
Genotyping-by-Sequencing Analysis Reveals Associations between Agronomic and Oil Traits in Gamma Ray-Derived Mutant Rapeseed (Brassica napus L.)

 , , , , , ,
, , , , , ,  , ,
, ,  , ,
, ,  and
and
Abstract
1. Introduction
2. Results
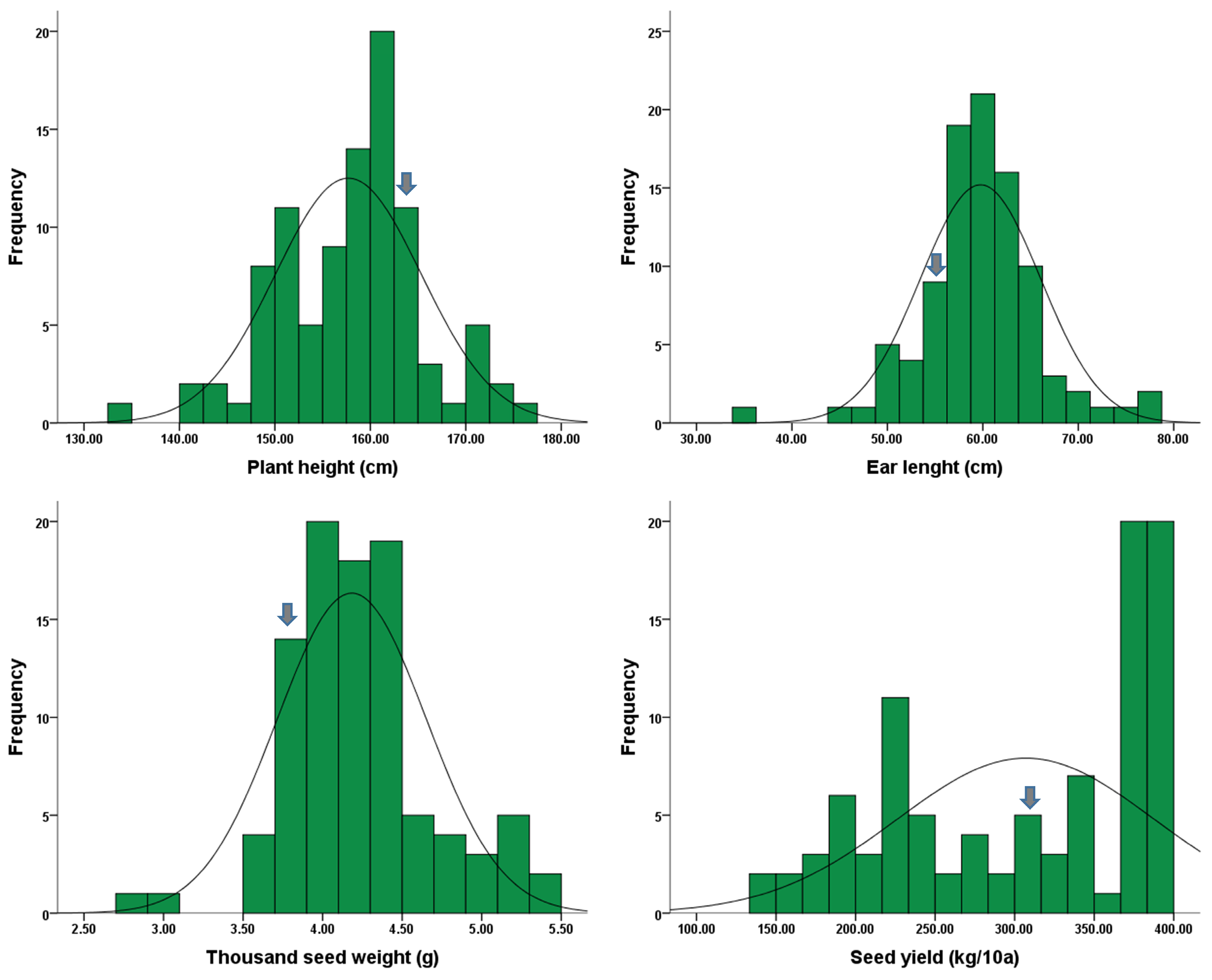
2.1. Assessment of Agronomic Traits
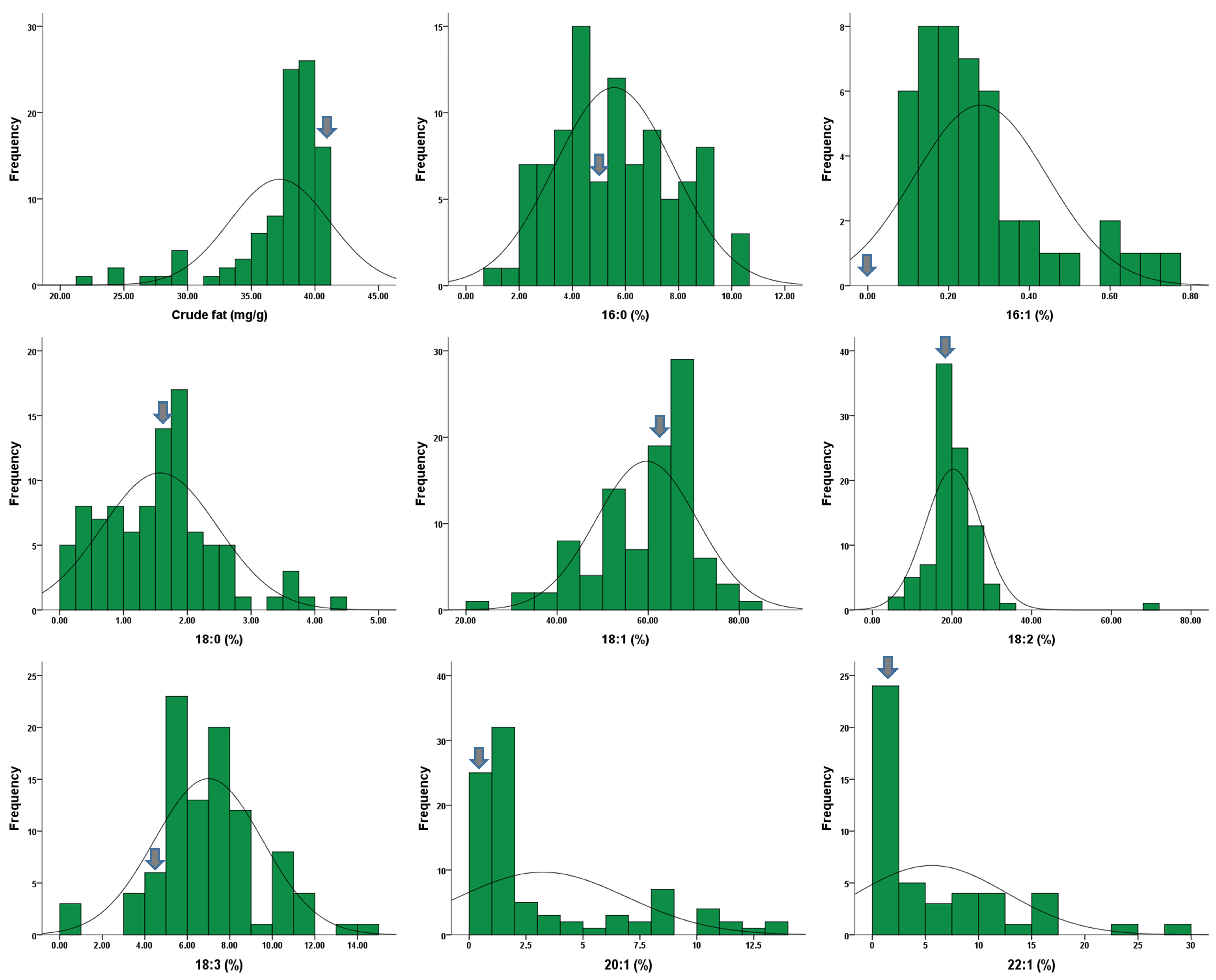
2.2. Fatty Acid Compositions and Crude Fat Contents
2.3. GBS Analysis of Rapeseed Mutant Lines
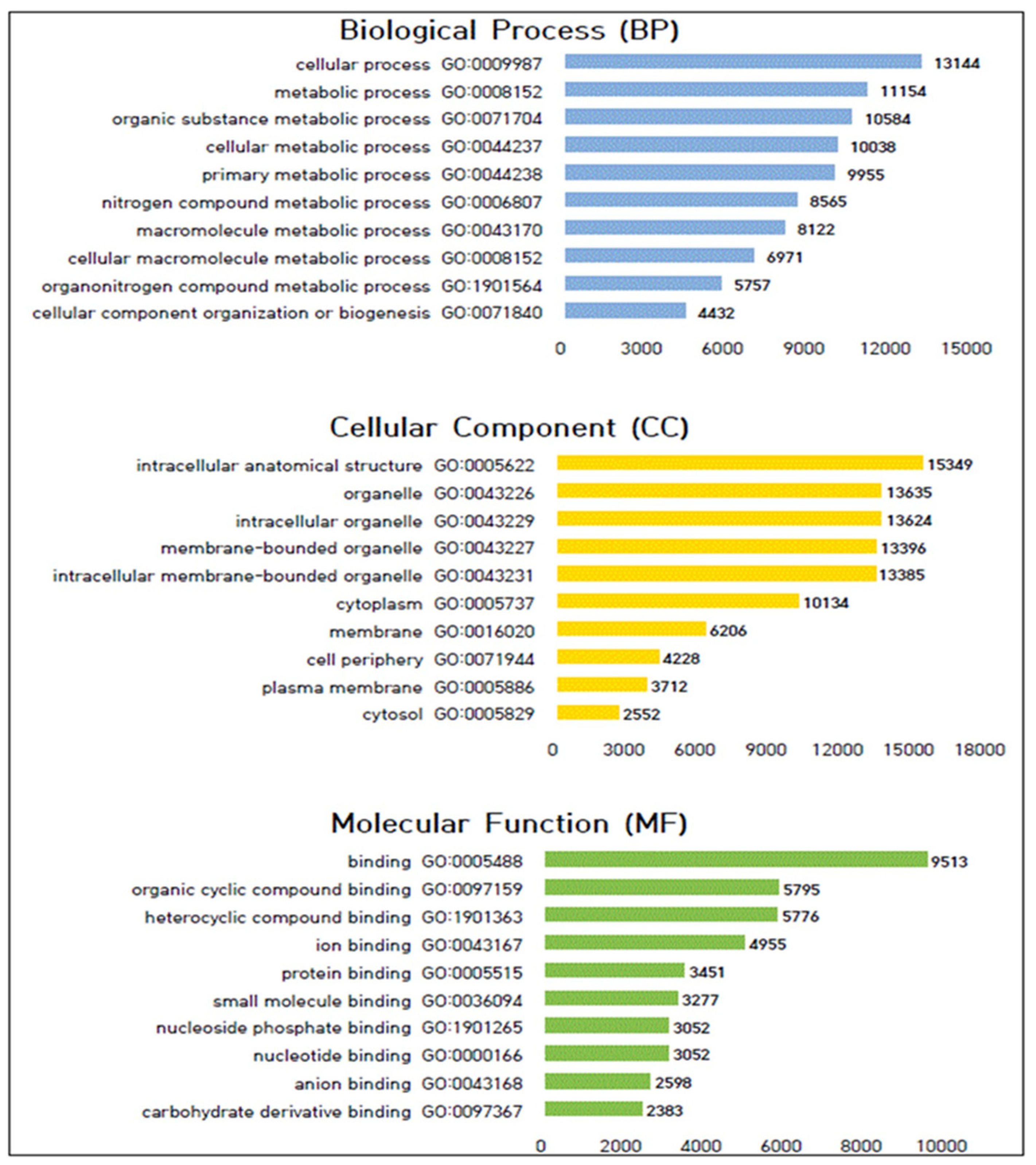
2.4. Gene Ontology (GO) Analysis of Genes with Ploymorphic SNPs
2.5. GWAS Reveals SNPs Associated with Agronomic Traits
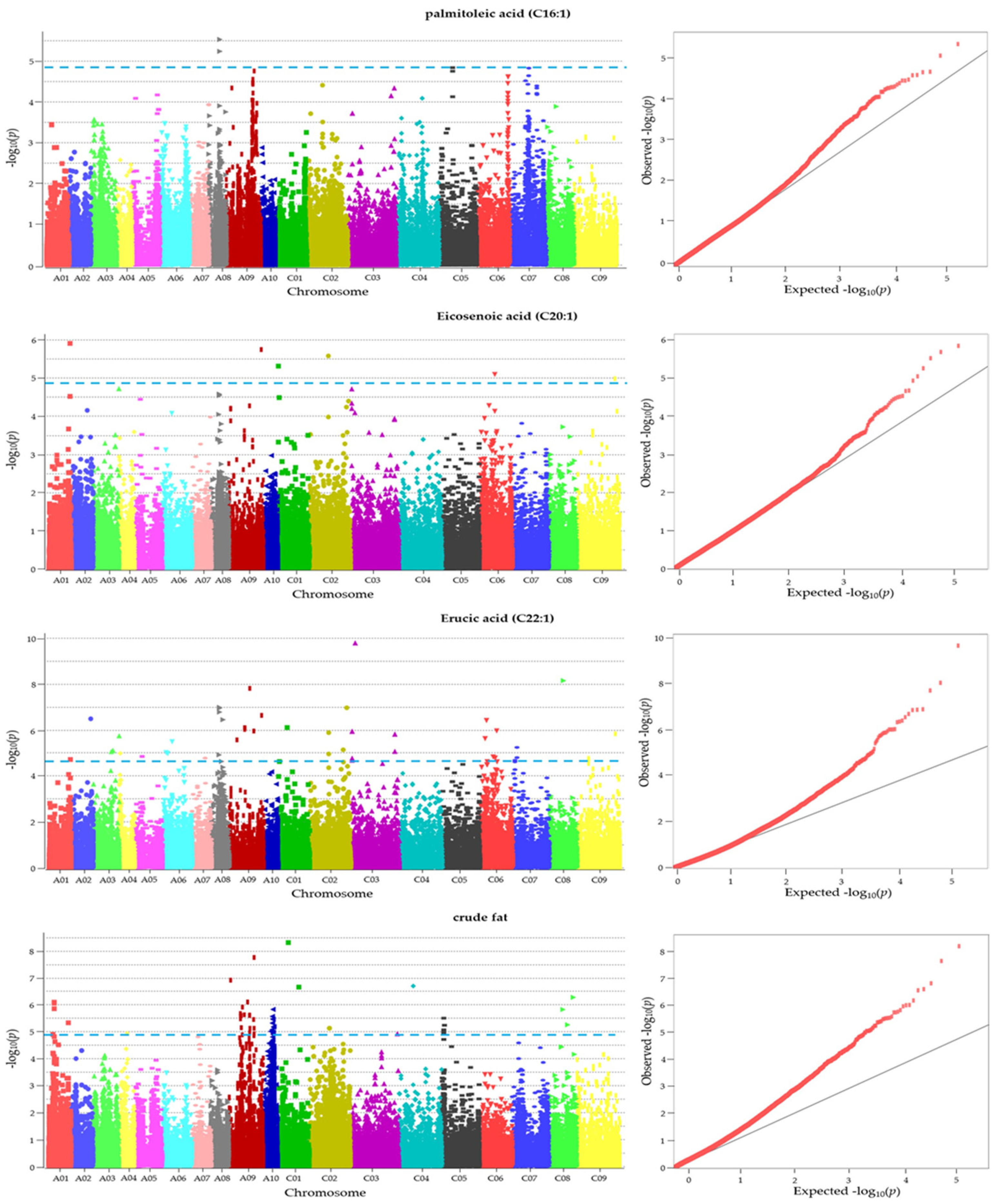
2.6. GWAS Exposed SNPs Associated with Fatty Acid Compositions and Crude Fat Content
3. Discussion
4. Materials and Methods
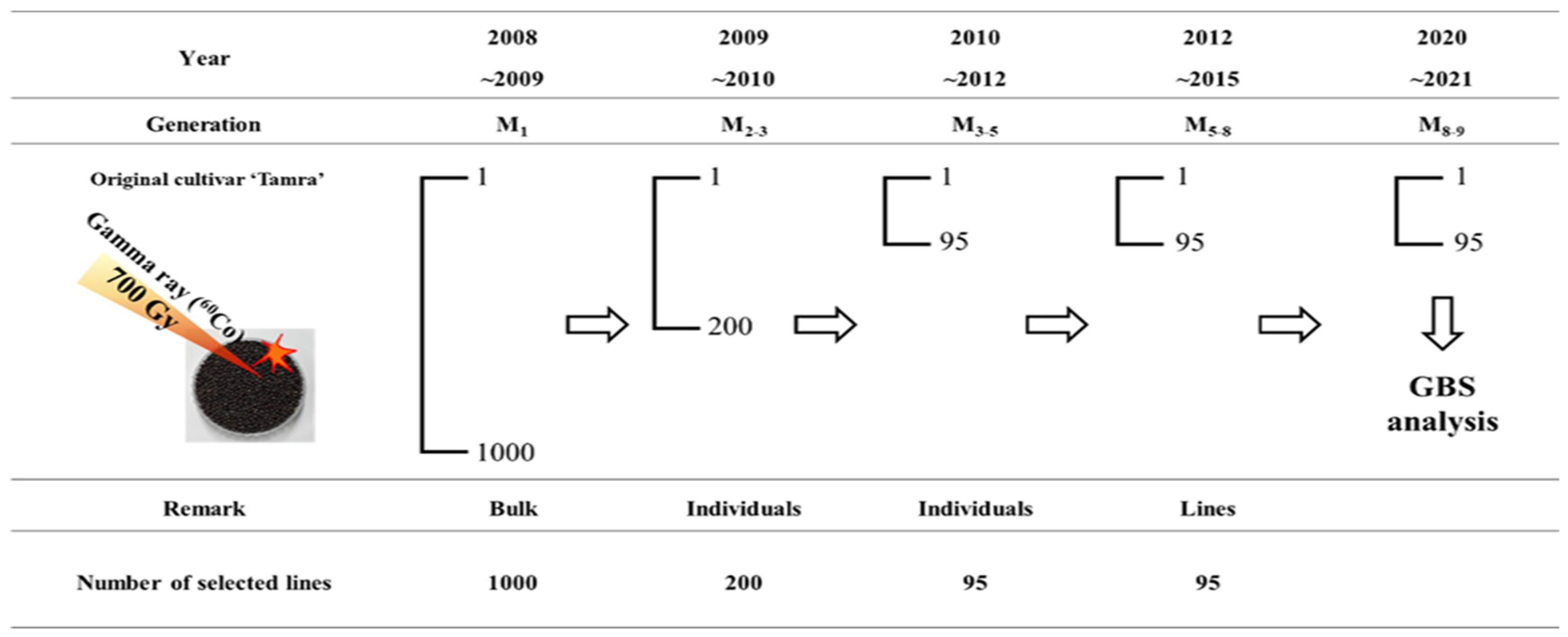
4.1. Plant Material
4.2. Determination of Fatty Acid Compositions and Crude Fat Contents
4.3. DNA Extraction
4.4. Library Construction and Genotyping-by-Sequencing (GBS) Analysis
4.5. Sequence Preprocessing and Alignment to Reference Genome Sequence
4.6. Raw SNP Detection and Consensus Sequence Extraction
4.7. Generate SNP Matrix
4.8. Gene Ontology (GO) Analysis of Genes with Polymorphic SNPs
4.9. GWAS with Agronomic Characteristics, Fatty Acid, and Crude Fat
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Cheung, F.; Trick, M.; Drou, N.; Lim, Y.P.; Park, J.-Y.; Kwon, S.-J.; Kim, J.-A.; Scott, R.; Pires, J.C.; Paterson, A.H. Comparative analysis between homoeologous genome segments of Brassica napus and its progenitor species reveals extensive sequence-level divergence. Plant Cell 2009, 21, 1912–1928. [Google Scholar] [CrossRef] [PubMed]
- Malek, M.A.; Ismail, M.R.; Rafii, M.Y.; Rahman, M. Synthetic Brassica napus L.: Development and Studies on Morphological Characters, Yield Attributes, and Yield. Sci. World J. 2012, 2012, 416901. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, E.J.; Sidebottom, C.H.D.; Koh, C.S.; MacInnes, T.; Sharpe, A.G.; Haughn, G.W. A Mutant Brassica napus (Canola) Population for the Identification of New Genetic Diversity via TILLING and Next Generation Sequencing. PLoS ONE 2013, 8, e84303. [Google Scholar] [CrossRef]
- Zhu, Q.; King, G.J.; Liu, X.; Shan, N.; Borpatragohain, P.; Baten, A.; Wang, P.; Luo, S.; Zhou, Q. Identification of SNP loci and candidate genes related to four important fatty acid composition in Brassica napus using genome wide association study. PLoS ONE 2019, 14, e0221578. [Google Scholar] [CrossRef] [PubMed]
- Nath, U.K.; Kim, H.T.; Khadiza Khatun, K.K.; Park, J.I.; Kang, K.K.; Nou, I.S. Modification of fatty acid profiles of rapeseed (Brassica napus L.) oil for using as food, industrial feed-stock and biodiesel. Plant Breed. Biotechnol. 2016, 4, 123–134. [Google Scholar] [CrossRef]
