
Integrated Transcriptome and Metabolome Analysis Reveals Molecular Mechanisms Underlying Resistance to Phytophthora Root Rot

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Overview of RNA Sequencing Data
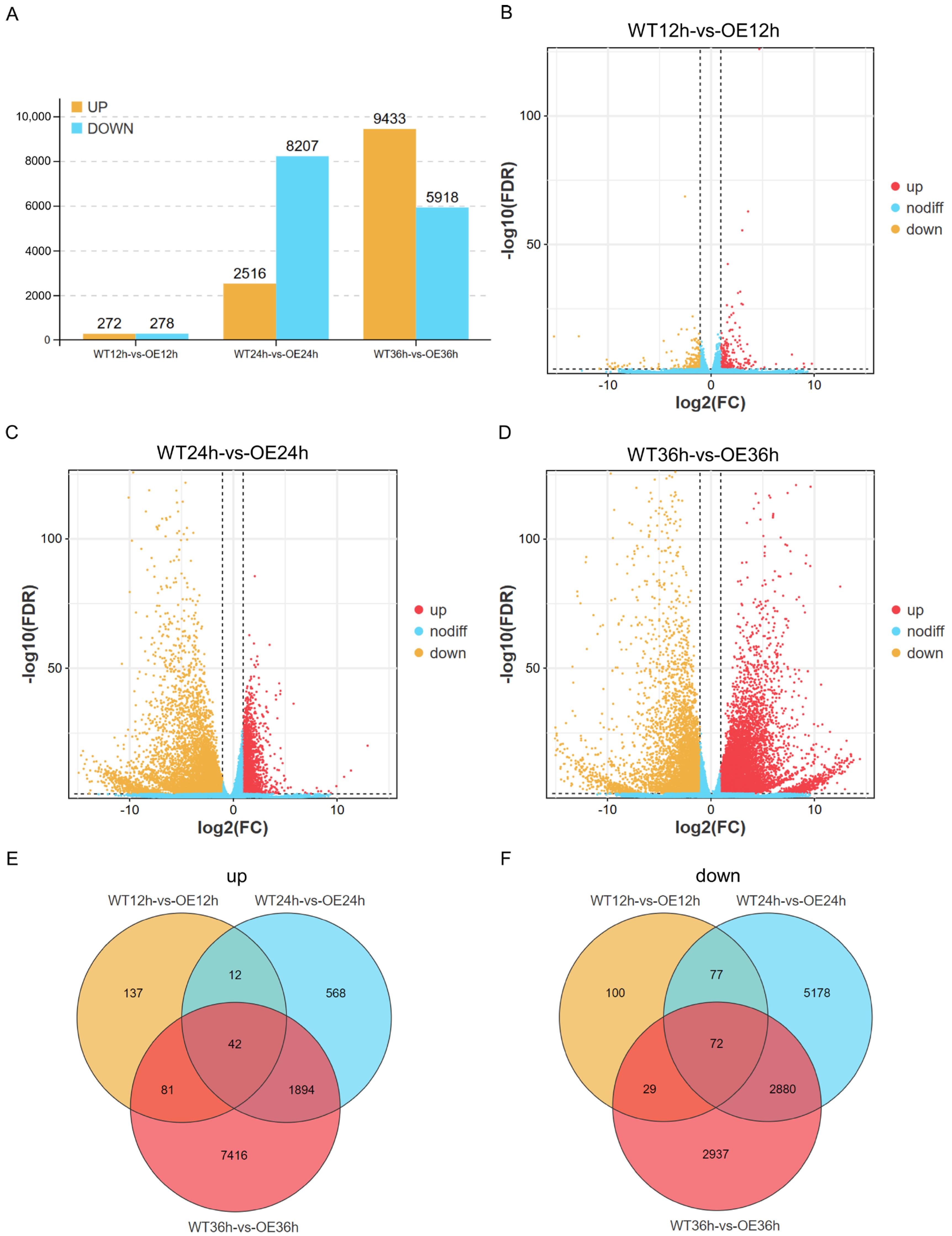
2.2. Differentially Expressed Genes in Response to Phytophthora sojae Infection
2.3. Validation of the Transcriptomic Data Based on the RT-qPCR Analysis
2.4. Functional Classification of DEGs of WT and OE Plants
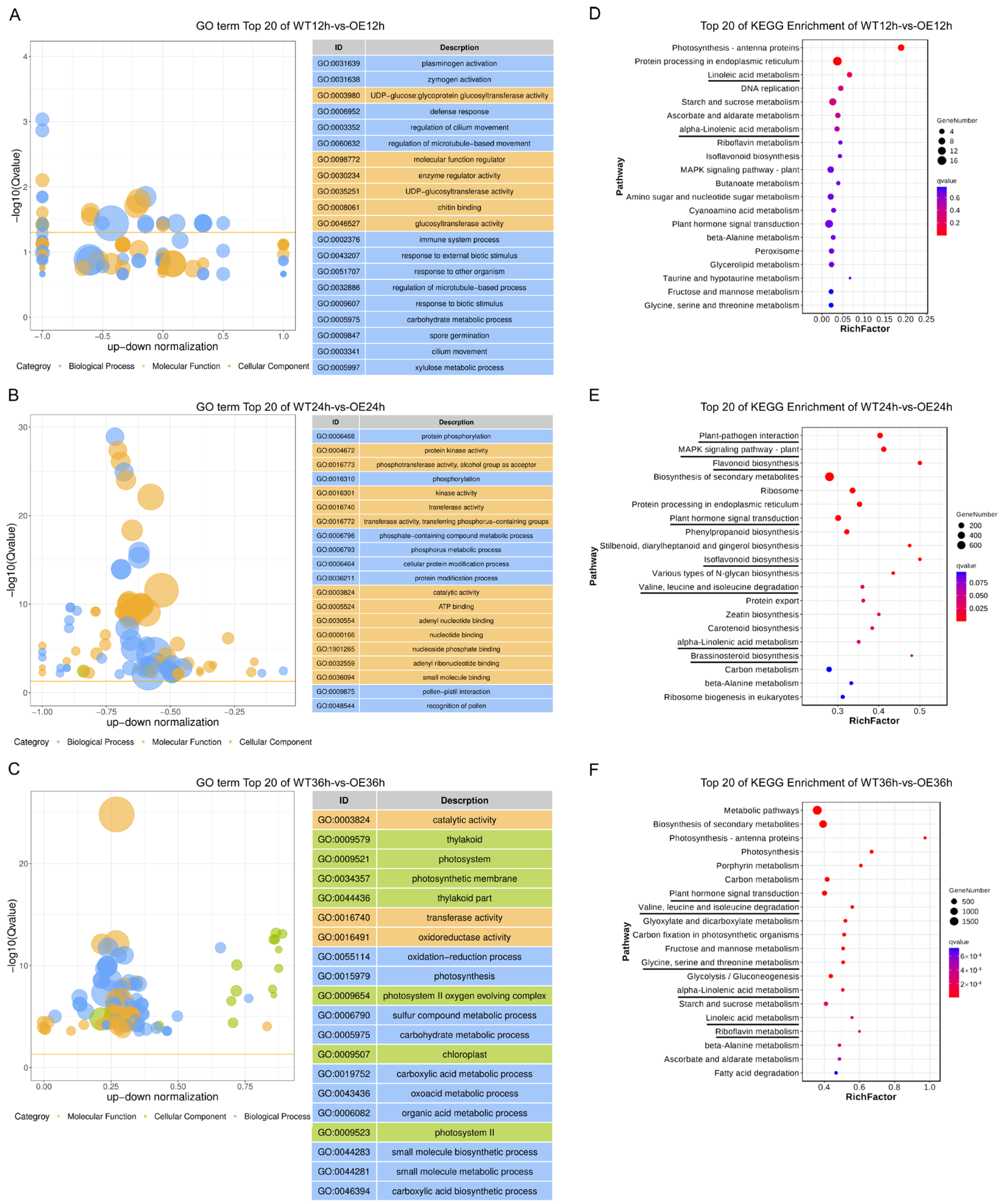
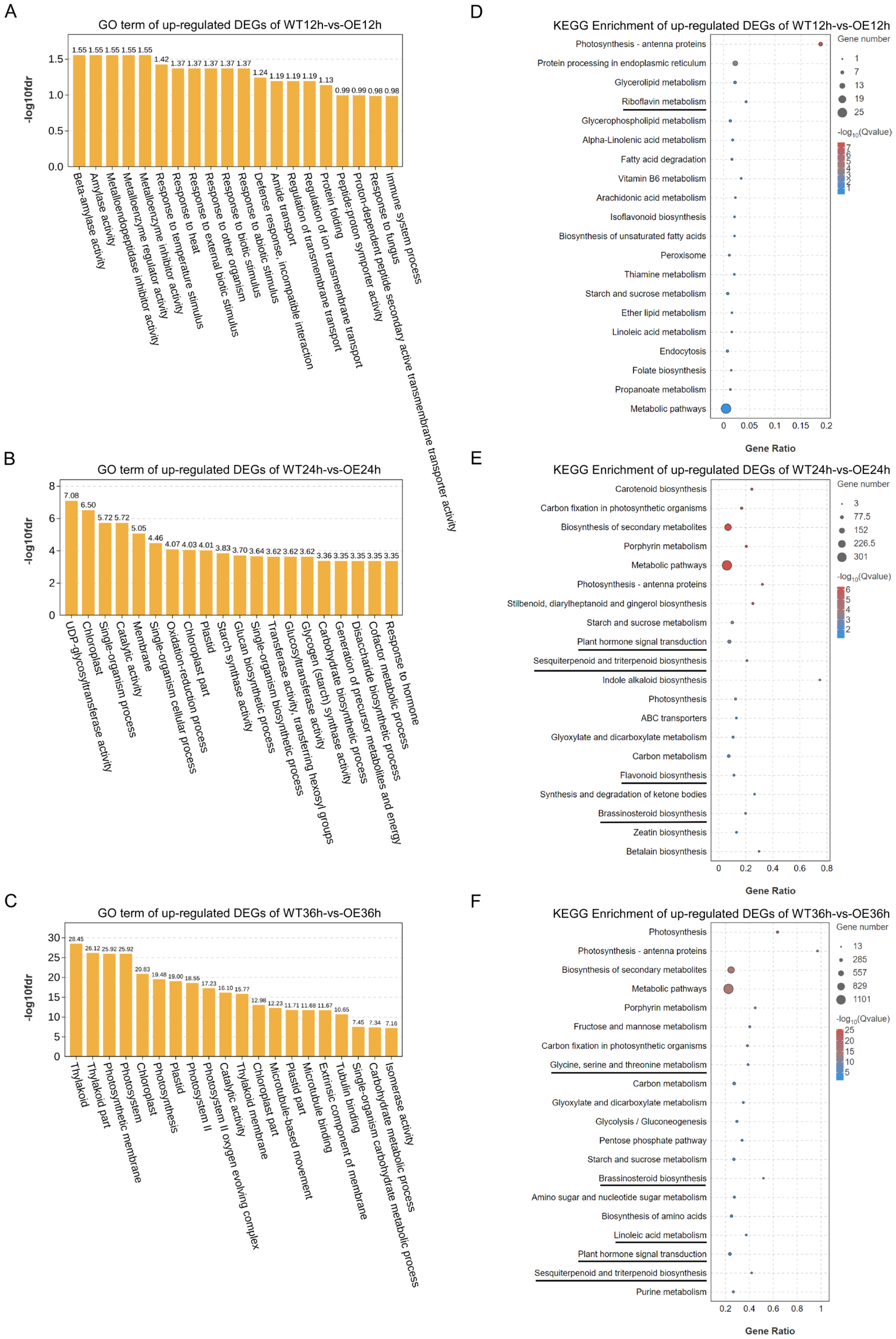
2.5. GO Term and KEGG Pathway Enrichment Analysis of Up-Regulated DEGs
2.6. Analysis of Gene Expression Patterns and Clustering and Functional Classification of DEGs
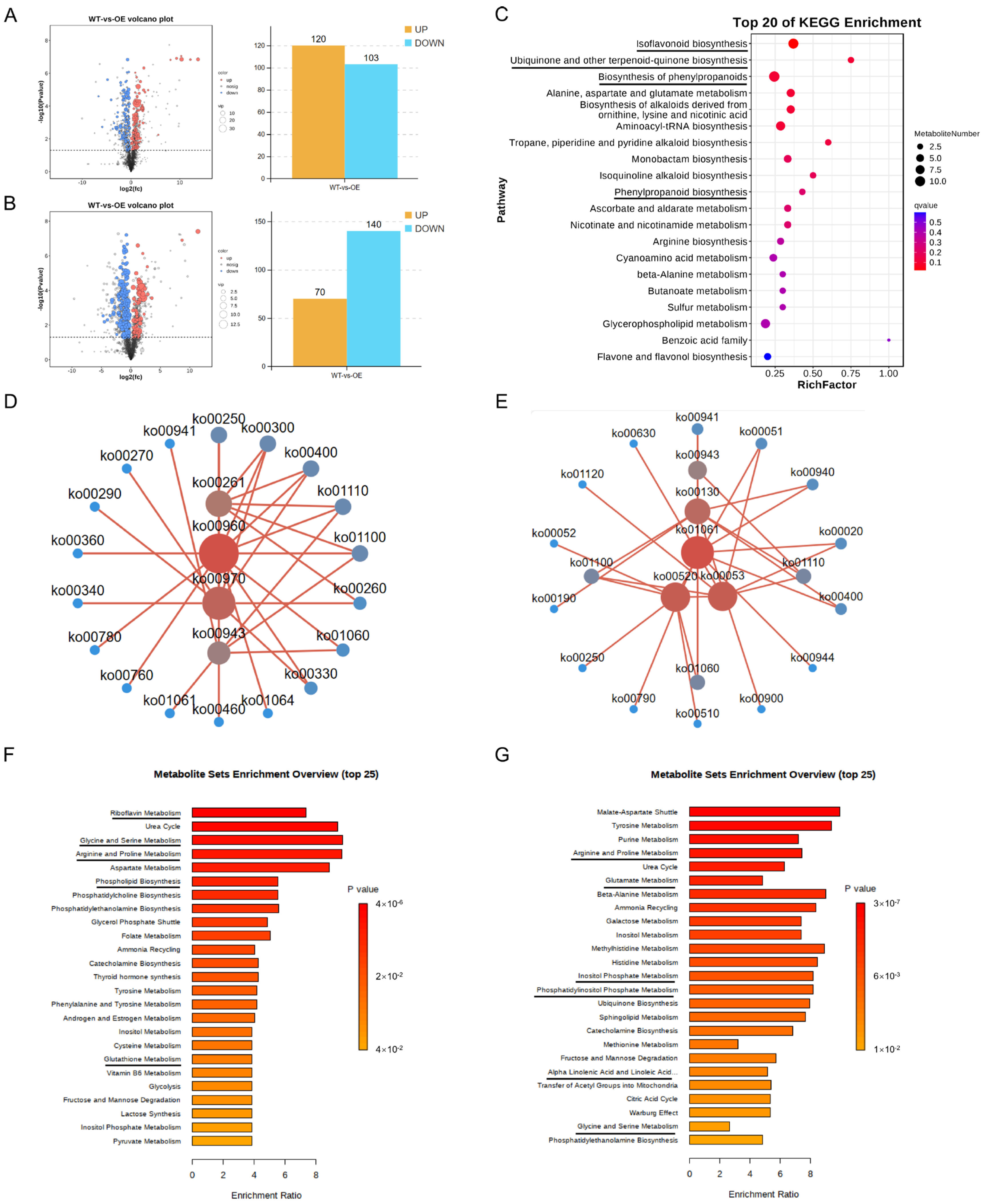
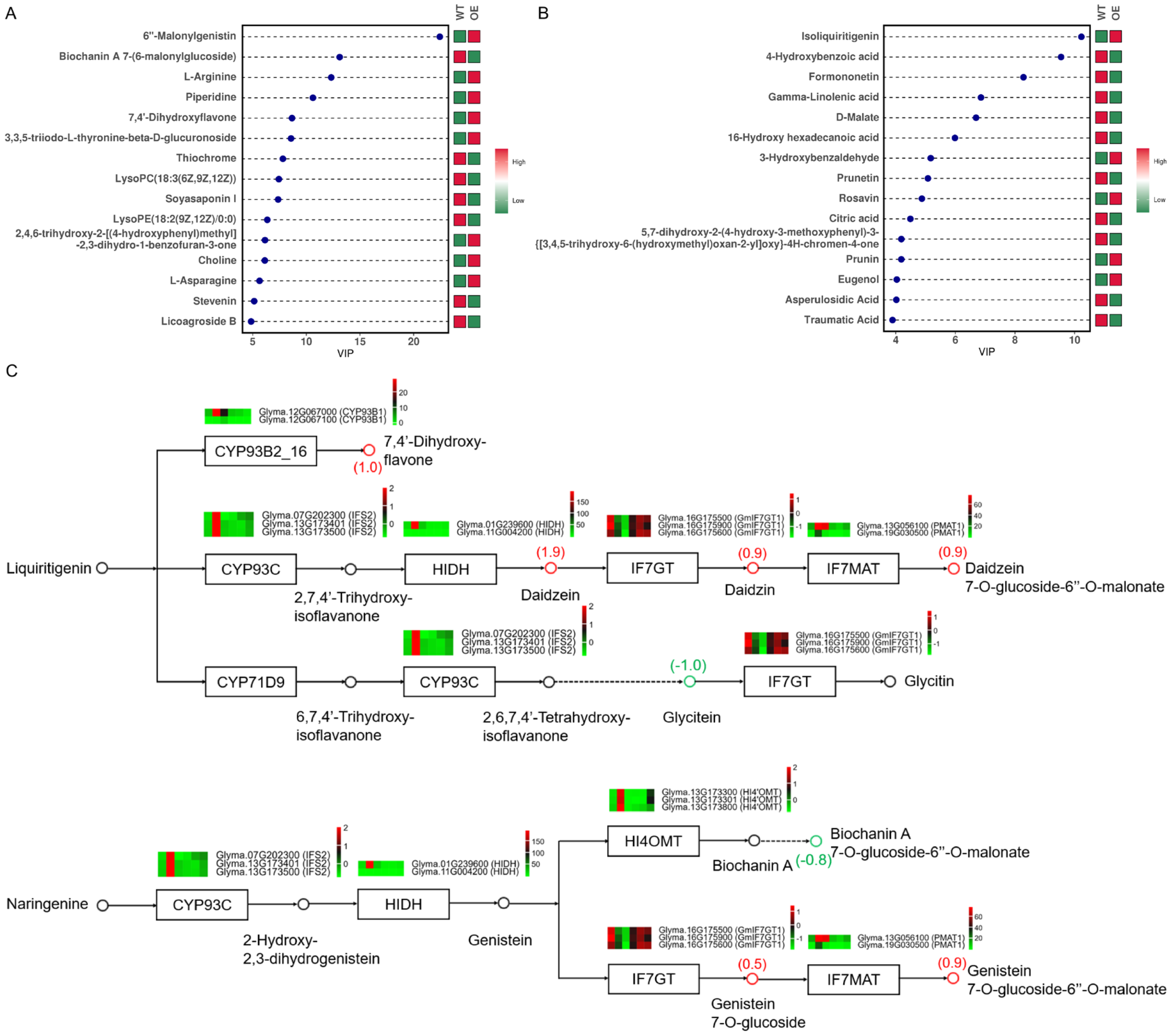
2.7. Differential Metabolites and Their Functional Analysis
2.8. Association Analysis of Transcriptome and Metabolome Reveals the Molecular Mechanism of OE Plants in Regulating Resistance
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Plant Materials, Inoculations, and Experimental Conditions
5.2. RNA Extraction, Library Construction, and Sequencing
5.3. Differential Expression Analysis and Functional Enrichment
5.4. Quantitative Real-Time Polymerase Chain Reaction Assays
5.5. Metabolites Extraction
5.6. LC-MS/MS Analysis
5.7. Screening and Identification of Differential Accumulation Metabolite (DAM)
5.8. Integrated Metabolome and Transcriptome Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tyler, B.M. Phytophthora sojae: Root rot pathogen of soybean and model oomycete. Mol. Plant Pathol. 2007, 8, 1–8. [Google Scholar] [CrossRef]
