
Integrative Transcriptomic and Metabolic Analyses Reveal That Flavonoid Biosynthesis Is the Key Pathway Regulating Pigment Deposition in Naturally Brown Cotton Fibers
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
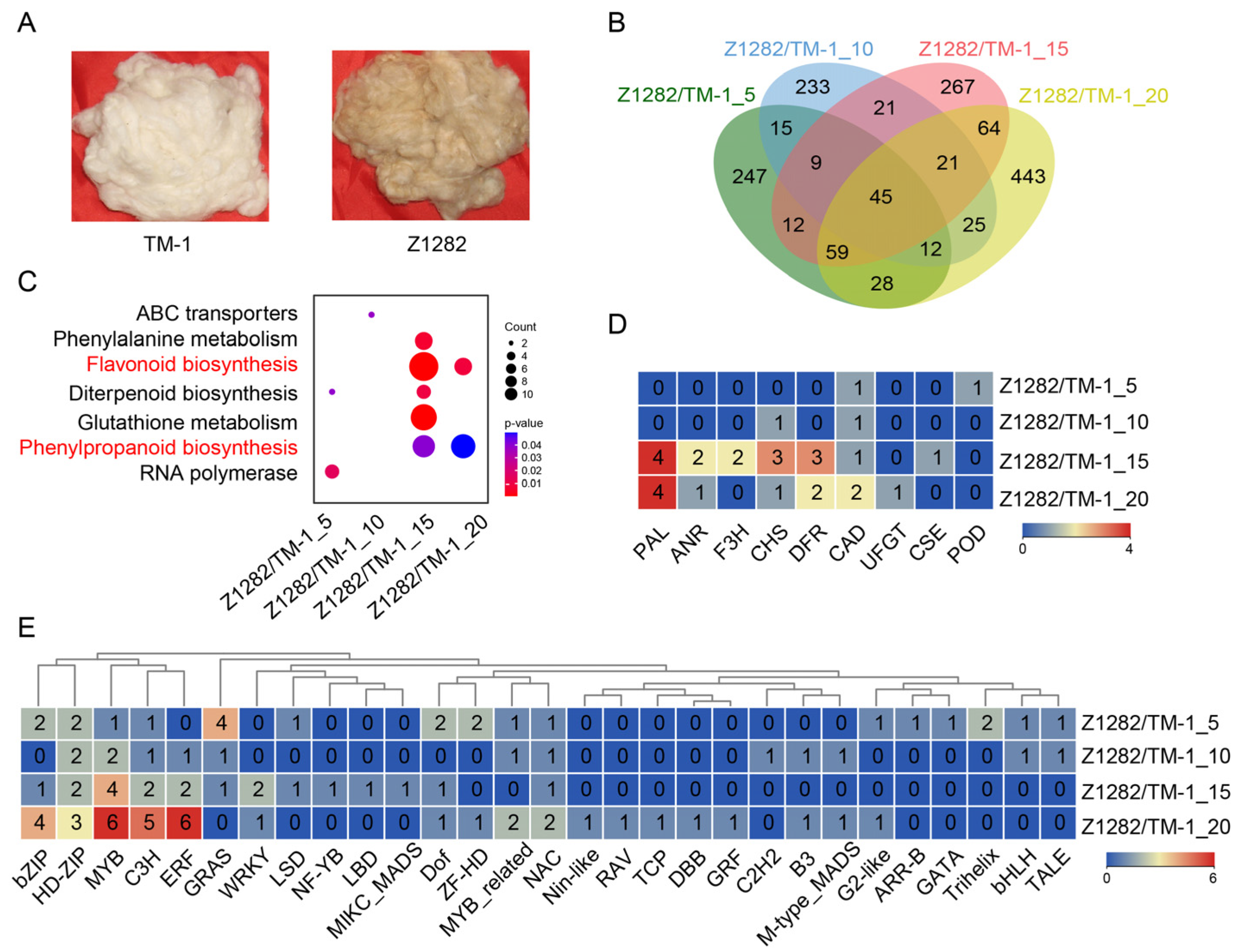
2.1. Comparative Transcriptomic Analysis between White and Brown Fibers
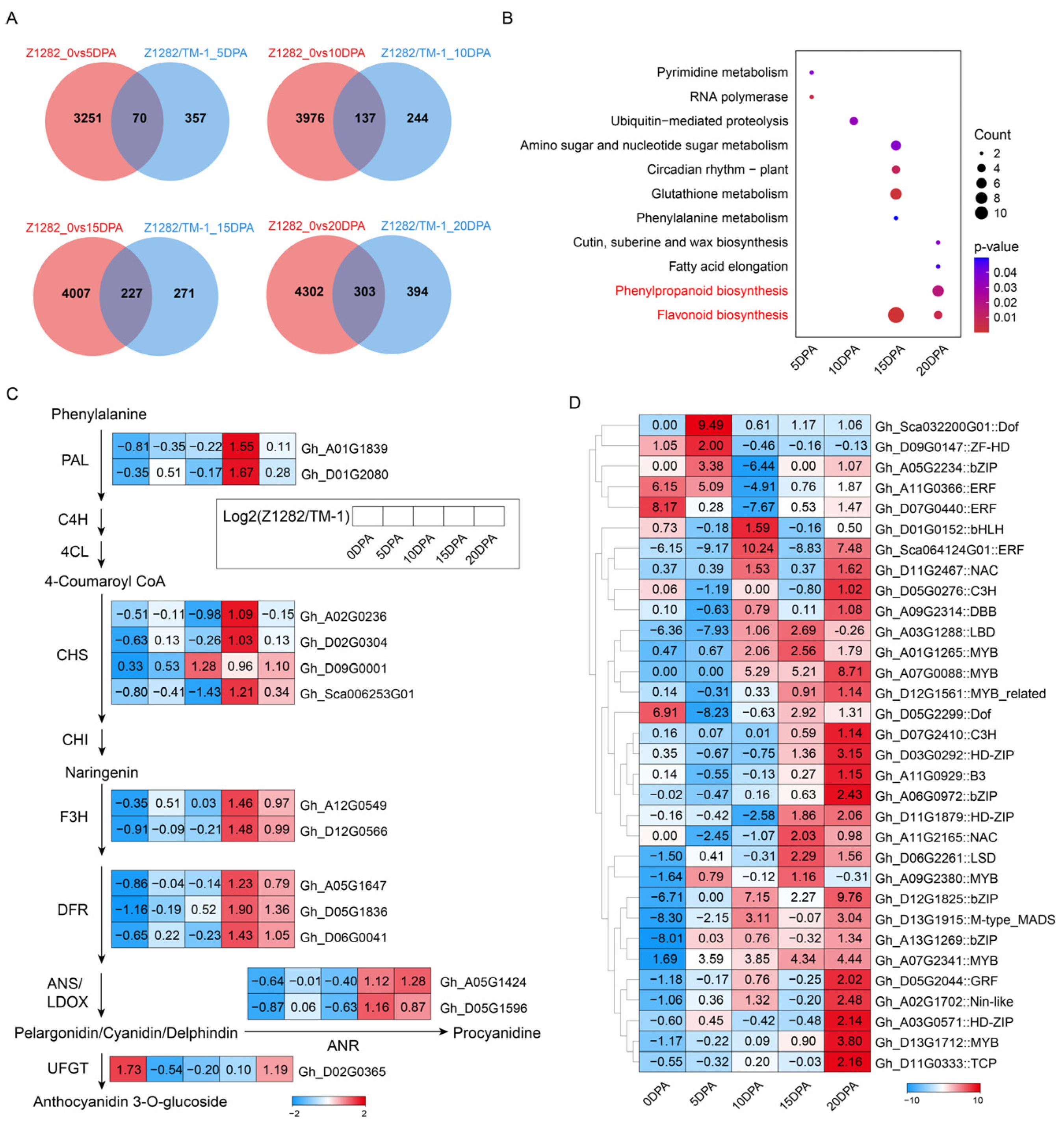
2.2. Transcriptomic Analysis of Brown Fibers at Different Development Periods
2.3. Comprehensive Transcriptomic Analysis of Brown/White Fibers and Brown Fiber Development
2.4. Comparative Metabolomic Analysis between White and Brown Fibers
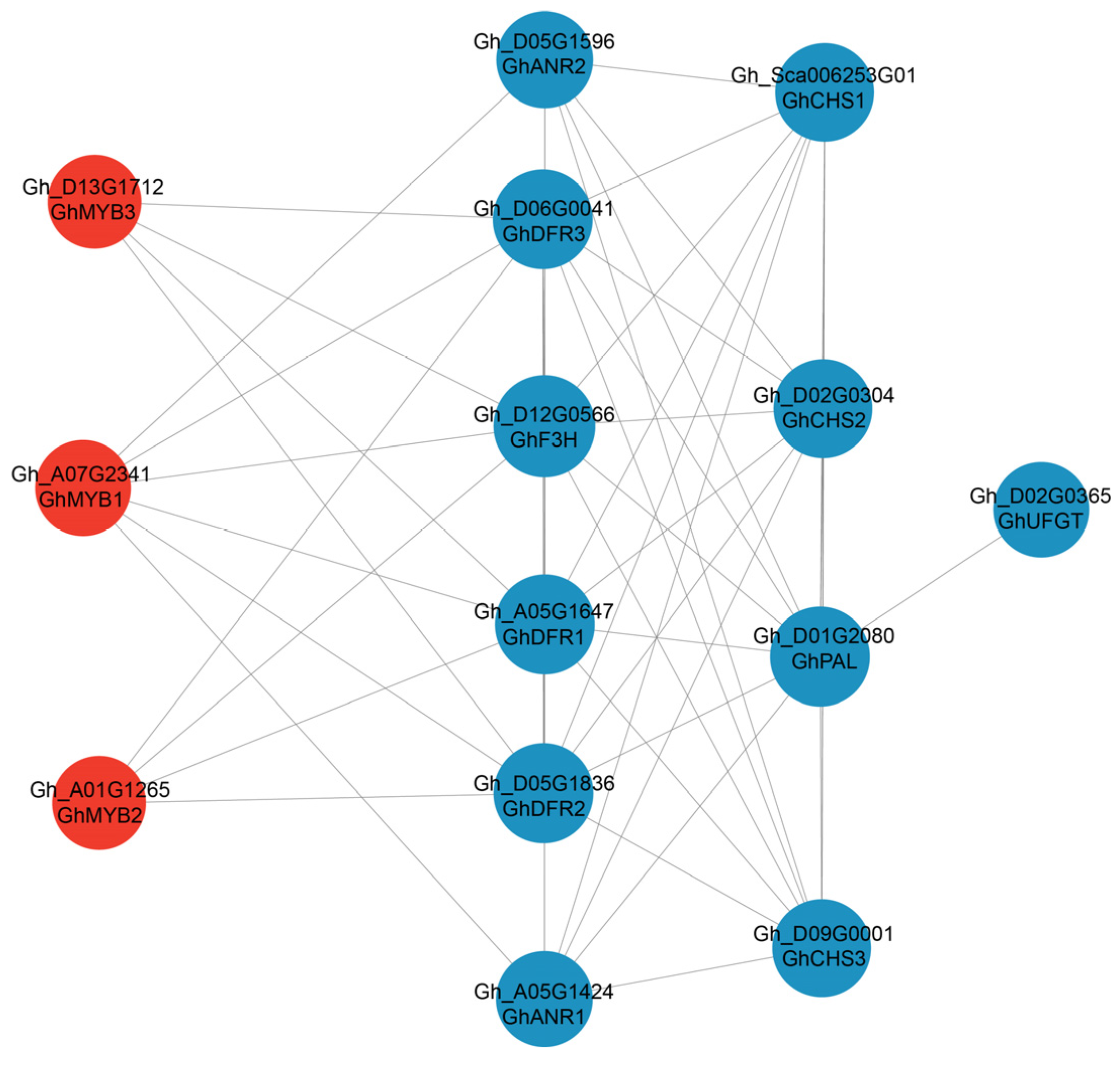
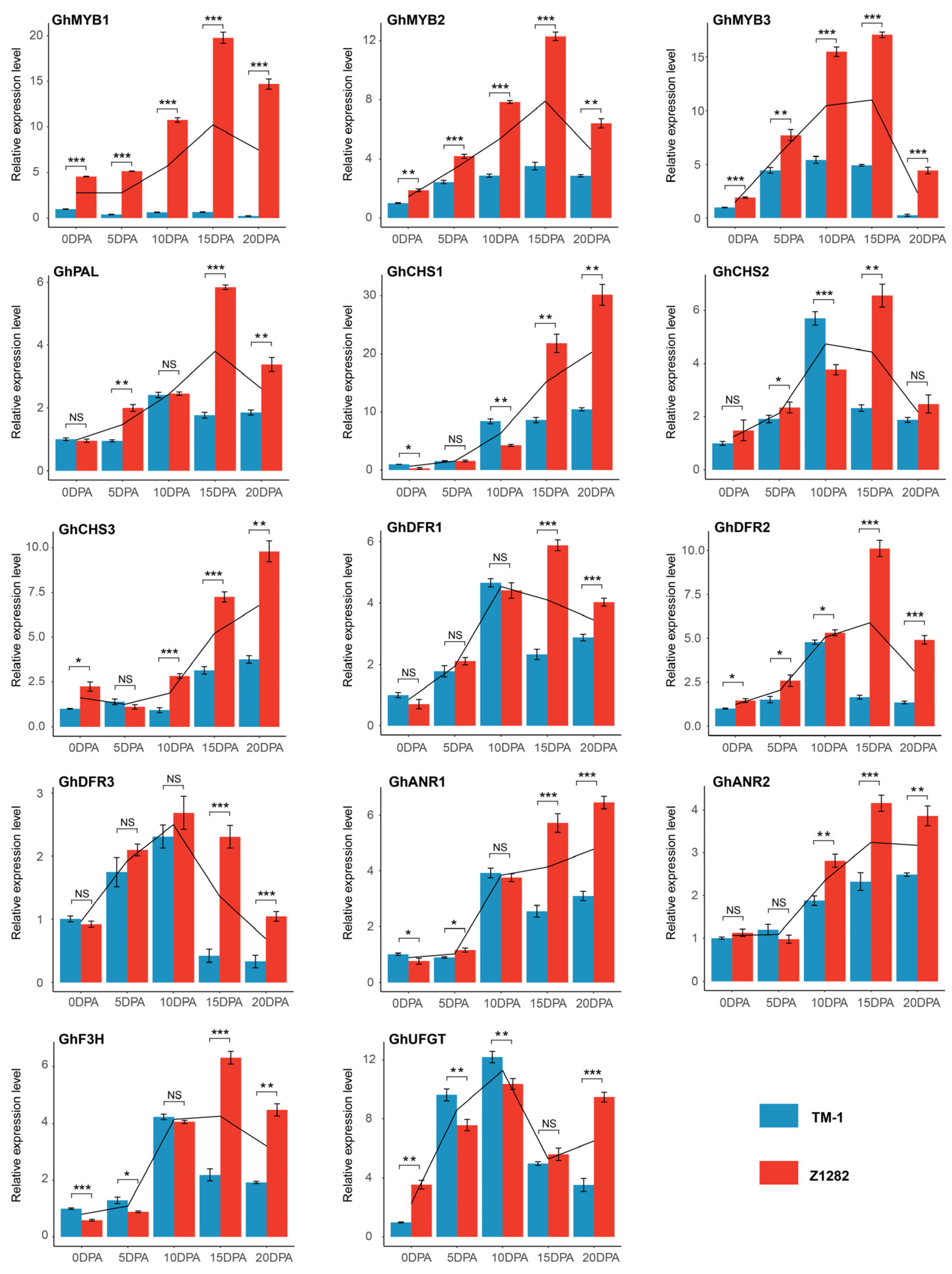
2.5. Conjoint Analysis of Transcriptomes and Metabolomes of Brown and White Fibers
3. Discussion
3.1. Flavonoid Biosynthesis and Phenylpropanoid Biosynthesis Pathways Are Enriched during the Development of BCFs
3.2. Flavonoid Biosynthesis Pathway Genes and MYBs Are the Main Regulatory Genes during the Development of BCFs
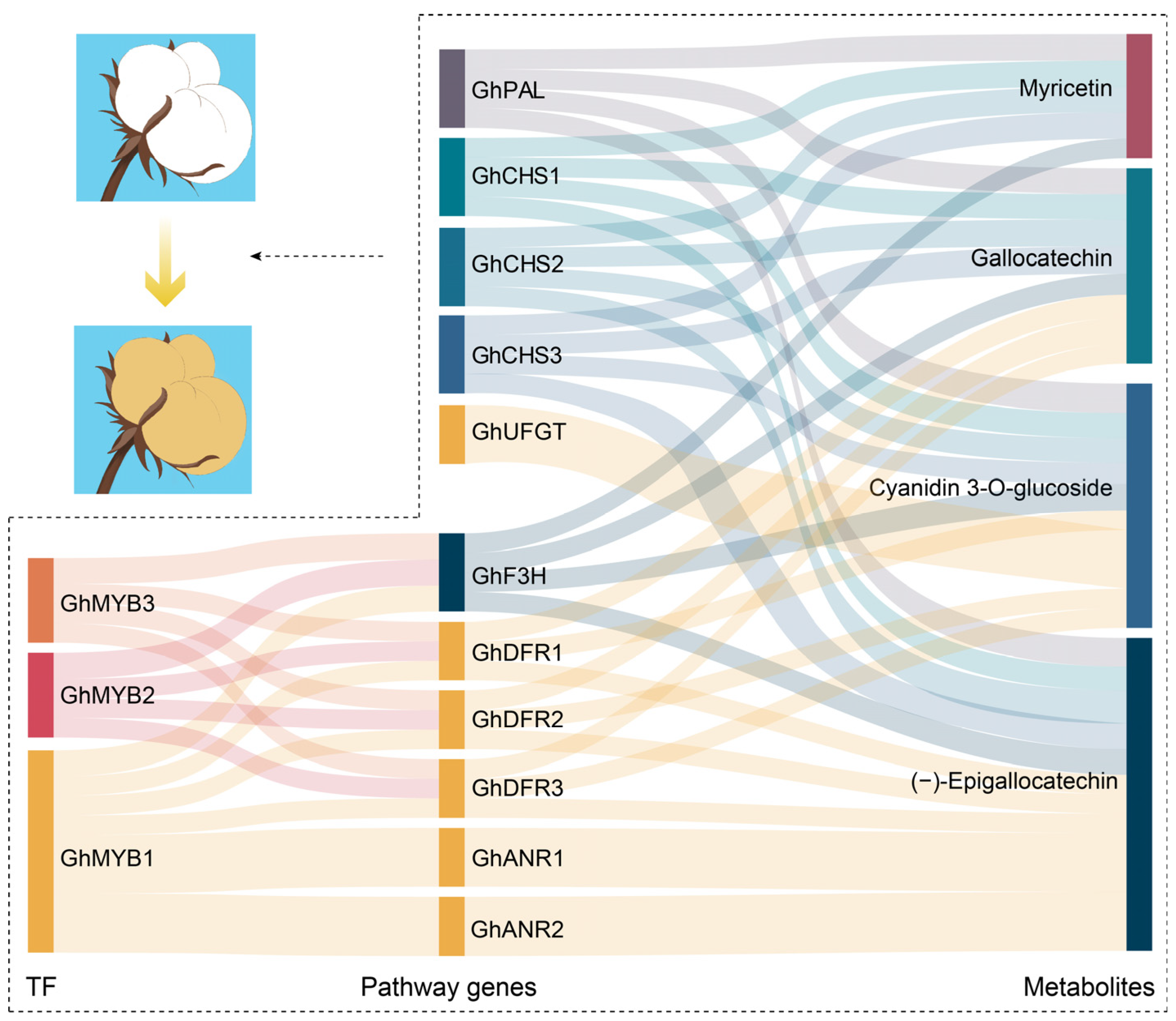
3.3. Regulatory Network of Flavonoid Pathway Genes and Metabolites Related to Pigment Synthesis and Deposition
4. Materials and Methods
4.1. Plant Materials
4.2. RNA Extraction, cDNA Library Construction, and Sequencing