- Ryu, J.; Lyu, J.I.; Kim, D.-G.; Koo, K.M.; Yang, B.; Jo, Y.D.; Kim, S.H.; Kwon, S.-J.; Ha, B.-K.; Kang, S.-Y.; et al. Single Nucleotide Polymorphism (SNP) Discovery and Association Study of Flowering Times, Crude Fat and Fatty Acid Composition in Rapeseed (Brassica napus L.) Mutant Lines Using Genotyping-by-Sequencing (GBS). Agronomy 2021, 11, 508. [Google Scholar] [CrossRef]
- Geng, X.; Dong, N.; Wang, Y.; Li, G.; Wang, L.; Guo, X.; Li, J.; Wen, Z.; Wei, W. RNA-seq transcriptome analysis of the immature seeds of two Brassica napus lines with extremely different thousand-seed weight to identify the candidate genes related to seed weight. PLoS ONE 2018, 13, e0191297. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.U.; Yangmiao, J.; Liu, S.; Zhang, K.; Khan, M.H.U.; Zhai, Y.; Olalekan, A.; Fan, C.; Zhou, Y. Genome-wide association studies in the genetic dissection of ovule number, seed number, and seed weight in Brassica napus L. Ind. Crops Prod. 2019, 142, 111877. [Google Scholar] [CrossRef]
- Li, N.; Peng, W.; Shi, J.; Wang, X.; Liu, G.; Wang, H. The Natural Variation of Seed Weight Is Mainly Controlled by Maternal Genotype in Rapeseed (Brassica napus L.). PLoS ONE 2015, 10, e0125360. [Google Scholar] [CrossRef]
- Long, W.; Hu, M.; Gao, J.; Chen, S.; Zhang, J.; Cheng, L.; Pu, H. Identification and Functional Analysis of Two New Mutant BnFAD2 Alleles That Confer Elevated Oleic Acid Content in Rapeseed. Front. Genet. 2018, 9, 399. [Google Scholar] [CrossRef]
- Wang, P.; Xiong, X.; Zhang, X.; Wu, G.; Liu, F. A Review of Erucic Acid Production in Brassicaceae Oilseeds: Progress and Prospects for the Genetic Engineering of High and Low-Erucic Acid Rapeseeds (Brassica napus). Front. Plant Sci. 2022, 13, 899076. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liao, X.; Zhang, C.; Xu, H. Influences of plant density on the seed yield and oil content of winter oilseed rape (Brassica napus L.). Ind. Crops Prod. 2012, 40, 27–32. [Google Scholar] [CrossRef]
- Sharafi, Y.; Majidi, M.M.; Goli, S.A.H.; Rashidi, F. Oil Content and Fatty Acids Composition in Brassica Species. Int. J. Food Prop. 2015, 18, 2145–2154. [Google Scholar] [CrossRef]
- Kim, S.H.; Ryu, J.; Kim, W.J.; Kang, R.; Seo, E.; Kim, G.; Kang, S.-Y.; Lee, J.-D.; Ha, B.-K. Identification of a new GmSACPD-C allele in high stearic acid mutant Hfa180 derived from gamma-ray irradiation. Mol. Breed. 2019, 39, 17. [Google Scholar] [CrossRef]
- Anwar, M.M.; Ali, S.E.; Nasr, E.H. Improving the nutritional value of canola seed by gamma irradiation. J. Radiat. Res. Appl. Sci. 2015, 8, 328–333. [Google Scholar] [CrossRef]
- Ha, B.-K.; Lee, K.J.; Velusamy, V.; Kim, J.-B.; Kim, S.H.; Ahn, J.-W.; Kang, S.-Y.; Kim, D.S. Improvement of soybean through radiation-induced mutation breeding techniques in Korea. Plant Genet. Resour. Charact. Util. 2014, 12, S54–S57. [Google Scholar] [CrossRef]