- Chandra, S.; Choudhary, M.; Bagaria, P.K.; Nataraj, V.; Kumawat, G.; Choudhary, J.R.; Sonah, H.; Gupta, S.; Wani, S.H.; Ratnaparkhe, M.B. Progress and prospectus in genetics and genomics of Phytophthora root and stem rot resistance in soybean (Glycine max L.). Front. Genet. 2022, 13, 939182. [Google Scholar] [CrossRef]
- Jiang, B.; Cheng, Y.; Cai, Z.; Li, M.; Jiang, Z.; Ma, R.; Yuan, Y.; Xia, Q.; Nian, H. Fine mapping of a Phytophthora-resistance locus RpsGZ in soybean using genotyping-by-sequencing. BMC Genom. 2020, 21, 280. [Google Scholar] [CrossRef]
- Li, L.; Liu, J.; Zhou, J.-M. From molecule to cell: The expanding frontiers of plant immunity. J. Genet. Genom. 2024, in press. [Google Scholar] [CrossRef]
- Jones, J.D.G.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef]
- Li, W.; Zheng, X.; Cheng, R.; Zhong, C.; Zhao, J.; Liu, T.H.; Yi, T.; Zhu, Z.; Xu, J.; Meksem, K.; et al. Soybean ZINC FINGER PROTEIN03 targets two SUPEROXIDE DISMUTASE1s and confers resistance to Phytophthora sojae. Plant Physiol. 2023, 192, 633–647. [Google Scholar] [CrossRef]
- Wang, W.; Chen, L.; Fengler, K.; Bolar, J.; Llaca, V.; Wang, X.; Clark, C.B.; Fleury, T.J.; Myrvold, J.; Oneal, D.; et al. A giant NLR gene confers broad-spectrum resistance to Phytophthora sojae in soybean. Nat. Commun. 2021, 12, 6263. [Google Scholar] [CrossRef]
- Li, Q.; Zhu, H.; Ai, G.; Yu, J.; Dou, D. Plant genes related to Phytophthora pathogens resistance. Phytopathol. Res. 2024, 6, 15. [Google Scholar] [CrossRef]
