Imran, A. Plant survival under drought stress: Implications, adaptive responses, and integrated rhizosphere management strategy for stress mitigation. Microbiol. Res. 2021, 242, 126626. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, D.; Pang, L.; Kobayashi, A.; Fozard, J.A.; Boudolf, V.; Bhosale, R.; Antoni, R.; Nguyen, T.; Hiratsuka, S.; Fujii, N. Root hydrotropism is controlled via a cortex-specific growth mechanism. Nat. Plants 2017, 3, 17057. [Google Scholar] [CrossRef] [PubMed]
- Rellán-Álvarez, R.; Lobet, G.; Dinneny, J.R. Environmental control of root system biology. Annu. Rev. Plant Biol. 2016, 67, 619–642. [Google Scholar] [CrossRef] [PubMed]
- Scharwies, J.D.; Dinneny, J.R. Water transport, perception, and response in plants. J. Plant Res. 2019, 132, 311–324. [Google Scholar] [CrossRef]
- Jia, A.; Xu, L.; Wang, Y. Venn diagrams in bioinformatics. Brief. Bioinform. 2021, 22, bbab108. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Liu, X.; Zhou, J.; Deng, H.; Zhang, G.; Xiao, Y.; Tang, W. WGCNA analysis identifies the hub genes related to heat stress in seedling of rice (Oryza sativa L.). Genes 2022, 13, 1020. [Google Scholar] [CrossRef]
- Yu, H.; Du, X.; Zhao, Q.; Yin, C.; Song, W. Weighted gene Co-expression network analysis (WGCNA) reveals a set of hub genes related to chlorophyll metabolism process in chlorella (Chlorella vulgaris) response androstenedione. Environ. Pollut. 2022, 306, 119360. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Majeed, A.; Mukhtar, S. Protein–Protein Interaction Network Exploration Using Cytoscape. In Protein-Protein Interactions: Methods and Protocols; Springer: Berlin/Heidelberg, Germany, 2023; pp. 419–427. [Google Scholar]
- Jiang, X.-W.; Lu, H.-Y.; Xu, Z.-H.; Zhang, Y.-S.; Zhao, Q.-C. Network pharmacology-based research uncovers cold resistance and thermogenesis mechanism of Cinnamomum cassia. Fitoterapia 2021, 149, 104824. [Google Scholar]
- Galway, M.E.; Eng, R.C.; Schiefelbein, J.W.; Wasteneys, G.O. Root hair-specific disruption of cellulose and xyloglucan in AtCSLD3 mutants, and factors affecting the post-rupture resumption of mutant root hair growth. Planta 2011, 233, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Seo, E.; Lee, H.; Jeon, J.; Park, H.; Kim, J.; Noh, Y.-S.; Lee, I. Crosstalk between cold response and flowering in Arabidopsis is mediated through the flowering-time gene SOC1 and its upstream negative regulator FLC. Plant Cell 2009, 21, 3185–3197. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Cubría-Radío, M.; Vavrdová, T.; De Winter, F.; Schilling, N.; Huysmans, M.; Nanda, A.K.; Melnyk, C.W.; Nowack, M.K. Repressive ZINC FINGER OF ARABIDOPSIS THALIANA proteins promote programmed cell death in the Arabidopsis columella root cap. Plant Physiol. 2023, 192, 1151–1167. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Xu, P.; Li, B.; Li, P.; Wen, X.; An, F.; Gong, Y.; Xin, Y.; Zhu, Z.; Wang, Y. Ethylene promotes root hair growth through coordinated EIN3/EIL1 and RHD6/RSL1 activity in Arabidopsis. Proc. Natl. Acad. Sci. USA 2017, 114, 13834–13839. [Google Scholar] [CrossRef] [PubMed]