- Morita, R.; Kusaba, M.; Iida, S.; Yamaguchi, H.; Nishio, T.; Nishimura, M. Molecular characterization of mutations induced by gamma irradiation in rice. Genes Genet. Syst. 2009, 84, 361–370. [Google Scholar] [CrossRef]
- Larik, A.; Memon, S.; Soomro, Z. Radiation induced polygenic mutations in Sorghum bicolor L. J. Agric. Res. 2009, 47, 03681157. [Google Scholar]
- He, J.; Zhao, X.; Laroche, A.; Lu, Z.-X.; Liu, H.K.; Li, Z. Genotyping-by-sequencing (GBS), an ultimate marker-assisted selection (MAS) tool to accelerate plant breeding. Front. Plant Sci. 2014, 5, 484. [Google Scholar] [CrossRef]
- Kim, D.-G.; Lyu, J.I.; Kim, J.M.; Seo, J.S.; Choi, H.-I.; Jo, Y.D.; Kim, S.H.; Eom, S.H.; Ahn, J.-W.; Bae, C.-H.; et al. Identification of Loci Governing Agronomic Traits and Mutation Hotspots via a GBS-Based Genome-Wide Association Study in a Soybean Mutant Diversity Pool. Int. J. Mol. Sci. 2022, 23, 10441. [Google Scholar] [CrossRef]
- Glaubitz, J.C.; Casstevens, T.M.; Lu, F.; Harriman, J.; Elshire, R.J.; Sun, Q.; Buckler, E.S. TASSEL-GBS: A High Capacity Genotyping by Sequencing Analysis Pipeline. PLoS ONE 2014, 9, e90346. [Google Scholar] [CrossRef]
- Kim, W.J.; Kang, B.H.; Moon, C.Y.; Kang, S.; Shin, S.; Chowdhury, S.; Jeong, S.-C.; Choi, M.-S.; Park, S.-K.; Moon, J.-K.; et al. Genome-Wide Association Study for Agronomic Traits in Wild Soybean (Glycine soja). Agronomy 2023, 13, 739. [Google Scholar] [CrossRef]
- Hwang, S.-G.; Lee, S.C.; Lee, J.; Lee, J.W.; Kim, J.-H.; Choi, S.Y.; Kim, J.-B.; Choi, H.-I.; Jang, C.S. Resequencing of a core rice mutant population induced by gamma-ray irradiation and its application in a genome-wide association study. J. Plant Biol. 2020, 63, 463–472. [Google Scholar] [CrossRef]
- Zhou, X.; Zhang, H.; Wang, P.; Liu, Y.; Zhang, X.; Song, Y.; Wang, Z.; Ali, A.; Wan, L.; Yang, G.; et al. BnaC7.ROT3, the causal gene of cqSL-C7, mediates silique length by affecting cell elongation in Brassica napus. J. Exp. Bot. 2022, 73, 154–167. [Google Scholar] [CrossRef]
- Kwon, D.-E.; Kim, K.-S.; Hwang, E.-J.; Park, J.-C.; Lee, J.-E.; Lee, Y.-H. Changes in growth and productivity characteristics by sowing date on spring sowing Rapeseed (Brassica napus L.) in paddy field of southern region of South Korea. Korean J. Crop Sci. 2021, 66, 80–86. [Google Scholar]
- Hase, Y.; Satoh, K.; Seito, H.; Oono, Y. Genetic Consequences of Acute/Chronic Gamma and Carbon Ion Irradiation of Arabidopsis thaliana. Front. Plant Sci. 2020, 11, 336. [Google Scholar] [CrossRef]
- Guan, M.; Shi, X.; Chen, S.; Wan, Y.; Tang, Y.; Zhao, T.; Gao, L.; Sun, F.; Yin, N.; Zhao, H.; et al. Comparative transcriptome analysis identifies candidate genes related to seed coat color in rapeseed. Front. Plant Sci. 2023, 14, 1154208. [Google Scholar] [CrossRef]