- Kumar, G.; Dasgupta, I. Comprehensive molecular insights into the stress response dynamics of rice (Oryza sativa L.) during rice tungro disease by RNA-seq-based comparative whole transcriptome analysis. J. Biosci. 2020, 45, 27. [Google Scholar] [CrossRef]
- Lin, F.; Zhao, M.; Baumann, D.D.; Ping, J.; Sun, L.; Liu, Y.; Zhang, B.; Tang, Z.; Hughes, E.; Doerge, R.W.; et al. Molecular response to the pathogen Phytophthora sojae among ten soybean near isogenic lines revealed by comparative transcriptomics. BMC Genom. 2014, 15, 18. [Google Scholar] [CrossRef]
- Xu, P.; Wu, J.; Xue, A.; Li, W.; Chen, W.; Wei, L.; Lv, H.; Lin, S.; Fan, S.; Li, N.; et al. Differentially Expressed Genes of Soybean During Infection by Phytophthora sojae. J. Integr. Agric. 2012, 11, 368–377. [Google Scholar] [CrossRef]
- Zhu, L.; Zhou, Y.; Li, X.; Zhao, J.; Guo, N.; Xing, H. Metabolomics Analysis of Soybean Hypocotyls in Response to Phytophthora sojae Infection. Front. Plant Sci. 2018, 9, 1530. [Google Scholar] [CrossRef]
- Sun, J.; Li, L.; Zhao, J.; Huang, J.; Yan, Q.; Xing, H.; Guo, N. Genetic analysis and fine mapping of, a novel resistance gene to Phytophthora sojae in soybean [Glycine max (L.) Merr.]. Theor. Appl. Genet. 2014, 127, 913–919. [Google Scholar] [CrossRef]
- Sahoo, D.K.; Das, A.; Huang, X.; Cianzio, S.; Bhattacharyya, M.K. Tightly linked Rps12 and Rps13 genes provide broad-spectrum Phytophthora resistance in soybean. Sci. Rep. 2021, 11, 16907. [Google Scholar] [CrossRef]