4.3. RNA Sequencing Data Analysis
4.4. TF Prediction
4.5. KEGG Enrichment and PPI Analysis of DEGs
4.6. Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR) Analysis
4.7. Metabolomic Analysis of Cotton Fibers
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Liu, H.F.; Luo, C.; Song, W.; Shen, H.; Li, G.; He, Z.G.; Chen, W.G.; Cao, Y.Y.; Huang, F.; Tang, S.W.; et al. Flavonoid biosynthesis controls fiber color in naturally colored cotton. PeerJ 2018, 6, e4537. [Google Scholar] [CrossRef] [PubMed]
- Hovav, R.; Udall, J.A.; Chaudhary, B.; Hovav, E.; Flagel, L.; Hu, G.; Wendel, J.F. The evolution of spinnable cotton fiber entailed prolonged development and a novel metabolism. PLoS Genet. 2008, 4, e25. [Google Scholar] [CrossRef]
- Li, Y.J.; Zhang, X.Y.; Wang, F.X.; Yang, C.L.; Liu, F.; Xia, G.X.; Sun, J. A comparative proteomic analysis provides insights into pigment biosynthesis in brown color fiber. J. Proteomics 2013, 78, 374–388. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Xiong, X.P.; Zhu, Q.; Li, Y.J.; Sun, J. Transcriptome Sequencing and Metabolome Analysis Reveal Genes Involved in Pigmentation of Green-Colored Cotton Fibers. Int. J. Mol. Sci. 2019, 20, 4838. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Hua, S.; Malik, W.; Bibi, N.; Wang, X. Physiological and biochemical dissection of fiber development in colored cotton. Euphytica 2012, 187, 215–226. [Google Scholar] [CrossRef]