- Tavridou, E.; Pireyre, M.; Ulm, R. Degradation of the transcription factors PIF4 and PIF5 under UV-B promotes UVR8-mediated inhibition of hypocotyl growth in Arabidopsis. Plant J. 2020, 101, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Afek, A.; Shi, H.; Rangadurai, A.; Sahay, H.; Senitzki, A.; Xhani, S.; Fang, M.; Salinas, R.; Mielko, Z.; Pufall, M.A. DNA mismatches reveal conformational penalties in protein–DNA recognition. Nature 2020, 587, 291–296. [Google Scholar] [CrossRef]
- Ponce, O.P.; Torres, Y.; Prashar, A.; Buell, R.; Lozano, R.; Orjeda, G.; Compton, L. Transcriptome profiling shows a rapid variety-specific response in two Andigenum potato varieties under drought stress. Front. Plant Sci. 2022, 13, 1003907. [Google Scholar] [CrossRef]
- Wu, Q.; Pan, Y.-B.; Su, Y.; Zou, W.; Xu, F.; Sun, T.; Grisham, M.P.; Yang, S.; Xu, L.; Que, Y. WGCNA identifies a comprehensive and dynamic gene co-expression network that associates with smut resistance in sugarcane. Int. J. Mol. Sci. 2022, 23, 10770. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Qing, C.; Liu, P.; Zou, C.; Yuan, G.; Pan, G.; Shen, Y.; Ma, L. Joint GWAS and WGCNA uncover the genetic control of calcium accumulation under salt treatment in maize seedlings. Physiol. Plant. 2022, 174, e13606. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ren, Y.; Zhang, D.; Chen, X.; Huang, J.; Xu, Y.; Aucapiña, C.B.; Zhang, Y.; Miao, Y. Transcriptome-based WGCNA analysis reveals regulated metabolite fluxes between floral color and scent in Narcissus tazetta flower. Int. J. Mol. Sci. 2021, 22, 8249. [Google Scholar] [CrossRef] [PubMed]
- DiLeo, M.V.; Strahan, G.D.; den Bakker, M.; Hoekenga, O.A. Weighted correlation network analysis (WGCNA) applied to the tomato fruit metabolome. PLoS ONE 2011, 6, e26683. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Ji, W.; McKnight, T.D. Candidate gene networks for acylsugar metabolism and plant defense in wild tomato Solanum pennellii. Plant Cell 2020, 32, 81–99. [Google Scholar] [CrossRef] [PubMed]
- Kuang, J.F.; Wu, C.J.; Guo, Y.F.; Walther, D.; Shan, W.; Chen, J.Y.; Chen, L.; Lu, W.J. Deciphering transcriptional regulators of banana fruit ripening by regulatory network analysis. Plant Biotechnol. J. 2021, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, J.; Wang, J. Identification of a comprehensive gene co-expression network associated with autotetraploid Potato (Solanum tuberosum L.) development using WGCNA Analysis. Genes 2023, 14, 1162. [Google Scholar] [CrossRef]
- Zhu, M.; Xie, H.; Wei, X.; Dossa, K.; Yu, Y.; Hui, S.; Tang, G.; Zeng, X.; Yu, Y.; Hu, P. WGCNA analysis of salt-responsive core transcriptome identifies novel hub genes in rice. Genes 2019, 10, 719. [Google Scholar] [CrossRef]
- De Silva, K.K.; Dunwell, J.M.; Wickramasuriya, A.M. Weighted Gene Correlation Network Analysis (WGCNA) of Arabidopsis somatic embryogenesis (SE) and identification of key gene modules to uncover SE-associated hub genes. Int. J. Genom. 2022, 2022, 7471063. [Google Scholar] [CrossRef]