- Lee, C.W.; Yoo, W.; Park, S.-H.; Le, L.T.H.L.; Jeong, C.-S.; Ryu, B.H.; Shin, S.C.; Kim, H.-W.; Park, H.; Kim, K.K.; et al. Structural and functional characterization of a novel cold-active S-formylglutathione hydrolase (SfSFGH) homolog from Shewanella frigidimarina, a psychrophilic bacterium. Microb. Cell Factories 2019, 18, 140. [Google Scholar] [CrossRef]
- Huang, Y.; Du, L.; Wang, M.; Ren, M.; Yu, S.; Yang, Q. Multifaceted roles of zinc finger proteins in regulating various agronomic traits in rice. Front. Plant Sci. 2022, 13, 974396. [Google Scholar] [CrossRef]
- Basim, H.; Basim, E.; Tombuloglu, H.; Unver, T. Comparative transcriptome analysis of resistant and cultivated tomato lines in response to Clavibacter michiganensis subsp. michiganensis. Genomics 2021, 113, 2455–2467. [Google Scholar] [CrossRef]
- Guo, C.; Yao, L.; You, C.; Wang, S.; Cui, J.; Ge, X.; Ma, H. MID1 plays an important role in response to drought stress during reproductive development. Plant J. 2016, 88, 280–293. [Google Scholar] [CrossRef]
- Huang, S.; Qu, X.; Zhang, R. Plant villins: Versatile actin regulatory proteins. J. Integr. Plant Biol. 2015, 57, 40–49. [Google Scholar] [CrossRef]
- Piombo, E.; Vetukuri, R.R.; Broberg, A.; Kalyandurg, P.B.; Kushwaha, S.; Jensen, D.F.; Karlsson, M.; Dubey, M. Role of Dicer-Dependent RNA Interference in Regulating Mycoparasitic Interactions. Microbiol. Spectr. 2021, 9, e01099-21. [Google Scholar] [CrossRef]
- Shimada, T.; Kunieda, T.; Sumi, S.; Koumoto, Y.; Tamura, K.; Hatano, K.; Ueda, H.; Hara-Nishimura, I. The AP-1 Complex is Required for Proper Mucilage Formation in Arabidopsis Seeds. Plant Cell Physiol. 2018, 59, 2331–2338. [Google Scholar] [CrossRef]
- Shahnejat-Bushehri, S.; Tarkowska, D.; Sakuraba, Y.; Balazadeh, S. Arabidopsis NAC transcription factor JUB1 regulates GA/BR metabolism and signalling. Nat. Plants 2016, 2, 16013. [Google Scholar] [CrossRef]
- Licatalosi, D.D. Roles of RNA-binding proteins and post-transcriptional regulation in driving male germ cell development in the mouse. In RNA Processing: Disease and Genome-Wide Probing; Springer: Cham, Switzerland, 2016; pp. 123–151. [Google Scholar]
- Lou, L.; Ding, L.; Wang, T.; Xiang, Y. Emerging Roles of RNA-Binding Proteins in Seed Development and Performance. Int. J. Mol. Sci. 2020, 21, 6822. [Google Scholar] [CrossRef]
- Li, X.L.; Huang, W.L.; Yang, H.H.; Jiang, R.C.; Sun, F.; Wang, H.C.; Zhao, J.; Xu, C.H.; Tan, B.C. EMP18 functions in mitochondrial atp6 and cox2 transcript editing and is essential to seed development in maize. New Phytol. 2019, 221, 896–907. [Google Scholar] [CrossRef]
- Scholz, P.; Anstatt, J.; Krawczyk, H.E.; Ischebeck, T. Signalling Pinpointed to the Tip: The Complex Regulatory Network That Allows Pollen Tube Growth. Plants 2020, 9, 1098. [Google Scholar] [CrossRef]
- Xiang, X.; Wu, Y.; Planta, J.; Messing, J.; Leustek, T. Overexpression of serine acetyltransferase in maize leaves increases seed-specific methionine-rich zeins. Plant Biotechnol. J. 2018, 16, 1057–1067. [Google Scholar] [CrossRef]
