
Oil/Water Biphasic Solvent System for the Eco-Extraction and Cosmetic Formulation of Bixa orellana L.
 , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Selection of Ultrasound-Assisted Extraction parameters
2.2. Oil Selection
2.2.1. Characterisation of Crude Oils
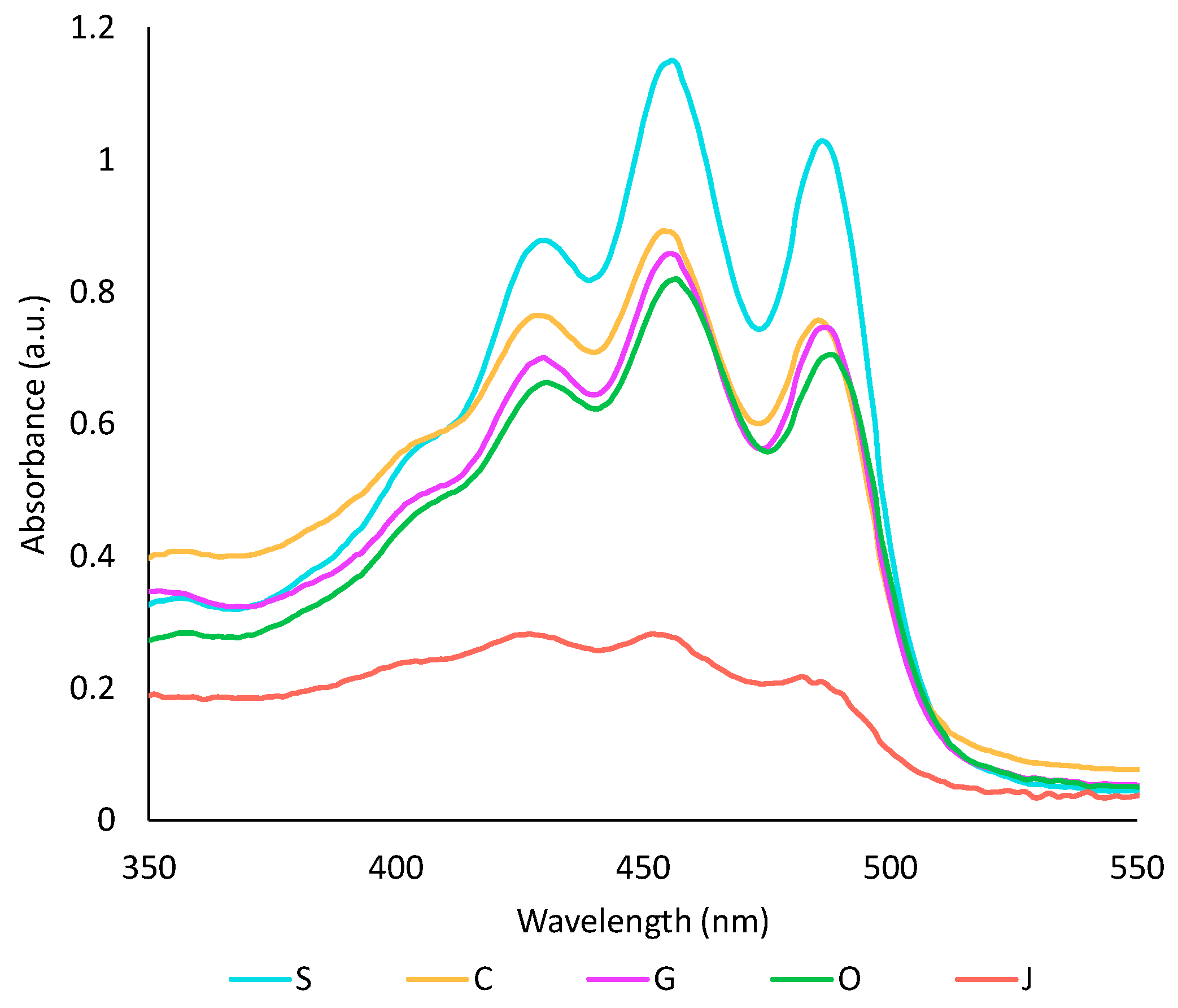
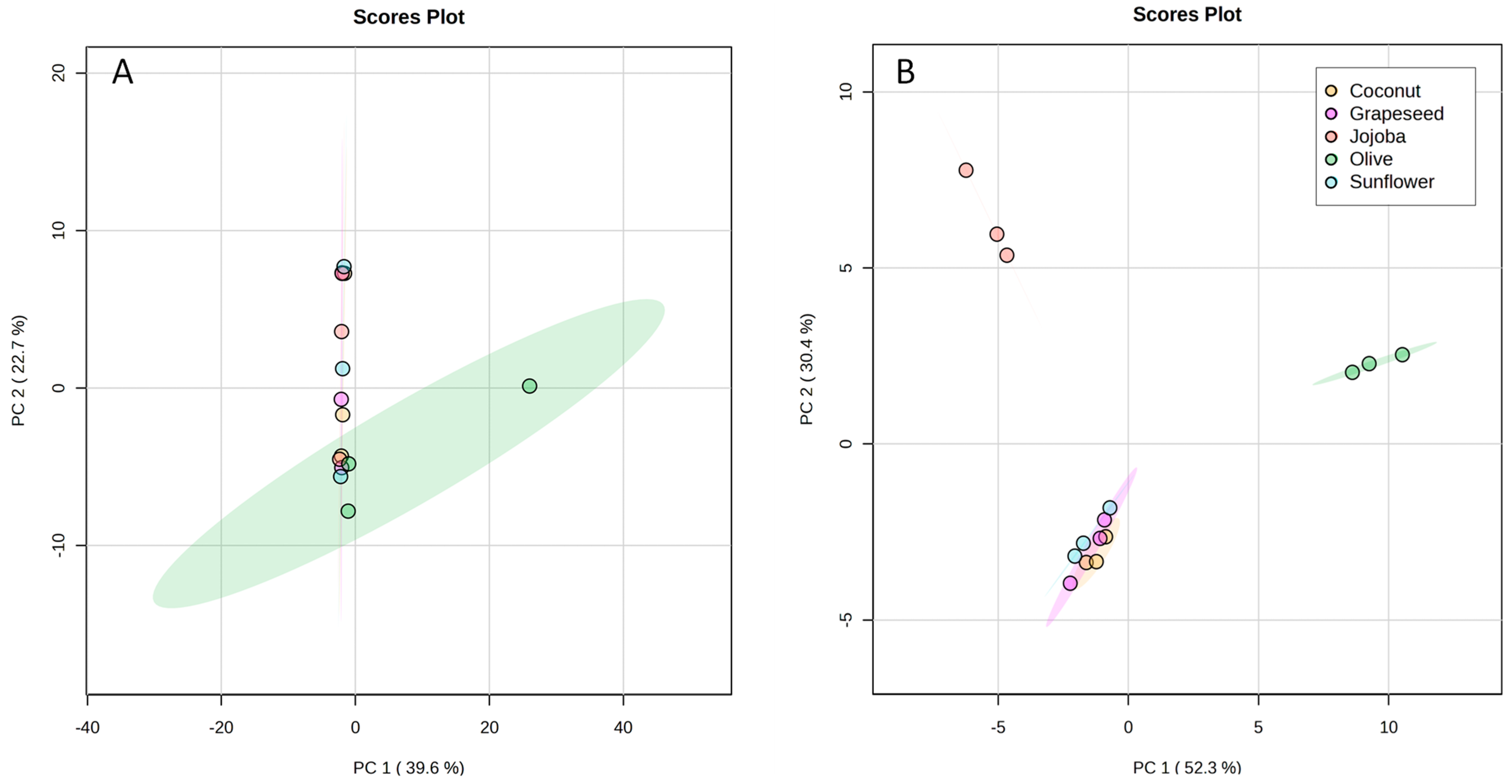
2.2.2. Comparison of Oil Phases
2.2.3. Impact of the Oil on the Aqueous Phase
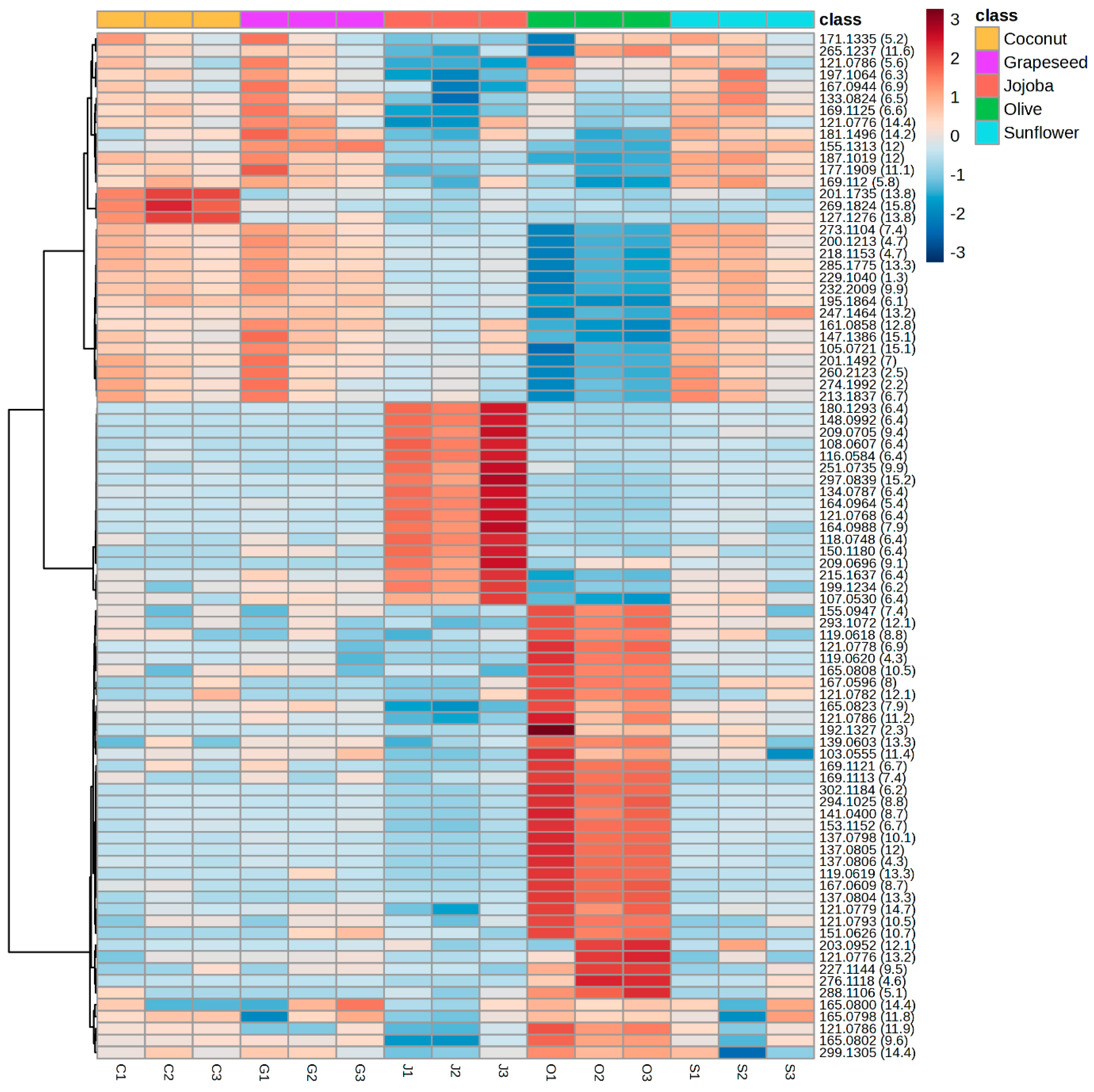
2.3. Characterisation of the Optimised Extract
2.3.1. Characterisation of the Oil Phase
2.3.2. Characterisation of the Aqueous Phase
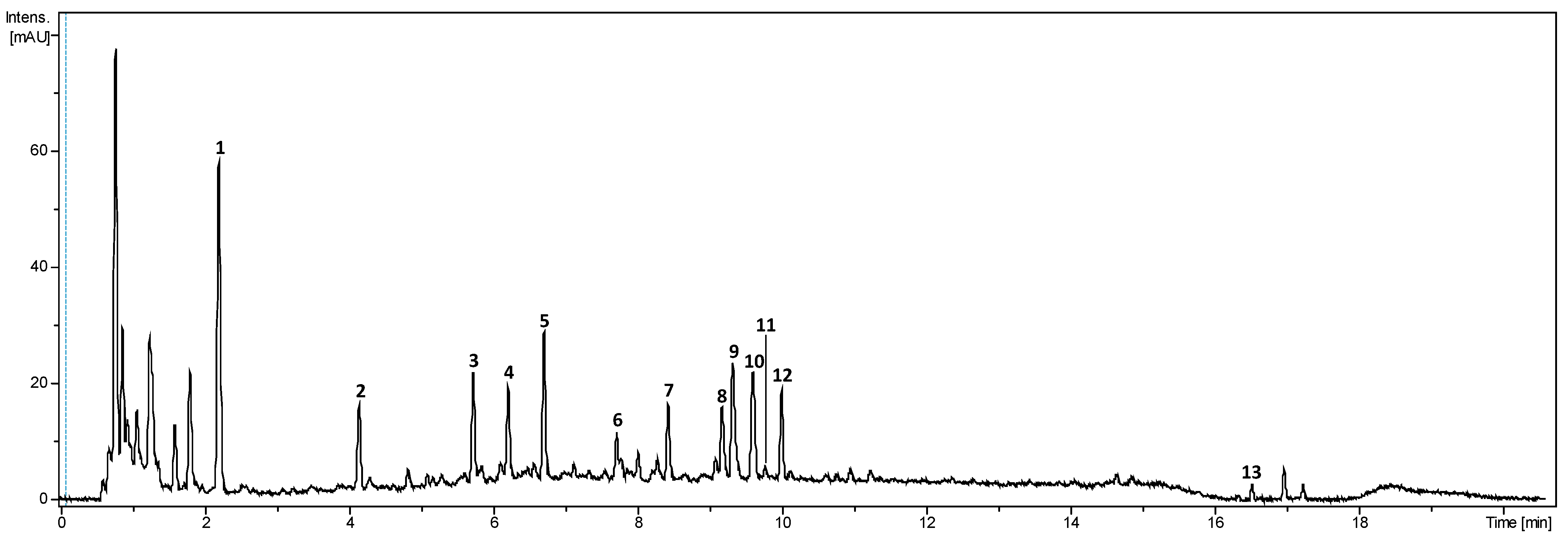
2.3.3. SPME-GC-MS of the Volatile Fraction
2.4. Formulation of the Extract
3. Materials and Methods
3.1. Plant Material, Standards and Chemicals
3.2. Biphasic Ultrasound-Assisted Extraction
3.2.1. Optimisation of the Parameters
3.2.2. Oil Selection as an Extraction Solvent
3.3. Physicochemical Analysis of the Oils
3.4. SFC-MS Analysis of the Oils
3.5. Carotenoid and Phenolic Contents
3.5.1. UV-Visible Spectrophotometry
3.5.2. Quantification of Apocarotenoids
3.5.3. Total Phenolic Content Determination
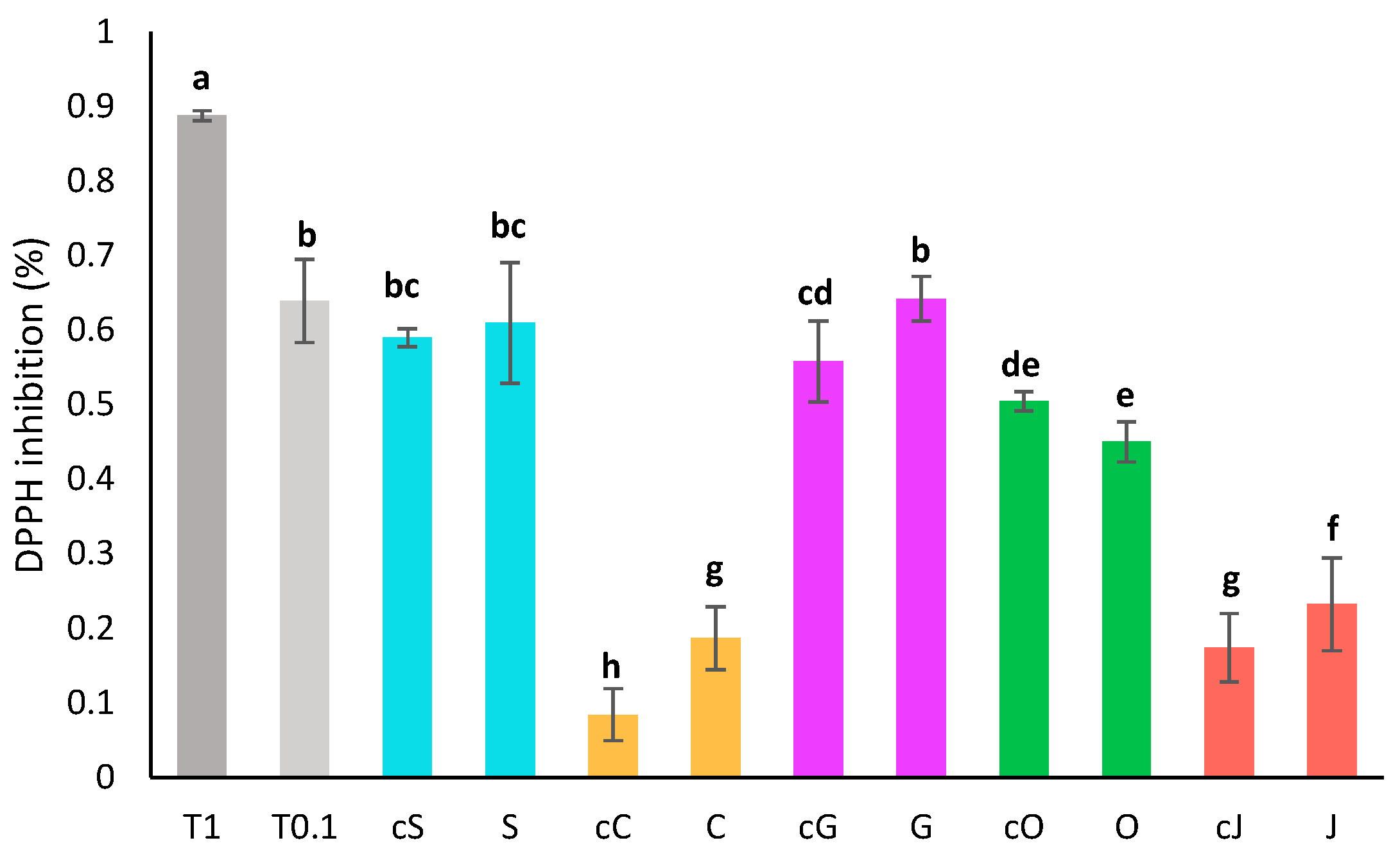
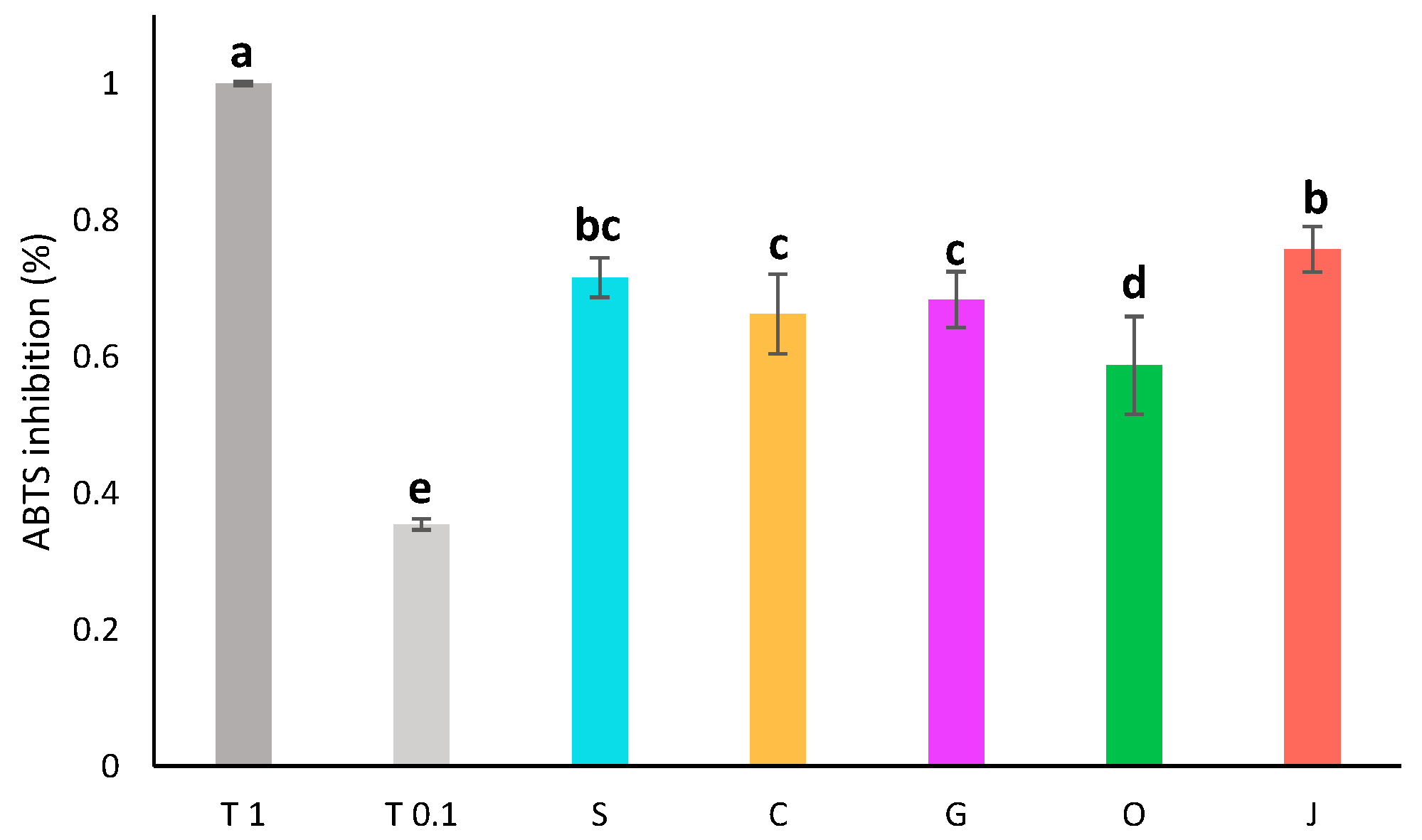
3.6. Determination of the Antioxidant Activity
3.6.1. DPPH Assay
3.6.2. ABTS Assay
3.7. Characterisation of Oil Phases
3.7.1. Liquid-Liquid Extraction of the Oil Phase
3.7.2. UHPLC-DAD-HRMS/MS Analysis
3.8. Characterisation and Comparison of Aqueous Phases
3.8.1. UHPLC-DAD-HRMS/MS Analysis
3.8.2. Statistical Analysis
3.9. Characterisation of the Volatil Compounds Using GC-MS Analysis
3.10. Formulation of the Optimised Extract
3.10.1. Base Formulation
3.10.2. Development of an Integrated Extraction-Cosmetic Formulation Process
3.10.3. Physicochemical Characterisation of the Emulsions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hirko, B.; Getu, A. Bixa orellana (Annatto Bixa): A Review on Use, Structure, Extraction Methods and Analysis. J. Agron. Technol. Eng. Manag. 2022, 5, 687–696. [Google Scholar]
- Raddatz-Mota, D.; Pérez-Flores, L.J.; Carrari, F.; Mendoza-Espinoza, J.A.; de León-Sánchez, F.D.; Pinzón-López, L.L.; Godoy-Hernández, G.; Rivera-Cabrera, F. Achiote (Bixa orellana L.): A Natural Source of Pigment and Vitamin E. J. Food Sci. Technol. 2017, 54, 1729–1741. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Madrid, R.; Aguilar-Espinosa, M.; Cárdenas-Conejo, Y.; Garza-Caligaris, L.E. Carotenoid Derivates in Achiote (Bixa orellana) Seeds: Synthesis and Health Promoting Properties. Front. Plant Sci. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, A.; Ijaz, M.U.; Muzammil, S.; Nazir, M.M.; Zafar, S.; Zihad, S.M.N.K.; Uddin, S.J.; Hasnain, M.S.; Nayak, A.K. The Role of Bixin as Antioxidant, Anti-Inflammatory, Anticancer, and Skin Protecting Natural Product Extracted from Bixa orellana L. Fitoterapia 2023, 169, 105612. [Google Scholar] [CrossRef] [PubMed]
- Pillai, S.; Soni, S.; Dhulap, S. Pharmacological and Cosmeceutical Applications of Bixa orellana L.: A Review of the Scientific and Patent Literature. Indian J. Nat. Prod. Resour. 2018, 9, 281–289. [Google Scholar]