- Zhao, H.; Du, X.; Zhang, K.; Liu, Y.; Wang, Y.; Liu, J.; He, Y.; Wang, X.; Zhang, Q. Weighted correlation network analysis (WGCNA) of Japanese flounder (Paralichthys olivaceus) embryo transcriptome provides crucial gene sets for understanding haploid syndrome and rescue by diploidization. J. Ocean. Univ. China 2018, 17, 1441–1450. [Google Scholar] [CrossRef]
- Meng, H.-L.; Sun, P.-Y.; Wang, J.-R.; Sun, X.-Q.; Zheng, C.-Z.; Fan, T.; Chen, Q.-F.; Li, H.-Y. Comparative physiological, transcriptomic, and WGCNA analyses reveal the key genes and regulatory pathways associated with drought tolerance in Tartary buckwheat. Front. Plant Sci. 2022, 13, 985088. [Google Scholar] [CrossRef] [PubMed]
- Irani, S.; Todd, C.D. Exogenous allantoin increases Arabidopsis seedlings tolerance to NaCl stress and regulates expression of oxidative stress response genes. J. Plant Physiol. 2018, 221, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Huang, L.-J.; Sun, X.-F.; Zhao, L.-L.; Wang, P.-C. Differential physiological, transcriptomic, and metabolomic responses of paspalum wettsteinii under high-temperature stress. Front. Plant Sci. 2022, 13, 865608. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Mubeen, B.; Hasnain, A.; Rizwan, M.; Adrees, M.; Naqvi, S.A.H.; Iqbal, S.; Kamran, M.; El-Sabrout, A.M.; Elansary, H.O. Role of promising secondary metabolites to confer resistance against environmental stresses in crop plants: Current scenario and future perspectives. Front. Plant Sci. 2022, 13, 881032. [Google Scholar] [CrossRef]
- Jiang, Q.; Niu, F.; Sun, X.; Hu, Z.; Li, X.; Ma, Y.; Zhang, H. RNA-seq analysis of unintended effects in transgenic wheat overexpressing the transcription factor GmDREB1. Crop J. 2017, 5, 207–218. [Google Scholar] [CrossRef]
- Dormatey, R.; Qin, T.; Wang, Y.; Karikari, B.; Dekomah, S.D.; Fan, Y.; Bi, Z.; Yao, P.; Ali, K.; Sun, C. Comparative Transcriptome Profiling Reveals Potential Candidate Genes, Transcription Factors, and Biosynthetic Pathways for Phosphite Response in Potato (Solanum tuberosum L.). Genes 2022, 13, 1379. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Wang, Q.; Zhao, F.; Liu, J.; Liu, H. Understanding the regulatory mechanisms of milk production using integrative transcriptomic and proteomic analyses: Improving inefficient utilization of crop by-products as forage in dairy industry. BMC Genom. 2018, 19, 403. [Google Scholar] [CrossRef]
- Zeng, W.; Peng, Y.; Zhao, X.; Wu, B.; Chen, F.; Ren, B.; Zhuang, Z.; Gao, Q.; Ding, Y. Comparative proteomics analysis of the seedling root response of drought-sensitive and drought-tolerant maize varieties to drought stress. Int. J. Mol. Sci. 2019, 20, 2793. [Google Scholar] [CrossRef] [PubMed]
- Mishra, D.C.; Arora, D.; Budhlakoti, N.; Solanke, A.U.; Mithra, S.A.C.; Kumar, A.; Pandey, P.; Srivastava, S.; Kumar, S.; Farooqi, M. Identification of potential cytokinin responsive key genes in rice treated with trans-zeatin through systems biology approach. Front. Genet. 2022, 12, 780599. [Google Scholar] [CrossRef]
- Dumont, S.; Rivoal, J. Consequences of oxidative stress on plant glycolytic and respiratory metabolism. Front. Plant Sci. 2019, 10, 432113. [Google Scholar] [CrossRef]