- Pal, L.; Sandhu, S.K.; Bhatia, D.; Sethi, S. Genome-wide association study for candidate genes controlling seed yield and its components in rapeseed (Brassica napus subsp. napus). Physiol. Mol. Biol. Plants 2021, 27, 1933–1951. [Google Scholar] [CrossRef]
- Fujiki, Y.; Teshima, H.; Kashiwao, S.; Kawano-Kawada, M.; Ohsumi, Y.; Kakinuma, Y.; Sekito, T. Functional identification of AtAVT3, a family of vacuolar amino acid transporters, in Arabidopsis. FEBS Lett. 2017, 591, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zhao, J.; Chen, D.; Wang, Y. E3 ubiquitin ligases: Styles, structures and functions. Mol. Biomed. 2021, 2, 23. [Google Scholar] [CrossRef] [PubMed]
- Ando, H.; Watabe, H.; Valencia, J.C.; Yasumoto, K.-I.; Furumura, M.; Funasaka, Y.; Oka, M.; Ichihashi, M.; Hearing, V.J. Fatty acids regulate pigmentation via proteasomal degradation of tyrosinase: A new aspect of ubiquitin-proteasome function. J. Biol. Chem. 2004, 279, 15427–15433. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ma, Z.-H.; Mao, J.; Chen, B.-H. Genome-wide identification and expression analysis of the EXO70 gene family in grape (Vitis vinifera L.). PeerJ 2021, 9, e11176. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Lopez, J.A.; James, D.E.; Hunziker, W. Snapin Interacts with the Exo70 Subunit of the Exocyst and Modulates GLUT4 Trafficking. J. Biol. Chem. 2008, 283, 324–331. [Google Scholar] [CrossRef]
- Wang, S.; Crisman, L.; Miller, J.; Datta, I.; Gulbranson, D.R.; Tian, Y.; Yin, Q.; Yu, H.; Shen, J. Inducible Exoc7/Exo70 knockout reveals a critical role of the exocyst in insulin-regulated GLUT4 exocytosis. J. Biol. Chem. 2019, 294, 19988–19996. [Google Scholar] [CrossRef] [PubMed]
- Vorwerk, S.; Schiff, C.; Santamaria, M.; Koh, S.; Nishimura, M.; Vogel, J.; Somerville, C.; Somerville, S. EDR2 negatively regulates salicylic acid-based defenses and cell death during powdery mildew infections of Arabidopsis thaliana. BMC Plant Biol. 2007, 7, 35. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Ade, J.; Frye, C.A.; Innes, R.W. Regulation of plant defense responses in Arabidopsis by EDR2, a PH and START domain-containing protein. Plant J. 2005, 44, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Omnus, D.J.; Cadou, A.; Thomas, F.B.; Bader, J.M.; Soh, N.; Chung, G.H.C.; Vaughan, A.N.; Stefan, C.J. A heat-sensitive Osh protein controls PI4P polarity. BMC Biol. 2020, 18, 28. [Google Scholar] [CrossRef]
- Delage, E.; Puyaubert, J.; Zachowski, A.; Ruelland, E. Signal transduction pathways involving phosphatidylinositol 4-phosphate and phosphatidylinositol 4,5-bisphosphate: Convergences and divergences among eukaryotic kingdoms. Prog. Lipid Res. 2013, 52, 1–14. [Google Scholar] [CrossRef]