- Cardarelli, C.R.; Benassi, M.d.T.; Mercadante, A.Z. Characterization of Different Annatto Extracts Based on Antioxidant and Colour Properties. LWT—Food Sci. Technol. 2008, 41, 1689–1693. [Google Scholar] [CrossRef]
- De Oliveira Júnior, R.G.; Bonnet, A.; Braconnier, E.; Groult, H.; Prunier, G.; Beaugeard, L.; Grougnet, R.; Da Silva Almeida, J.R.G.; Ferraz, C.A.A.; Picot, L. Bixin, an Apocarotenoid Isolated from Bixa orellana L., Sensitizes Human Melanoma Cells to Dacarbazine-Induced Apoptosis through ROS-Mediated Cytotoxicity. Food Chem. Toxicol. 2019, 125, 549–561. [Google Scholar] [CrossRef]
- Quintero Quiroz, J.; Naranjo Duran, A.M.; Silva Garcia, M.; Ciro Gomez, G.L.; Rojas Camargo, J.J. Ultrasound-Assisted Extraction of Bioactive Compounds from Annatto Seeds, Evaluation of Their Antimicrobial and Antioxidant Activity, and Identification of Main Compounds by LC/ESI-MS Analysis. Int. J. Food Sci. 2019, 2019, 1–9. [Google Scholar] [CrossRef]
- Vilar, D.d.A.; Vilar, M.S.d.A.; e Moura, T.F.A.d.L.; Raffin, F.N.; de Oliveira, M.R.; Franco, C.F.d.O.; de Athayde-Filho, P.F.; Diniz, M.d.F.F.M.; Barbosa-Filho, J.M. Traditional Uses, Chemical Constituents, and Biological Activities of Bixa orellana L.: A Review. Sci. World J. 2014, 2014, 1–11. [Google Scholar] [CrossRef]
- Ghazali, N.I.; Mohd Rais, R.Z.; Makpol, S.; Chin, K.Y.; Yap, W.N.; Goon, J.A. Effects of Tocotrienol on Aging Skin: A Systematic Review. Front. Pharmacol. 2022, 13, 1006198. [Google Scholar] [CrossRef]
- Guzmán, E.; Lucia, A. Essential Oils and Their Individual Components in Cosmetic Products. Cosmetics 2021, 8, 114. [Google Scholar] [CrossRef]
- Osorio-Tobón, J.F. Recent Advances and Comparisons of Conventional and Alternative Extraction Techniques of Phenolic Compounds. J. Food Sci. Technol. 2020, 57, 4299–4315. [Google Scholar] [CrossRef] [PubMed]
- Saini, R.K.; Keum, Y.-S. Tocopherols and Tocotrienols in Plants and Their Products: A Review on Methods of Extraction, Chromatographic Separation, and Detection. Food Res. Int. 2016, 82, 59–70. [Google Scholar] [CrossRef]
- Chemat, F.; Vian, M.A.; Cravotto, G. Green Extraction of Natural Products: Concept and Principles. Int. J. Mol. Sci. 2012, 13, 8615–8627. [Google Scholar] [CrossRef]
- Anastas, P.T.; Warner, J.C. Green Chemistry: Theory and Practice; Oxford University Press: Oxford, UK, 2000; ISBN 978-0-19-770405-9. [Google Scholar]
- Chemat, F.; Abert Vian, M.A.; Ravi, H.K.; Khadhraoui, B.; Hilali, S.; Perino, S.; Fabiano Tixier, A.-S. Review of Alternative Solvents for Green Extraction of Food and Natural Products: Panorama, Principles, Applications and Prospects. Molecules 2019, 24, 3007. [Google Scholar] [CrossRef] [PubMed]
- Yara-Varón, E.; Li, Y.; Balcells, M.; Canela-Garayoa, R.; Fabiano-Tixier, A.-S.; Chemat, F. Vegetable Oils as Alternative Solvents for Green Oleo-Extraction, Purification and Formulation of Food and Natural Products. Molecules 2017, 22, 1474. [Google Scholar] [CrossRef] [PubMed]