- Plaxton, W.C.; Carswell, M.C. Metabolic aspects of the phosphate starvation response in plants. In Plant Responses to Environmental Stresses; Routledge: London, UK, 2018; pp. 349–372. [Google Scholar]
- Singh, P.K.; Srivastava, D.; Tiwari, P.; Tiwari, M.; Verma, G.; Chakrabarty, D. Drought tolerance in plants: Molecular mechanism and regulation of signaling molecules. In Plant Signaling Molecules; Elsevier: Amsterdam, The Netherlands, 2019; pp. 105–123. [Google Scholar]
- Tiwari, J.K.; Buckseth, T.; Challam, C.; Natarajan, N.; Singh, R.K.; Kumar, M. Omics approaches for biotic, abiotic, and quality traits improvement in potato (Solanum tuberosum L.). In Bioinformatics in Agriculture; Elsevier: Amsterdam, The Netherlands, 2022; pp. 341–359. [Google Scholar]
- Chen, Y.; Li, C.; Yi, J.; Yang, Y.; Lei, C.; Gong, M. Transcriptome response to drought, rehydration and re-dehydration in potato. Int. J. Mol. Sci. 2019, 21, 159. [Google Scholar] [CrossRef]
- Liang, Y.; Li, X.; Lei, F.; Yang, R.; Bai, W.; Yang, Q.; Zhang, D. Transcriptome Profiles Reveals ScDREB10 from Syntrichia caninervis Regulated Phenylpropanoid Biosynthesis and Starch/Sucrose Metabolism to Enhance Plant Stress Tolerance. Plants 2024, 13, 205. [Google Scholar] [CrossRef] [PubMed]
- Karlova, R.; Boer, D.; Hayes, S.; Testerink, C. Root plasticity under abiotic stress. Plant Physiol. 2021, 187, 1057–1070. [Google Scholar] [CrossRef]
- Fromm, H. Root plasticity in the pursuit of water. Plants 2019, 8, 236. [Google Scholar] [CrossRef]
- Kamanga, R.; Mbega, E.; Ndakidemi, P. Drought tolerance mechanisms in plants: Physiological responses associated with water deficit stress in Solanum lycopersicum. Adv. Crop Sci. Technol. 2018, 362, 1000362. [Google Scholar] [CrossRef]
- Guo, Y.; Yu, H.; Yang, M.; Kong, D.; Zhang, Y. Effect of drought stress on lipid peroxidation, osmotic adjustment and antioxidant enzyme activity of leaves and roots of Lycium ruthenicum Murr. seedling. Russ. J. Plant Physiol. 2018, 65, 244–250. [Google Scholar] [CrossRef]
- Shafi, A.; Zahoor, I.; Mushtaq, U. Proline accumulation and oxidative stress: Diverse roles and mechanism of tolerance and adaptation under salinity stress. In Salt Stress, Microbes, and Plant Interactions: Mechanisms and Molecular Approaches; Springer: Berlin/Heidelberg, Germany, 2019; Volume 2, pp. 269–300. [Google Scholar]
- Per, T.S.; Khan, N.A.; Reddy, P.S.; Masood, A.; Hasanuzzaman, M.; Khan, M.I.R.; Anjum, N.A. Approaches in modulating proline metabolism in plants for salt and drought stress tolerance: Phytohormones, mineral nutrients and transgenics. Plant Physiol. Biochem. 2017, 115, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Singh, D.; Pandey, H.; Jatav, R.B.; Singh, V.; Pandey, D. An overview of roles of enzymatic and nonenzymatic antioxidants in plant. In Antioxidant Defense in Plants: Molecular Basis of Regulation; Springer: Berlin/Heidelberg, Germany, 2022; pp. 1–13. [Google Scholar]