- Vreeland, J.; James, M. The revival of colored cotton. Sci. Am. 1999, 4, 112–118. [Google Scholar] [CrossRef]
- Dutt, Y.; Wang, X.D.; Zhu, Y.G.; Li, Y.Y. Breeding for high yield and fibre quality in coloured cotton. Plant Breed. 2004, 123, 145–151. [Google Scholar] [CrossRef]
- Hua, S.; Wang, X.; Yuan, S.; Shao, M.; Zhao, X.; Zhu, S.; Jiang, L. Characterization of Pigmentation and Cellulose Synthesis in Colored Cotton Fibers. Crop Sci. 2007, 47, 1540–1546. [Google Scholar] [CrossRef]
- Khatri, A.; Peerzada, M.H.; Mohsin, M.; White, M. A review on developments in dyeing cotton fabrics with reactive dyes for reducing effluent pollution. J. Clean. Prod. 2015, 87, 50–57. [Google Scholar] [CrossRef]
- Ma, M.; Hussain, M.; Memon, H.; Zhou, W. Structure of pigment compositions and radical scavenging activity of naturally green-colored cotton fiber. Cellulose 2015, 23, 955–963. [Google Scholar] [CrossRef]
- Kohel, R.J. Genetic Analysis of Fiber Color Variants in Cotton. Crop Sci. 1985, 5, 793–797. [Google Scholar] [CrossRef]
- Murthy, M. The story of coloured cotton. Resonance 2001, 12, 29–35. [Google Scholar] [CrossRef]