- Massimo, A.; Maura, S.; Nicoletta, L.; Giulia, M.; Valentina, M.; Elena, C.; Alessandro, P.; Rosaria, B.; Sandro, S. Current and Novel Aspects on the Non-lysosomal β-Glucosylceramidase GBA2. Neurochem. Res. 2016, 41, 210–220. [Google Scholar] [CrossRef]
- Gao, Y.; Li, H.; Deng, D.; Chen, S.; Jiang, W.; Chen, J. Characterization and expression analysis of the maize RING-H2 finger protein gene ZmXERICO responsive to plant hormones and abiotic stresses. Acta Physiol. Plant. 2012, 34, 1529–1535. [Google Scholar] [CrossRef]
- Zeng, D.-E.; Hou, P.; Xiao, F.; Liu, Y. Overexpressing a novel RING-H2 finger protein gene, OsRHP1, enhances drought and salt tolerance in rice (Oryza sativa L.). J. Plant Biol. 2014, 57, 357–365. [Google Scholar] [CrossRef]
- Qiu, Z.; Zhuang, K.; Liu, Y.; Ge, X.; Chen, C.; Hu, S.; Han, H. Functional characterization of C-TERMINALLY ENCODED PEPTIDE (CEP) family in Brassica rapa L. Plant Signal. Behav. 2022, 17, 2021365. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Cox, M.P.; Peterson, D.A.; Biggs, P.J. SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinform. 2010, 11, 485. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows—Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Kim, J.-E.; Oh, S.-K.; Lee, J.-H.; Lee, B.-M.; Jo, S.-H. Genome-Wide SNP Calling Using Next Generation Sequencing Data in Tomato. Mol. Cells 2014, 37, 36–42. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Huang, Y.; Hou, L.; Ma, J.; Chen, C.; Ai, H.; Huang, L.; Ren, J. Genome-wide detection of genetic markers associated with growth and fatness in four pig populations using four approaches. Genet. Sel. Evol. 2017, 49, 21. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Min | Max | SD | Mean | CV(%) | Skew | Kur |
|---|---|---|---|---|---|---|---|
| Plant height (cm) | 134.00 | 175.00 | 7.62 | 157.69 | 4.83 | −0.23 | 0.34 |
| Ear length (cm) | 35.00 | 77.50 | 6.27 | 59.76 | 10.49 | −0.24 | 2.78 |
| Thousand-seed weight (g) | 2.80 | 5.40 | 0.47 | 4.18 | 11.15 | 0.33 | 1.04 |
| Seed yield (kg/10a) | 144.00 | 398.00 | 80.26 | 306.93 | 26.15 | −0.45 | −1.28 |
| Trait | Min | Max | SD | Mean | CV(%) | Skew | Kur |
|---|---|---|---|---|---|---|---|
| Crude fat (mg/g) | 21.31 | 40.98 | 3.88 | 37.22 | 10.43 | −2.19 | 4.76 |
| C16:0 (%) | 0.88 | 10.50 | 2.22 | 5.57 | 39.83 | 0.21 | −0.71 |
| C16:1 | 0.10 | 0.76 | 0.16 | 0.28 | 58.63 | 1.41 | 1.36 |
| C18:0 | 0.15 | 4.42 | 0.90 | 1.57 | 57.40 | 0.68 | 0.61 |
| C18:1 | 20.93 | 81.73 | 11.07 | 59.65 | 18.55 | −0.84 | 0.84 |
| C18:2 | 7.66 | 71.09 | 7.01 | 20.34 | 34.46 | 3.92 | 27.86 |
| C18:3 | 0.43 | 14.68 | 2.53 | 7.00 | 36.17 | 0.25 | 0.97 |
| C20:1 | 0.40 | 13.10 | 3.65 | 3.23 | 113.14 | 1.37 | 0.45 |
| C22:1 | 0.08 | 29.12 | 6.94 | 5.61 | 123.83 | 1.53 | 1.95 |
| C16:0 | C16:1 | C18:0 | C18:1 | C18:2 | C18:3 | C20:1 | |
|---|---|---|---|---|---|---|---|
| C16:1 | 0.588 ** | ||||||
| C18:0 | 0.625 ** | 0.268 | |||||