- Rajput, V.D.; Harish; Singh, R.K.; Verma, K.K.; Sharma, L.; Quiroz-Figueroa, F.R.; Meena, M.; Gour, V.S.; Minkina, T.; Sushkova, S. Recent developments in enzymatic antioxidant defence mechanism in plants with special reference to abiotic stress. Biology 2021, 10, 267. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Ramirez-Gonzalez, R.H.; Bonnal, R.; Caccamo, M.; MacLean, D. Bio-samtools: Ruby bindings for SAMtools, a library for accessing BAM files containing high-throughput sequence alignments. Source Code Biol. Med. 2012, 7, 6. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Putri, G.H.; Anders, S.; Pyl, P.T.; Pimanda, J.E.; Zanini, F. Analysing high-throughput sequencing data in Python with HTSeq 2.0. Bioinformatics 2022, 38, 2943–2945. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wang, Z.; Zhu, R.; Wang, F.; Cheng, Y.; Liu, Y. Three differential expression analysis methods for RNA sequencing: Limma, EdgeR, DESeq2. J. Vis. Exp. 2021, 175, e62528. [Google Scholar]
- Ghosh, D. Wavelet-based Benjamini-Hochberg procedures for multiple testing under dependence. Math. Biosci. Eng. 2019, 17, 56–72. [Google Scholar] [CrossRef] [PubMed]
- Demichev, V.; Szyrwiel, L.; Yu, F.; Teo, G.C.; Rosenberger, G.; Niewienda, A.; Ludwig, D.; Decker, J.; Kaspar-Schoenefeld, S.; Lilley, K.S. dia-PASEF data analysis using FragPipe and DIA-NN for deep proteomics of low sample amounts. Nat. Commun. 2022, 13, 3944. [Google Scholar] [CrossRef]
- Demichev, V.; Messner, C.B.; Vernardis, S.I.; Lilley, K.S.; Ralser, M. DIA-NN: Neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 2020, 17, 41–44. [Google Scholar] [CrossRef]
- Zhang, F.; Ge, W.; Ruan, G.; Cai, X.; Guo, T. Data-independent acquisition mass spectrometry-based proteomics and software tools: A glimpse in 2020. Proteomics 2020, 20, 1900276. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ooi, B.C.; Tan, K.-L.; Ong, T.-H.; Zhou, L. BLAST++: BLASTing queries in batches. Bioinformatics 2003, 19, 2323–2324. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Tang, Y.; Li, J.; Song, Q.; Cheng, Q.; Tan, Q.; Zhou, Q.; Nong, Z.; Lv, P. Transcriptome and WGCNA reveal hub genes in sugarcane tiller seedlings in response to drought stress. Sci. Rep. 2023, 13, 12823. [Google Scholar] [CrossRef] [PubMed]
- Blum, T.; Briesemeister, S.; Kohlbacher, O. MultiLoc2: Integrating phylogeny and Gene Ontology terms improves subcellular protein localization prediction. BMC Bioinform. 2009, 10, 274. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, Y.-H.; Wang, S.; Zhang, Y.; Huang, T.; Cai, Y.-D. Prediction and analysis of essential genes using the enrichments of gene ontology and KEGG pathways. PLoS ONE 2017, 12, e0184129. [Google Scholar] [CrossRef] [PubMed]
- Hodges, D.M.; DeLong, J.M.; Forney, C.F.; Prange, R.K. Improving the thiobarbituric acid-reactive-substances assay for estimating lipid peroxidation in plant tissues containing anthocyanin and other interfering compounds. Planta 1999, 207, 604–611. [Google Scholar] [CrossRef]
- Bates, L.S.; Waldren, R.; Teare, I. Rapid determination of free proline for water-stress studies. Plant Soil 1973, 39, 205–207. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Varieties | Module | Hub Gene | Hub Gene | Gene Function |