- Kanjilal, P.B.; Singh, R.S. Agronomic Evaluation of Annatto (Bixa orellana L.). J. Herbs Spices Med. Plants 1996, 3, 13–17. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, J.; Xing, L.; Zhang, W. Applications and Effects of Ultrasound Assisted Emulsification in the Production of Food Emulsions: A Review. Trends Food Sci. Technol. 2021, 110, 493–512. [Google Scholar] [CrossRef]
- Das, P.; Nayak, P.K.; Kesavan, R.K. Ultrasound Assisted Extraction of Food Colorants: Principle, Mechanism, Extraction Technique and Applications: A Review on Recent Progress. Food Chem. Adv. 2022, 1, 100144. [Google Scholar] [CrossRef]
- Chemat, F.; Abert-Vian, M.; Fabiano-Tixier, A.S.; Strube, J.; Uhlenbrock, L.; Gunjevic, V.; Cravotto, G. Green Extraction of Natural Products. Origins, Current Status, and Future Challenges. TrAC Trends Anal. Chem. 2019, 118, 248–263. [Google Scholar] [CrossRef]
- Li, Y.; Fabiano-Tixier, A.S.; Ruiz, K.; Rossignol Castera, A.; Bauduin, P.; Diat, O.; Chemat, F. Comprehension of Direct Extraction of Hydrophilic Antioxidants Using Vegetable Oils by Polar Paradox Theory and Small Angle X-Ray Scattering Analysis. Food Chem. 2015, 173, 873–880. [Google Scholar] [CrossRef]
- Li, Y.; Fabiano-Tixier, A.S.; Ginies, C.; Chemat, F. Direct Green Extraction of Volatile Aroma Compounds Using Vegetable Oils as Solvents: Theoretical and Experimental Solubility Study. LWT—Food Sci. Technol. 2014, 59, 724–731. [Google Scholar] [CrossRef]
- Teramukai, K.; Kakui, S.; Beppu, F.; Hosokawa, M.; Miyashita, K. Effective Extraction of Carotenoids from Brown Seaweeds and Vegetable Leaves with Edible Oils. Innov. Food Sci. Emerg. Technol. 2020, 60, 102302. [Google Scholar] [CrossRef]
- Chemat, F.; Rombaut, N.; Sicaire, A.-G.; Meullemiestre, A.; Fabiano-Tixier, A.-S.; Abert-Vian, M. Ultrasound Assisted Extraction of Food and Natural Products. Mechanisms, Techniques, Combinations, Protocols and Applications. A Review. Ultrason. Sonochem. 2017, 34, 540–560. [Google Scholar] [CrossRef] [PubMed]
- Bharadvaja, N.; Gautam, S.; Singh, H. Natural Polyphenols: A Promising Bioactive Compounds for Skin Care and Cosmetics. Mol. Biol. Rep. 2023, 50, 1817–1828. [Google Scholar] [CrossRef] [PubMed]
- Bibi Sadeer, N.; Montesano, D.; Albrizio, S.; Zengin, G.; Mahomoodally, M.F. The Versatility of Antioxidant Assays in Food Science and Safety—Chemistry, Applications, Strengths, and Limitations. Antioxidants 2020, 9, 709. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Shang, K.; Lin, C.; Wang, C.; Shi, X.; Wang, H.; Li, H. Processing Technologies, Phytochemical Constituents, and Biological Activities of Grape Seed Oil (GSO): A Review. Trends Food Sci. Technol. 2021, 116, 1074–1083. [Google Scholar] [CrossRef]
- Gotor, A.A.; Rhazi, L. Effects of Refining Process on Sunflower Oil Minor Components: A Review. OCL 2016, 23, D207. [Google Scholar] [CrossRef]
- Kiritsakis, A.; Markakis, P. Olive Oil: A Review. In Advances in Food Research; Elsevier: Amsterdam, The Netherlands, 1988; Volume 31, pp. 453–482. ISBN 978-0-12-016431-8. [Google Scholar]
- Deen, A.; Visvanathan, R.; Wickramarachchi, D.; Marikkar, N.; Nammi, S.; Jayawardana, B.C.; Liyanage, R. Chemical Composition and Health Benefits of Coconut Oil: An Overview. J. Sci. Food Agric. 2021, 101, 2182–2193. [Google Scholar] [CrossRef]