| C18:1 | −0.311 ** | −0.54 ** | −0.236 * | ||||
| C18:2 | 0.443 ** | 0.299 * | 0.210 * | −0.169 | |||
| C18:3 | 0.420 ** | 0.377 ** | 0.332 ** | −0.569 ** | 0.253 * | ||
| C20:1 | −0.292 ** | 0.650 | −0.234 * | −0.639 ** | −0.235 * | 0.007 | |
| C22:1 | −0.379 ** | 0.311 | −0.312 * | −0.633 ** | −0.255 | 0.125 | 0.871 ** |
| Total | Average/Plant | |
|---|---|---|
| Raw data | ||
| Reads | 715,259,878 | 7,450,624 |
| Bases (bp) | 108,004,241,578 | 1,125,044,183 |
| After trimming | ||
| Reads | 655,243,166 | 6,825,450 |
| Bases (bp) | 74,621,891,073 | 777,311,365 |
| Mapped reads on reference genome | ||
| Reads | 651,288,040 | 6,784,250 |
| Bases (bp) | 3,163,629,539 | 32,954,474 |
| Reference genome coverage (%) | 3.57% |
| Chr | Position | −log10(p) | Gene ID | Description | TAIR ID | Allele |
|---|---|---|---|---|---|---|
| A05 | 1,312,542 | 12.37 | BnaA05p02350D | S-formylglutathione hydrolase-like | AT2G41530.1 | A/G |
| C01 | 45,614,214 | 8.75 | BnaC01p49650D | zinc finger BED domain-containing protein RICESLEEPER 1-like | AT3G14800.2 | G/T |
| C02 | 16,544,482 | 8.44 | BnaC02p22490D | midasin-like | AT1G67120.2 | G/A |
| C08 | 30,501,410 | 6.41 | BnaC08p32830D | villin-3-like isoform X2 | AT3G57410.9 | A/G |
| C03 | 21,138,185 | 6.40 | BnaC03p36140D | death-associated inhibitor of apoptosis 1 | AT4G03965.1 | C/T |
| C05 | 4,473,754 | 5.90 | BnaC05p08830D | ubiquitin-like-specific protease 1C | AT1G10570.3 | G/T |
| A05 | 2,029,410 | 5.58 | BnaA05p03850D | chitinase 1 | AT2G43570.1 | T/A |
| A05 | 1,197,170 | 5.46 | BnaA05p02100D | amino acid transporter AVT1A | AT2G41190.2 | A/C |
| C05 | 439,211 | 5.30 | BnaC05p00900D | endoribonuclease Dicer homolog 1-like | AT1G01040.2 | T/C |
| C05 | 4,629,489 | 5.25 | BnaC05p09110D | AP-1 complex subunit mu-2 | AT1G10730.1 | C/T |
| C08 | 28,339,798 | 5.12 | BnaC08p29480D | RNA-binding protein 5 | AT3G54230.5 | G/A |
| A03 | 20,827,042 | 5.07 | BnaA03p44720D | ATPase 6, plasma membrane-type | AT2G07560.1 | T/A |
| A03 | 5,465,396 | 5.02 | BnaA03p13200D | serine acetyltransferase 5 | AT5G56760.1 | G/A |
| C09 | 4,097,062 | 4.91 | BnaC09p07010D | U2 snRNP-associated SURP motif-containing protein isoform X2 | AT5G25060.1 | T/G |
| Trait | Chr_Position | −log10(p) | Gene ID | Description | TAIR ID | Allele |
|---|---|---|---|---|---|---|
| C16:1 | A08_16,580,415 | 5.24 | BnaA08p17150D | Encodes a putative E3 ubiquitin ligase | AT4G34100.1 | C/T |
| C20:1 | C01_1,257,401 | 5.32 | BnaC01p02330D | probable Xaa-Pro aminopeptidase P isoform X2 | AT4G36760.2 | C/A |
| C22:1 | C03_6,878,724 | 9.78 | BnaC03p14350D | EXO70 gene family | AT5G58430.1 | C/T |
| C22:1 | C08_21,249,315 | 8.15 | BnaC08p19880D | paramyosin-like | AT1G19980.1 | C/T |
| C22:1 | A08_12,298,481 | 6.98 | BnaA08p10580D | mediator of RNA polymerase II transcription subunit 34 | AT1G31360.3 | C/A |