|---|---|---|---|---|
| Atl | brown | Soltu.DM.04G030030 | AT4G08950 | Phosphate-responsive 1 family protein |
| Atl | brown | Soltu.DM.03G016820 | AT5G52060 | BCL-2-associated athanogene |
| Atl | brown | Soltu.DM.03G020780 | AT3G03050 | cellulose synthase-like D3 |
| Atl | brown | Soltu.DM.11G004580 | #N/A | Protein of unknown function (DUF1635) |
| Atl | brown | Soltu.DM.03G016740 | AT4G25480 | dehydration response element B1A |
| Atl | brown | Soltu.DM.07G024430 | AT5G53760 | Seven transmembrane MLO family protein |
| Atl | green | Soltu.DM.04G037350 | AT5G42220 | Ubiquitin-like superfamily protein |
| Atl | green | Soltu.DM.06G031840 | AT1G07400 | HSP20-like chaperones superfamily protein |
| Atl | green | Soltu.DM.10G002820 | #N/A | phosphoenolpyruvate carboxylase |
| Atl | green | Soltu.DM.12G010920 | #N/A | HSP20-like chaperones superfamily protein |
| Atl | green | Soltu.DM.06G031830 | AT1G18640 | 3-phosphoserine phosphatase |
| Atl | green | Soltu.DM.09G004920 | AT5G03555 | permease, cytosine/purines, uracil, thiamine, allantoin family protein |
| Atl | turquoise | Soltu.DM.01G028350 | AT2G44380 | Cysteine/Histidine-rich C1 domain family protein |
| Atl | turquoise | Soltu.DM.11G023040 | #N/A | plasma membrane intrinsic protein |
| Atl | turquoise | Soltu.DM.10G019060 | #N/A | Peroxidase superfamily protein |
| Atl | turquoise | Soltu.DM.01G051390 | AT1G20330 | sterol methyltransferase |
| Atl | turquoise | Soltu.DM.01G001830 | AT1G50480 | 10-formyltetrahydrofolate synthetase |
| Atl | turquoise | Soltu.DM.03G033470 | AT3G18080 | B-S glucosidase |
| Atl | turquoise | Soltu.DM.06G021870 | AT1G51630 | O-fucosyltransferase family protein |
| Atl | turquoise | Soltu.DM.09G020340 | AT3G22830 | heat shock transcription factor A6B |
| Atl | yellow | Soltu.DM.04G029560 | AT4G08850 | Leucine-rich repeat receptor-like protein kinase family protein |
| Atl | yellow | Soltu.DM.02G010790 | AT3G27170 | chloride channel B |
| Atl | yellow | Soltu.DM.01G028700 | AT2G44260 | Plant protein of unknown function (DUF946) |
| Atl | yellow | Soltu.DM.04G027350 | AT4G08850 | Leucine-rich repeat receptor-like protein kinase family protein |
| Atl | yellow | Soltu.DM.04G029550 | AT1G35710 | Protein kinase family protein with leucine-rich repeat domain |
| Atl | yellow | Soltu.DM.03G037250 | AT1G53903 | Protein of unknown function (DUF581) |
| Atl | yellow | Soltu.DM.04G029480 | AT4G08850 | Leucine-rich repeat receptor-like protein kinase family protein |
| Q9 | brown | Soltu.DM.02G006090 | AT5G25610 | BURP domain-containing protein |
| Q9 | brown | Soltu.DM.01G003450 | AT1G70520 | cysteine-rich RLK (RECEPTOR-like protein kinase) |
| Q9 | brown | Soltu.DM.10G022490 | #N/A | PYR1-like |
| Q9 | brown | Soltu.DM.03G017170 | AT5G52190 | Sugar isomerase (SIS) family protein |
| Q9 | brown | Soltu.DM.02G010510 | AT5G40510 | Sucrase/ferredoxin-like family protein |
| Q9 | brown | Soltu.DM.07G027920 | AT1G55690 | Sec14p-like phosphatidylinositol transfer family protein |