- Sun, R.; Ding, W.; Fan, X.; Han, A.; Deng, S.; Zhang, Y.; Wang, C.; Guo, N.; Xing, H.; Zhao, J. The Soybean NLR Gene GmRPP13 Confers Resistance to Phytophthora sojae; Nanjing Agricultural University: Nanjing, China, 2024. [Google Scholar]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Ernst, J.; Bar-Joseph, Z. STEM: A tool for the analysis of short time series gene expression data. BMC Bioinform. 2006, 7, 191. [Google Scholar] [CrossRef]
- Yadav, V.; Wang, Z.; Wei, C.; Amo, A.; Ahmed, B.; Yang, X.; Zhang, X. Phenylpropanoid Pathway Engineering: An Emerging Approach towards Plant Defense. Pathogens 2020, 9, 312. [Google Scholar] [CrossRef]
- Liu, X.; Matsumoto, H.; Lv, T.; Zhan, C.; Fang, H.; Pan, Q.; Xu, H.; Fan, X.; Chu, T.; Chen, S.; et al. Phyllosphere microbiome induces host metabolic defence against rice false-smut disease. Nat. Microbiol. 2023, 8, 1419–1433. [Google Scholar] [CrossRef]
- Kadotani, N.; Akagi, A.; Takatsuji, H.; Miwa, T.; Igarashi, D. Exogenous proteinogenic amino acids induce systemic resistance in rice. BMC Plant Biol. 2016, 16, 60. [Google Scholar] [CrossRef]
- Wang, L.; Chen, M.; Lam, P.Y.; Dini-Andreote, F.; Dai, L.; Wei, Z. Multifaceted roles of flavonoids mediating plant-microbe inter-actions. Microbiome 2022, 10, 233. [Google Scholar] [CrossRef]
- Weston, L.A.; Mathesius, U. Flavonoids: Their structure, biosynthesis and role in the rhizosphere, including allelopathy. J. Chem. Ecol. 2013, 39, 283–297. [Google Scholar] [CrossRef]
- Liu, J.; Deng, J.; Zhang, K.; Wu, H.; Yang, C.; Zhang, X.; Du, J.; Shu, K.; Yang, W. Pod Mildew on Soybeans Can Mitigate the Damage to the Seed Arising from Field Mold at Harvest Time. J. Agric. Food Chem. 2016, 64, 9135–9142. [Google Scholar] [CrossRef]