- Gad, H.A.; Roberts, A.; Hamzi, S.H.; Gad, H.A.; Touiss, I.; Altyar, A.E.; Kensara, O.A.; Ashour, M.L. Jojoba Oil: An Updated Comprehensive Review on Chemistry, Pharmaceutical Uses, and Toxicity. Polymers 2021, 13, 1711. [Google Scholar] [CrossRef]
- Busson-Breysse, J.; Farines, M.; Soulier, J. Jojoba Wax: Its Esters and Some of Its Minor Components. J. Am. Oil Chem. Soc. 1994, 71, 999–1002. [Google Scholar] [CrossRef]
- El-Mahrab-Robert, M.; Rosilio, V.; Bolzinger, M.A.; Chaminade, P.; Grossiort, J.L. Assessment of Oil Polarity: Comparison of Evaluation Methods. Int. J. Pharm. 2008, 348, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Chutia, H.; Mahanta, C.L. Green Ultrasound and Microwave Extraction of Carotenoids from Passion Fruit Peel Using Vegetable Oils as a Solvent: Optimization, Comparison, Kinetics, and Thermodynamic Studies. Innov. Food Sci. Emerg. Technol. 2021, 67, 102547. [Google Scholar] [CrossRef]
- Nour, V.; Corbu, A.R.; Rotaru, P.; Karageorgou, I.; Lalas, S. Effect of Carotenoids, Extracted from Dry Tomato Waste, on the Stability and Characteristics of Various Vegetable Oils. Grasas Aceites 2018, 69, 238. [Google Scholar] [CrossRef]
- Corbu, A.R.; Rotaru, A.; Nour, V. Edible Vegetable Oils Enriched with Carotenoids Extracted from By-Products of Sea Buckthorn (Hippophae Rhamnoides Ssp. Sinensis): The Investigation of Some Characteristic Properties, Oxidative Stability and the Effect on Thermal Behaviour. J. Therm. Anal. Calorim. 2020, 142, 735–747. [Google Scholar] [CrossRef]
- Sabudak, T.; Ozturk, M.; Goren, A.C.; Kolak, U.; Topcu, G. Fatty Acids and Other Lipid Composition of Five Trifolium Species with Antioxidant Activity. Pharm. Biol. 2009, 47, 137–141. [Google Scholar] [CrossRef]
- Singh, A.; Bajpai, V.; Kumar, S.; Sharma, K.R.; Kumar, B. Profiling of Gallic and Ellagic Acid Derivatives in Different Plant Parts of Terminalia Arjuna by HPLC-ESI-QTOF-MS/MS. Nat. Prod. Commun. 2016, 11, 1934578X1601100. [Google Scholar] [CrossRef]
- Formato, M.; Piccolella, S.; Zidorn, C.; Pacifico, S. UHPLC-HRMS Analysis of Fagus Sylvatica (Fagaceae) Leaves: A Renewable Source of Antioxidant Polyphenols. Antioxidants 2021, 10, 1140. [Google Scholar] [CrossRef]
- Sánchez-Fernández, J.A.; Peña-Parás, L.; Vidaltamayo, R.; García-Acosta, G.E. Synthesis of Chromophores Based on Carbazole and Azobenzenes as Electron Donors and Acceptors with Nonlinear Optoelectrical and Optical Properties and Applications in Energy Storage Devices. In Proceedings-AES-ATEMA 2016: International Conference Series-MONTREAL 2016 AES-ATEMA, Proceedings of the 28th International Conference, Montréal, QC, Canada, 20–24 June 2016; Advanced Engineering Solutions: Springboro, OH, USA, 2016. [Google Scholar]
- Youn, U.-J.; Lee, J.; Nam, J.-W.; Lee, Y.; Seo, E.-K. Identification of a New Isomer of Dihydrophaseic Acid 3′-O-β-D-Glucopyranoside from Nelumbo Nucifera. Bull. Korean Chem. Soc. 2011, 32, 4083–4085. [Google Scholar] [CrossRef]
- Fang, C.; Fernie, A.R.; Luo, J. Exploring the Diversity of Plant Metabolism. Trends Plant Sci. 2019, 24, 83–98. [Google Scholar] [CrossRef]