| C22:1 | C02_56,355,748 | 6.97 | BnaC02p56750D | cyclin-C1-1 | AT5G48640.1 | A/G |
| C22:1 | C02_51,025,897 | 5.14 | BnaC02p51020D | ribonuclease T2 family | AT2G02990.1 | G/A |
| C22:1 | A08_11,469,852 | 4.92 | BnaA08p09630D | protein ENHANCED DISEASE RESISTANCE 2-like isoform X2 | AT5G35180.4 | G/A |
| Crude fat | A09_3,387,964 | 6.92 | BnaA09p05630D | Part of a nanodomain complex that tethers PI4Kα1 to the plasma membrane | AT5G26850.1 | C/A |
| Crude fat | A09_18,251,187 | 5.59 | BnaA09p27860D | 1-aminocyclopropane-1-carboxylate synthase 6 | AT4G11280.1 | A/G |
| Crude fat | C05_2,572,075 | 5.51 | BnaC05p05120D | mitoferrin | AT1G07030.1 | G/C |
| Crude fat | A09_38,265,711 | 5.46 | BnaA09p39540D | Transmembrane amino acid transporter family protein | AT1G25530.1 | C/T |
| Crude fat | A10_15,664,715 | 5.20 | BnaA10p21730D | chromo domain-containing protein LHP1-like | AT5G17690.3 | T/C |
| Crude fat | A09_32,798,569 | 5.14 | BnaA09p32690D | nonlysosomal glucosylceramidase | AT1G33700.1 | T/G |
| Crude fat | A09_39,092,505 | 5.14 | BnaA09p40920D | RING-H2 finger protein ATL47-like | AT1G23980.1 | C/T |
| Crude fat | A09_32,277,805 | 4.98 | BnaA09p32130D | embryogenesis-associated protein EMB8 | AT1G34340.1 | T/C |
| Crude fat | C05_2,503,939 | 4.97 | BnaC05p04980D | CEP DOWNSTREAM 1 | AT1G06830.1 | A/G |
| Crude fat | C03_71,451,788 | 4.93 | BnaC03p90410D | protein CHUP1, chloroplastic-like | AT1G52080.1 | A/T |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, W.J.; Yang, B.; Kim, D.-G.; Kim, S.H.; Lee, Y.-J.; Kim, J.; Baek, S.H.; Kang, S.-Y.; Ahn, J.-W.; Choi, Y.-J.; et al. Genotyping-by-Sequencing Analysis Reveals Associations between Agronomic and Oil Traits in Gamma Ray-Derived Mutant Rapeseed (Brassica napus L.). Plants 2024, 13, 1576. https://doi.org/10.3390/plants13111576
Kim WJ, Yang B, Kim D-G, Kim SH, Lee Y-J, Kim J, Baek SH, Kang S-Y, Ahn J-W, Choi Y-J, et al. Genotyping-by-Sequencing Analysis Reveals Associations between Agronomic and Oil Traits in Gamma Ray-Derived Mutant Rapeseed (Brassica napus L.). Plants. 2024; 13(11):1576. https://doi.org/10.3390/plants13111576
Chicago/Turabian StyleKim, Woon Ji, Baul Yang, Dong-Gun Kim, Sang Hoon Kim, Ye-Jin Lee, Juyoung Kim, So Hyeon Baek, Si-Yong Kang, Joon-Woo Ahn, Yu-Jin Choi, and et al. 2024. "Genotyping-by-Sequencing Analysis Reveals Associations between Agronomic and Oil Traits in Gamma Ray-Derived Mutant Rapeseed (Brassica napus L.)" Plants 13, no. 11: 1576. https://doi.org/10.3390/plants13111576
APA StyleKim, W. J., Yang, B., Kim, D.-G., Kim, S. H., Lee, Y.-J., Kim, J., Baek, S. H., Kang, S.-Y., Ahn, J.-W., Choi, Y.-J., Bae, C.-H., Iwar, K., Kim, S.-H., & Ryu, J. (2024). Genotyping-by-Sequencing Analysis Reveals Associations between Agronomic and Oil Traits in Gamma Ray-Derived Mutant Rapeseed (Brassica napus L.). Plants, 13(11), 1576. https://doi.org/10.3390/plants13111576