- Mathesius, U. Flavonoid Functions in Plants and Their Interactions with Other Organisms. Plants 2018, 7, 30. [Google Scholar] [CrossRef]
- Polturak, G.; Misra, R.C.; El-Demerdash, A.; Owen, C.; Steed, A.; McDonald, H.P.; Wang, J.; Saalbach, G.; Martins, C.; Chartrain, L.; et al. Discovery of isoflavone phytoalexins in wheat reveals an alternative route to isoflavonoid biosynthesis. Nat. Commun. 2023, 14, 6977. [Google Scholar] [CrossRef]
- Bi, G.; Su, M.; Li, N.; Liang, Y.; Dang, S.; Xu, J.; Hu, M.; Wang, J.; Zou, M.; Deng, Y.; et al. The ZAR1 resistosome is a calcium-permeable channel triggering plant immune signaling. Cell 2021, 184, 3528–3541.e12. [Google Scholar] [CrossRef]
- Hetherington, A.M.; Brownlee, C. The generation of Ca2+ signals in plants. Annu. Rev. Plant Biol. 2004, 55, 401–427. [Google Scholar] [CrossRef]
- Saand, M.A.; Xu, Y.-P.; Li, W.; Wang, J.-P.; Cai, X.-Z. Cyclic nucleotide gated channel gene family in tomato: Genome-wide identification and functional analyses in disease resistance. Front. Plant Sci. 2015, 6, 303. [Google Scholar] [CrossRef]
- Kaplan, B.; Sherman, T.; Fromm, H. Cyclic nucleotide-gated channels in plants. FEBS Lett. 2007, 581, 2237–2246. [Google Scholar] [CrossRef]
- Wang, J.; Hu, M.; Wang, J.; Qi, J.; Han, Z.; Wang, G.; Qi, Y.; Wang, H.-W.; Zhou, J.-M.; Chai, J. Reconstitution and structure of a plant NLR resistosome conferring immunity. Science 2019, 364, eaav5870. [Google Scholar] [CrossRef]