| Q9 | brown | Soltu.DM.01G042680 | AT1G71900 | Protein of unknown function (DUF803) |
| Q9 | blue | Soltu.DM.01G046560 | AT1G27730 | salt tolerance zinc finger |
| Q9 | blue | Soltu.DM.06G031610 | AT5G59480 | Haloacid dehalogenase-like hydrolase (HAD) superfamily protein |
| Q9 | blue | Soltu.DM.03G024660 | AT3G48520 | cytochrome P450, family 94, subfamily B, polypeptide |
| Q9 | blue | Soltu.DM.03G028100 | AT1G60190 | ARM repeat superfamily protein |
| Q9 | blue | Soltu.DM.03G024690 | AT3G48520 | cytochrome P450, family 94, subfamily B, polypeptide |
| Q9 | blue | Soltu.DM.03G002340 | AT1G61260 | Protein of unknown function (DUF761) |
| Q9 | turquoise | Soltu.DM.06G017960 | AT2G35710 | Nucleotide-diphospho-sugar transferases superfamily protein |
| Q9 | turquoise | Soltu.DM.01G029490 | AT1G05160 | ent-kaurenoic acid hydroxylase |
| Q9 | turquoise | Soltu.DM.02G034460 | AT3G02040 | senescence-related gene |
| Q9 | turquoise | Soltu.DM.06G024860 | AT1G70700 | jasmonate-zim-domain protein |
| Q9 | turquoise | Soltu.DM.06G026270 | AT1G74790 | catalytics |
| Q9 | turquoise | Soltu.DM.04G012230 | AT1G76690 | 12-oxophytodienoate reductase |
| Q9 | turquoise | Soltu.DM.10G019340 | #N/A | PYR1-like |
| Q9 | turquoise | Soltu.DM.07G013340 | #N/A | Bax inhibitor-1 family protein |
| Q9 | yellow | Soltu.DM.07G014300 | AT2G43010 | phytochrome interacting factor 3-like |
| Q9 | yellow | Soltu.DM.11G004020 | #N/A | Plant stearoyl-acyl-carrier-protein desaturase family protein |
| Q9 | yellow | Soltu.DM.04G020610 | AT3G48310 | cytochrome P450, family 71, subfamily A, polypeptide |
| Q9 | yellow | Soltu.DM.02G027580 | AT2G22910 | N-acetyl-l-glutamate synthase |
| Q9 | yellow | Soltu.DM.08G020150 | AT4G19230 | cytochrome P450, family 707, subfamily A, polypeptide |
| Q9 | yellow | Soltu.DM.06G034620 | AT3G22600 | Bifunctional inhibitor/lipid-transfer protein/seed storage 2S albumin superfamily protein |
| Q9 | yellow | Soltu.DM.09G025790 | AT1G06760 | winged-helix DNA-binding transcription factor family protein |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, T.; Wang, Y.; Pu, Z.; Shi, N.; Dormatey, R.; Wang, H.; Sun, C. Comprehensive Transcriptome and Proteome Analyses Reveal the Drought Responsive Gene Network in Potato Roots. Plants 2024, 13, 1530. https://doi.org/10.3390/plants13111530
Qin T, Wang Y, Pu Z, Shi N, Dormatey R, Wang H, Sun C. Comprehensive Transcriptome and Proteome Analyses Reveal the Drought Responsive Gene Network in Potato Roots. Plants. 2024; 13(11):1530. https://doi.org/10.3390/plants13111530
Chicago/Turabian StyleQin, Tianyuan, Yihao Wang, Zhuanfang Pu, Ningfan Shi, Richard Dormatey, Huiqiong Wang, and Chao Sun. 2024. "Comprehensive Transcriptome and Proteome Analyses Reveal the Drought Responsive Gene Network in Potato Roots" Plants 13, no. 11: 1530. https://doi.org/10.3390/plants13111530
APA StyleQin, T., Wang, Y., Pu, Z., Shi, N., Dormatey, R., Wang, H., & Sun, C. (2024). Comprehensive Transcriptome and Proteome Analyses Reveal the Drought Responsive Gene Network in Potato Roots. Plants, 13(11), 1530. https://doi.org/10.3390/plants13111530