- Guo, W.; Jiang, Y.; Chen, X.; Yu, P.; Wang, M.; Wu, X.; Zhang, D. Identification and Quantitation of Major Phenolic Compounds from Penthorum Chinense Pursh. by HPLC with Tandem Mass Spectrometry and HPLC with Diode Array Detection. J. Sep. Sci. 2015, 38, 2789–2796. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Xia, W. Analysis of Phenolic Compounds in Chinese Olive (Canarium Album L.) Fruit by RPHPLC–DAD–ESI–MS. Food Chem. 2007, 105, 1307–1311. [Google Scholar] [CrossRef]
- Rodrigues, D.B.; Veríssimo, L.; Finimundy, T.; Rodrigues, J.; Oliveira, I.; Gonçalves, J.; Fernandes, I.P.; Barros, L.; Heleno, S.A.; Calhelha, R.C. Chemical and Bioactive Screening of Green Polyphenol-Rich Extracts from Chestnut By-Products: An Approach to Guide the Sustainable Production of High-Added Value Ingredients. Foods 2023, 12, 2596. [Google Scholar] [CrossRef]
- Agrawal, O.D.; Kulkarni, Y.A. Mini-Review of Analytical Methods Used in Quantification of Ellagic Acid. Rev. Anal. Chem. 2020, 39, 31–44. [Google Scholar] [CrossRef]
- Boulekbache-Makhlouf, L.; Meudec, E.; Chibane, M.; Mazauric, J.-P.; Slimani, S.; Henry, M.; Cheynier, V.; Madani, K. Analysis by High-Performance Liquid Chromatography Diode Array Detection Mass Spectrometry of Phenolic Compounds in Fruit of Eucalyptus Globulus Cultivated in Algeria. J. Agric. Food Chem. 2010, 58, 12615–12624. [Google Scholar] [CrossRef]
- Owczarek, A.; Różalski, M.; Krajewska, U.; Olszewska, M. Rare Ellagic Acid Sulphate Derivatives from the Rhizome of Geum Rivale L.—Structure, Cytotoxicity, and Validated HPLC-PDA Assay. Appl. Sci. 2017, 7, 400. [Google Scholar] [CrossRef]
- Harborne, J.B. Flavonoid Bisulphates and Their Co-Occurrences with Ellagic Acid in the Bixaceae, Frankeniaceae and Related Families. Phytochemistry 1975, 14, 1331–1337. [Google Scholar] [CrossRef]
- Zillich, O.V.; Schweiggert-Weisz, U.; Eisner, P.; Kerscher, M. Polyphenols as Active Ingredients for Cosmetic Products. Int. J. Cosmet. Sci. 2015, 37, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Evtyugin, D.D.; Magina, S.; Evtuguin, D.V. Recent Advances in the Production and Applications of Ellagic Acid and Its Derivatives. A Review. Molecules 2020, 25, 2745. [Google Scholar] [CrossRef]
- Garcia, S.D.V.; Alves, P.E.S.; Dias, L.D.S.; Santos, L.J.D.C.; Lustosa, S.M.C.; Viana, D.D.S.F.; Viana, V.G.F. Synthesis and Determination of the Sun Protection Factor of a Cosmetic Gel-Cream Based on Norbixin. In Global Health Trends and Perspectives in Health Sciences; Seven Editora: São José dos Pinhais, Brazil, 2023. [Google Scholar]
- Franklin, V.A.; Bach Hi, E.M.; Wadt, N.S.Y.; Bach, E.E. Aqueous Extract from Urucum (Bixa orellana L.): Antimicrobial, Antioxidant, and Healing Activity. Porto Biomed. J. 2023, 8, e183. [Google Scholar] [CrossRef]
- Galindo-Cuspinera, V.; Lubran, M.B.; Rankin, S.A. Comparison of Volatile Compounds in Water- and Oil-Soluble Annatto (Bixa orellana L.) Extracts. J. Agric. Food Chem. 2002, 50, 2010–2015. [Google Scholar] [CrossRef] [PubMed]
- Pino, J.A.; Correa, M.T. Chemical Composition of the Essential Oil from Annatto (Bixa orellana L.) Seeds. J. Essent. Oil Res. 2003, 15, 66–67. [Google Scholar] [CrossRef]