- Jacob, P.; Kim, N.H.; Wu, F.; El-Kasmi, F.; Chi, Y.; Walton, W.G.; Furzer, O.J.; Lietzan, A.D.; Sunil, S.; Kempthorn, K.; et al. Plant “helper” immune receptors are Ca2+-permeable nonse-lective cation channels. Science 2021, 373, 420–425. [Google Scholar] [CrossRef]
- Pandey, S.P.; Somssich, I.E. The Role of WRKY Transcription Factors in Plant Immunity. Plant Physiol. 2009, 150, 1648–1655. [Google Scholar] [CrossRef]
- Ding, L.-N.; Li, Y.-T.; Wu, Y.-Z.; Li, T.; Geng, R.; Cao, J.; Zhang, W.; Tan, X.-L. Plant Disease Resistance-Related Signaling Pathways: Recent Progress and Future Prospects. Int. J. Mol. Sci. 2022, 23, 16200. [Google Scholar] [CrossRef]
- Yan, C.; Xie, D. Jasmonate in plant defence: Sentinel or double agent? Plant Biotechnol. J. 2015, 13, 1233–1240. [Google Scholar] [CrossRef]
- Thaler, J.S.; Owen, B.; Higgins, V.J. The role of the jasmonate response in plant susceptibility to diverse pathogens with a range of lifestyles. Plant Physiol. 2004, 135, 530–538. [Google Scholar] [CrossRef]
- Valenzuela-Riffo, F.; Zúñiga, P.E.; Morales-Quintana, L.; Lolas, M.; Cáceres, M.; Figueroa, C.R. Priming of Defense Systems and Upregulation of MYC2 and JAZ1 Genes after Botrytis cinerea Inoculation in Methyl Jasmonate-Treated Strawberry Fruits. Plants 2020, 9, 447. [Google Scholar] [CrossRef]
- Nolan, T.M.; Vukašinović, N.; Liu, D.; Russinova, E.; Yin, Y. Brassinosteroids: Multidimensional Regulators of Plant Growth, Development, and Stress Responses. Plant Cell 2020, 32, 295–318. [Google Scholar] [CrossRef]
- Yu, M.-H.; Zhao, Z.-Z.; He, J.-X. Brassinosteroid Signaling in Plant–Microbe Interactions. Int. J. Mol. Sci. 2018, 19, 4091. [Google Scholar] [CrossRef]