- Feng, H.; Li, Y.; Wang, S.; Zhang, L.; Liu, Y.; Xue, F.; Sun, Y.; Wang, Y.; Sun, J. Molecular analysis of proanthocyanidins related to pigmentation in brown cotton fibre (Gossypium hirsutum L.). J. Exp. Bot 2014, 65, 5759–5769. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, X.; He, S.; Rehman, A.; Jia, Y.; Li, H.; Pan, Z.; Geng, X.; Gao, Q.; Wang, L.; et al. Transcriptome Co-expression Network and Metabolome Analysis Identifies Key Genes and Regulators of Proanthocyanidins Biosynthesis in Brown Cotton. Front. Plant Sci. 2022, 12, 822198. [Google Scholar] [CrossRef]
- Wen, T.; Wu, M.; Shen, C.; Gao, B.; Zhu, D.; Zhang, X.; You, C.; Lin, Z. Linkage and association mapping reveals the genetic basis of brown fibre (Gossypium hirsutum). Plant Biotechnol. J. 2018, 16, 1654–1666. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Wang, Y.; Li, Q.; Zhang, Z.; Ding, H.; Zhang, Y.; Liu, H.; Luo, M.; Liu, D.; Song, W.; et al. Up-regulation of GhTT2-3A in cotton fibres during secondary wall thickening results in brown fibres with improved quality. Plant Biotechnol. J. 2018, 16, 1735–1747. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, H.; Li, X.; Xiao, X.; Ai, X.; Luo, C.; Zhu, L.; Li, X. Genetic mapping of fiber color genes on two brown cotton cultivars in Xinjiang. SpringerPlus 2014, 3, 480. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Vimolmangkang, S.; Soria-Guerra, R.E.; Rosales-Mendoza, S.; Zheng, D.; Lygin, A.V.; Korban, S.S. Ectopic Expression of Apple F3′H Genes Contributes to Anthocyanin Accumulation in the Arabidopsis tt7 Mutant Grown Under Nitrogen Stress. Plant Physiol. 2010, 153, 806–820. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Tian, X.; Liu, Y.; Li, Y.; Zhang, X.; Jones, B.J.; Sun, Y.; Sun, J. Analysis of flavonoids and the flavonoid structural genes in brown fiber of upland cotton. PLoS ONE 2013, 8, e58820. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.; He, S.; Tian, J.; Sun, J.; Pan, Z.; Jia, Y.; Sun, G.; Du, X. Comparison of the transcriptome between two cotton lines of different fiber color and quality. PLoS ONE 2014, 9, e112966. [Google Scholar] [CrossRef]
- Hinchliffe, D.J.; Condon, B.D.; Thyssen, G.; Naoumkina, M.; Madison, C.A.; Reynolds, M.; Delhom, C.D.; Fang, D.D.; Li, P.; McCarty, J. The GhTT2_A07 gene is linked to the brown colour and natural flame retardancy phenotypes of Lc1 cotton (Gossypium hirsutum L.) fibres. J. Exp. Bot. 2016, 67, 5461–5471. [Google Scholar] [CrossRef]