- Shi, J.; Cao, C.; Xu, J.; Zhou, C. Research Advances on Biosynthesis, Regulation, and Biological Activities of Apocarotenoid Aroma in Horticultural Plants. J. Chem. 2020, 2020, 1–11. [Google Scholar] [CrossRef]
- Gros, Q.; Wolniaczyk, M.; Duval, J.; West, C.; Horie, S.; Toyota, Y.; Funada, Y.; Lesellier, E. Comparison of the Triglyceride Composition of Vegetable Samples with Ultra-High Efficiency / Low-Pressure Supercritical Fluid Chromatography—Mass Spectrometry. J. Food Compos. Anal. 2023, 115, 104960. [Google Scholar] [CrossRef]
- Lesellier, E.; Latos, A.; De Oliveira, A.L. Ultra High Efficiency/Low Pressure Supercritical Fluid Chromatography with Superficially Porous Particles for Triglyceride Separation. J. Chromatogr. A 2014, 1327, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Food & Agriculture Organization. Compendium of Food Additive Specifications: Joint FAO/WHO Expert Committee on Food Additives: 67th Meeting 2006; Food & Agriculture Organization: Rome, Italy, 2006; ISBN 978-92-5-105559-5. [Google Scholar]
- Albuquerque, C.L.C.; Meireles, M.A.A. Defatting of Annatto Seeds Using Supercritical Carbon Dioxide as a Pretreatment for the Production of Bixin: Experimental, Modeling and Economic Evaluation of the Process. J. Supercrit. Fluids 2012, 66, 86–95. [Google Scholar] [CrossRef]
- Grigoras, C.G.; Destandau, E.; Fougère, L.; Elfakir, C. Evaluation of Apple Pomace Extracts as a Source of Bioactive Compounds. Ind. Crops Prod. 2013, 49, 794–804. [Google Scholar] [CrossRef]
- Lee, S.K.; Mbwambo, Z.H.; Chung, H.S.; Luyengi, L.; Gamez, E.J.C.; Mehta, R.G.; Kinghorn, A.D.; Pezzuto, J.M. Evaluation of the Antioxidant Potential of Natural Products. Comb. Chem. High Throughput Screen. 1998, 1, 35–46. [Google Scholar] [CrossRef]
- Tagliazucchi, D.; Verzelloni, E.; Bertolini, D.; Conte, A. In Vitro Bio-Accessibility and Antioxidant Activity of Grape Polyphenols. Food Chem. 2010, 120, 599–606. [Google Scholar] [CrossRef]
- van Den Dool, H.; Kratz, P.D. A Generalization of the Retention Index System Including Linear Temperature Programmed Gas—Liquid Partition Chromatography. J. Chromatogr. A 1963, 11, 463–471. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sunflower | Coconut | Grapeseed | Olive | Jojoba | |
|---|---|---|---|---|---|
| Esters | 0.0% | 0.0% | 0.0% | 0.0% | 99.2% |
| Monoglycerides | 0.3% | 0.4% | 0.4% | 0.4% | 0.8% |
| Diglycerides | 1.1% | 1.5% | 4.2% | 1.8% | 0.0% |
| Triglycerides | 98.6% | 98.4% | 95.3% | 97.9% | 0.0% |
| Triglycerides composition | |||||
| Only SFAs | 0.0% | 83.3% | 0.0% | 0.0% | - |
| SFAs and MUFAs | 15.5% | 13.2% | 7.3% | 70.7% | - |
| ≥1 PUFAs | 84.5% | 3.0% | 92.7% | 29.3% | - |
| Sunflower | Coconut | Grapeseed | Olive | Jojoba | |
|---|---|---|---|---|---|
| Density | 0.92 | 0.91 | 0.93 | 0.91 | 0.87 |
| Viscosity (mPa.s) | 50.2 | 40.8 | 43.9 | 54.5 | 32.8 |
| Interfacial tension (mN/m) | 18.6 ± 0.1 | 12.5 ± 0.1 | 22.2 ± 0.2 | 17.6 ± 0.1 | 24.6 ± 0.3 |