- Lozano-Durán, R.; Macho, A.P.; Boutrot, F.; Segonzac, C.; Somssich, I.E.; Zipfel, C. The transcriptional regulator BZR1 mediates trade-off between plant innate immunity and growth. Elife 2013, 2, e00983. [Google Scholar] [CrossRef]
- Meng, F.; Zheng, X.; Wang, J.; Qiu, T.; Yang, Q.; Fang, K.; Bhadauria, V.; Peng, Y.; Zhao, W. The GRAS protein OsDLA involves in brassinosteroid signalling and positively regulates blast resistance by forming a module with GSK2 and OsWRKY53 in rice. Plant Biotechnol. J. 2024, 22, 363–378. [Google Scholar] [CrossRef]
- Song, L.; Chen, W.; Yao, Q.; Guo, B.; Valliyodan, B.; Wang, Z.; Nguyen, H.T. Genome-wide transcriptional profiling for elucidating the effects of brassinosteroids on Glycine max during early vegetative development. Sci. Rep. 2019, 9, 16085. [Google Scholar] [CrossRef]
- Khan, M.; Luo, B.; Hu, M.; Fu, S.; Liu, J.; Jiang, M.; Zhao, Y.; Huang, S.; Wang, S.; Wang, X. Brassinosteroid Signaling Downstream Suppressor BIN2 Interacts with SLFRIGIDA-LIKE to Induce Early Flowering in Tomato. Int. J. Mol. Sci. 2022, 23, 11264. [Google Scholar] [CrossRef]
- Gong, C.; Diao, W.; Zhu, H.; Umer, M.J.; Zhao, S.; He, N.; Lu, X.; Yuan, P.; Anees, M.; Yang, D.; et al. Metabolome and Transcriptome Integration Reveals Insights Into Flavor Formation of ‘Crimson’ Watermelon Flesh During Fruit Development. Front. Plant Sci. 2021, 12, 629361. [Google Scholar] [CrossRef]
- Wang, H.; Wei, X.; Mo, C.; Wei, M.; Li, Y.; Fan, Y.; Gu, X.; Zhang, X.; Zhang, Y.; Kong, Q. Integrated full-length transcriptome and metabolome analysis reveals the defence response of melon to gummy stem blight. Plant Cell Environ. 2024, 47, 1997–2010. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Liu, Y.; Du, H.; Li, P.; Shen, Y.; Peng, H.; Liu, S.; Zhou, G.-A.; Zhang, H.; Liu, Z.; Shi, M.; et al. Pan-Genome of Wild and Cultivated Soybeans. Cell 2020, 182, 162–176.e13. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, R.; Han, A.; Wang, H.; Wang, C.; Lu, Y.; Ni, D.; Guo, N.; Xing, H.; Zhao, J. Integrated Transcriptome and Metabolome Analysis Reveals Molecular Mechanisms Underlying Resistance to Phytophthora Root Rot. Plants 2024, 13, 1705. https://doi.org/10.3390/plants13121705
Sun R, Han A, Wang H, Wang C, Lu Y, Ni D, Guo N, Xing H, Zhao J. Integrated Transcriptome and Metabolome Analysis Reveals Molecular Mechanisms Underlying Resistance to Phytophthora Root Rot. Plants. 2024; 13(12):1705. https://doi.org/10.3390/plants13121705
Chicago/Turabian StyleSun, Ruidong, Anan Han, Haitang Wang, Congcong Wang, Yang Lu, Danqing Ni, Na Guo, Han Xing, and Jinming Zhao. 2024. "Integrated Transcriptome and Metabolome Analysis Reveals Molecular Mechanisms Underlying Resistance to Phytophthora Root Rot" Plants 13, no. 12: 1705. https://doi.org/10.3390/plants13121705
APA StyleSun, R., Han, A., Wang, H., Wang, C., Lu, Y., Ni, D., Guo, N., Xing, H., & Zhao, J. (2024). Integrated Transcriptome and Metabolome Analysis Reveals Molecular Mechanisms Underlying Resistance to Phytophthora Root Rot. Plants, 13(12), 1705. https://doi.org/10.3390/plants13121705