| # | tR (min) | Meas. m/z ([M+H]+) | Error (ppm) | Meas. m/z ([M-H]−) | Error (ppm) | Molecular Formula | MS/MS+ (Intensity) | MS/MS− (Intensity) | λmax (nm) | Putative Identification |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2.23 | 171.0291 | −1.7 | 169.0138 | 2.7 | C7H6O5 | 153.0182 (77309) | 270 | Gallic acid | |
| 135.0078 (7878) | ||||||||||
| 127.0388 (50839) | ||||||||||
| 125.0227 (38830) | ||||||||||
| 109.0281 (35417) | ||||||||||
| 107.0099 (10599) | ||||||||||
| 2 | 4.16 | 155.0338 | 0.7 | 153.0185 | 3 | C7H6O4 | 137.0237 (29126) | 258, 292 | Protocatechuic acid | |
| 111.0164 17475) | ||||||||||
| 93.0334 (3482) | ||||||||||
| 3 | 5.75 | 445.2069 | −0.2 | 443.1921 | 0.4 | C21H32O10 | 265.1438 (18095) | 281.1369 (1045) | 266 | dihydroxyphaseic acid glucopyranoside |
| 247.1308 (3248) | 263.1292 (500) | |||||||||
| 237.1489 (1975) | ||||||||||
| 219.1361 (2263) | ||||||||||
| 201.1274 (504) | ||||||||||
| 189.1251 (1281) | ||||||||||
| 179.0558 (729) | ||||||||||
| 161.0446 (1535) | ||||||||||
| 119.0352 (1532) | ||||||||||
| 113.0244 (2084) | ||||||||||
| 4 | 6.22 | 293.0302 | −3.6 | 291.0133 | 4.6 | C13H8O8 | 275.0184 (9895) | 247.0238 (97693) | 276, 357 | Brevifolincarboxylic acid |
| 265.0333 (7850) | 219.0280 (16915) | |||||||||
| 247.0236 (44737) | 191.0339 (47419) | |||||||||
| 219.0289 (112482) | 173.0224 (11496) | |||||||||
| 205.0133 (16005) | 163.0394 (6121) | |||||||||
| 191.0339 (35272) | 147.0455 (7879) | |||||||||
| 177.0184 (21638) | 145.0291 (10327) | |||||||||
| 163.0390 (45035) | ||||||||||
| 5 | 6.72 | 635.0853 | 4.1 | 633.0729 | 0.7 | C27H22O18 | 465.0663 (169038) | 463.0499 (110807) | 270 | Galloyl-HHDP-glucose |
| 321.024 (65207) | 419.0604 (16835) | |||||||||
| 303.0082 (42157) | 300.9978 (139141) | |||||||||
| 277.0345 (120888) | 275.0182 (46930) | |||||||||
| 6 | 7.73 | 383.2067 | −0.6 | 381.1901 | 4.7 | C20H30O7 | 365.1954 (8265) | 337.2004 (14030) | 285 | Unidentified |
| 347.1864 (2982) | ||||||||||
| 283.1672 (2909) | ||||||||||
| 165.0916 (2074) | ||||||||||
| 7 | 8.44 | 303.0142 | −2.3 | 300.9990 | 4.2 | C14H6O8 | 285.0039 (10599) | 283.9928 (16311) | 252, 295 (sh), 359 | Ellagic acid |
| 275.0208 (15689) | 273.0033 (2226) | |||||||||
| 257.0081 (25410) | 257.0066 (7373) | |||||||||
| 247.0256 (9045) | 245.0088 (8087) | |||||||||
| 229.0133 (12687) | 229.0135 (14017) | |||||||||
| 201.0183 (14626) | 217.0133 (3027) | |||||||||
| 173.0234 (8093) | 201.0159 (8841) | |||||||||
| 145.0282 (3723) | 185.0240 (7621) | |||||||||
| 173.0261 (5121) | ||||||||||
| 157.0288 (3054) | ||||||||||
| 145.0293 (2019) | ||||||||||
| 129.0343 (2037) | ||||||||||
| 8 | 9.19 | 396.9862 | −0.4 | 394.9706 | -2.0 | C15H8O11S | 317.0292 (24927) | 315.0105 (187933) | 248, 359 | O-methylellagic acid sulfate |
| 302.0057 (7488) | 299.9902 (190471) | |||||||||
| 257.0080 (2220) | ||||||||||
| 9 | 9.33 | 396.9860 | −1.0 | 394.9701 | 3.4 | C15H8O11S | 317.0289 (22803) | 315.0105 (120225) | 248, 359 | O-methylellagic acid sulfate |
| 284.9989 (1076) | 299.9902 (138397) | |||||||||
| 257.0063 (4280) | ||||||||||
| 222.1122 (2321) | ||||||||||
| 10 | 9.63 | 463.0878 | −1.4 | 461.0706 | 4.3 | C21H18O12 | 317.0294 (150666) | 315.0132 (95391) | 247, 364 | methylellagic acid deoxyhexose |
| 147.0652 (5958) | 299.9895 (68927) | |||||||||
| 129.0545 (20771) | ||||||||||
| 11 | 9.78 | 317.0297 | −1.6 | 315.0146 | 4.1 | C15H8O8 | 302.0055 (11216) | 299.9893 (2786) | 236, 254, 364 | O-methylellagic acid |
| 246.0156 (2008) | ||||||||||
| 12 | 10.01 | 317.0299 | −2.0 | 315.0133 | 4.2 | C15H8O8 | 302.0051 (4169) | 299.9903 (124255) | 235, 250, 366 | O-methylellagic acid |
| 285.0031 (7753) | ||||||||||
| 257.0087 (9174) | ||||||||||
| 246.0156 (1446) | ||||||||||
| 218.0217 (1318) | ||||||||||
| 201.0165 (2570) | ||||||||||
| 13 | 16.51 | 381.2058 | 0.7 | - | C24H28O4 | 363.1950 (1923) | 429, 455, 485 | Norbixin | ||
| 145.1008 (10344) |
| Compound | Match a | Ref. RI a | Exp.RI | Signal Intensity b |
|---|---|---|---|---|
| Heterocycles | ||||
| 1,3-bis(1,1-dimethylethyl)-benzene * | 859 | 1249 | 1246 | + |
| Alkanes | ||||
| 4-methyl-octane * | 863 | 861 | 864 | + |
| 2-methyl-nonane * | 746 | 964 | 967 | + |
| Decane * | 894 | 1000 | 1002 | + |
| 2,5-Dimethylnonane * | 759 | 1021 | 1020 | tr |
| 2,6-dimethyl-nonane * | 724 | 1018 | 1023 | + |
| Undecane * | 776 | 1100 | 110 | + |
| Dodecane * | 892 | 1200 | 1200 | ++ |
| 2,6-dimethyl-undecane * | 742 | 1210 | 1213 | + |
| 2,6,11-trimethyldodecane * | 770 | 1275 | 1274 | + |
| 4,6-dimethyldodecane * | 773 | 1325 | 1321 | + |
| Tetradecane * | 827 | 1400 | 1400 | + |
| Monoterpenes | ||||
| α-pinene | 889 | 937 | 933 | ++ |
| Sabinene | 845 | 974 | 975 | + |
| Verbenone | 707 | 1204 | 1204 | + |
| Sobrerol | 775 | 1388 | 1378 | + |
| Geranyl-acetone | 771 | 1453 | 1446 | + |
| Sesquiterpenes | ||||
| Ishwarane | 808 | 1458 | 1463 | + |
| Spathulenol | 841 | 1576 | 1571 | + |
| Isospathulenol | 907 | 1638 | 1623 | +++ |
| Neointermedeol | 754 | 1660 | 1653 | + |
| Phase | INCI Name | Amount (%) | Function |
|---|---|---|---|
| Phase A (aqueous phase) | Aqua | q.s. 100.0 | Solvent |
| Sorbic acid | 0.5 | Preservative | |
| Phase B | Glycerine | 3.0 | Emollient |
| Xanthan gum | 1.0 | Emulsifier | |
| Phase C (oil phase) | Helianthus annuus seed oil | 8.5 | Solvent |
| Glyceryl stearate | 4.0 | Emulsifier | |
| Cetearyl Wheat Straw Glycosides and Cetearyl alcohol | 2.0 | Emulsifier | |
| Cetearyl alcohol | 2.0 | Emulsifier | |
| Butyrospermum parkii butter | 1.0 | Viscosity modifier |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chambaud, M.; Fournier, A.; De Saint Jores, C.; Caux, B.; Colas, C.; Destandau, E. Oil/Water Biphasic Solvent System for the Eco-Extraction and Cosmetic Formulation of Bixa orellana L. Plants 2024, 13, 1940. https://doi.org/10.3390/plants13141940
Chambaud M, Fournier A, De Saint Jores C, Caux B, Colas C, Destandau E. Oil/Water Biphasic Solvent System for the Eco-Extraction and Cosmetic Formulation of Bixa orellana L. Plants. 2024; 13(14):1940. https://doi.org/10.3390/plants13141940
Chicago/Turabian StyleChambaud, Marine, Ariane Fournier, Clément De Saint Jores, Benjamin Caux, Cyril Colas, and Emilie Destandau. 2024. "Oil/Water Biphasic Solvent System for the Eco-Extraction and Cosmetic Formulation of Bixa orellana L." Plants 13, no. 14: 1940. https://doi.org/10.3390/plants13141940