- Peng, Z.; Gao, Q.; Luo, C.; Gong, W.; Tang, S.; Zhang, X.; Song, W.; Wang, Z.; Liu, H.; Du, X.; et al. Flavonoid biosynthetic and starch and sucrose metabolic pathways are involved in the pigmentation of naturally brown-colored cotton fibers. Ind. Crop. Prod. 2020, 158, 113045. [Google Scholar] [CrossRef]
- Gao, J.; Shen, L.; Yuan, J.; Zheng, H.; Su, Q.; Yang, W.; Zhang, L.; Nnaemeka, V.E.; Sun, J.; Ke, L.; et al. Functional analysis of GhCHS, GhANR and GhLAR in colored fiber formation of Gossypium hirsutum L. BMC Plant Biol. 2019, 19, 455. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.H.; Zhang, Z.S.; Yin, M.H.; Luo, M.; Li, X.B.; Hou, L.; Pei, Y. Cotton flavonoid structural genes related to the pigmentation in brown fibers. Biochem. Biophys. Res. Commun. 2007, 358, 73–78. [Google Scholar] [CrossRef]
- Hua, S.; Yuan, S.; Shamsi, I.H.; Zhao, X.; Zhang, X.; Liu, Y.; Wen, G.; Wang, X.; Zhang, H. A Comparison of Three Isolines of Cotton Differing in Fiber Color for Yield, Quality, and Photosynthesis. Crop Sci. 2009, 49, 983–989. [Google Scholar] [CrossRef]
- Winkel-Shirley, B. Flavonoid biosynthesis. A colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant Physiol. 2001, 2, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Sasaki, N.; Ohmiya, A. Biosynthesis of plant pigments: Anthocyanins, betalains and carotenoids. Plant J. 2008, 54, 733–749. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Jiang, Q.; Shui, Z.; An, P.; Shi, S.; Liu, T.; Zhang, H.; Huang, S.; Jing, B.; Xiao, E.; et al. HaMYBA-HabHLH1 regulatory complex and HaMYBF fine-tune red flower coloration in the corolla of sunflower (Helianthus annuus L.). Plant Sci. 2024, 338, 111901. [Google Scholar] [CrossRef]
- Xiao, Y.H.; Yan, Q.; Ding, H.; Luo, M.; Hou, L.; Zhang, M.; Yao, D.; Liu, H.S.; Li, X.; Zhao, J.; et al. Transcriptome and biochemical analyses revealed a detailed proanthocyanidin biosynthesis pathway in brown cotton fiber. PLoS ONE 2014, 9, e86344. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, X.; Tu, L.; Zhu, L.; Guo, X. Isolation by suppression-subtractive hybridization of genes preferentially expressed during early and late fiber development stages in cotton. Mol. Biol. 2006, 40, 741–749. [Google Scholar] [CrossRef]
- Haigler, C.H.; Betancur, L.; Stiff, M.R.; Tuttle, J.R. Cotton fiber: A powerful single-cell model for cell wall and cellulose research. Front. Plant Sci. 2012, 3, 104. [Google Scholar] [CrossRef]
- Huang, G.; Huang, J.Q.; Chen, X.Y.; Zhu, Y.X. Recent Advances and Future Perspectives in Cotton Research. Annu. Rev. Plant Biol. 2021, 72, 437–462. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.M. Research progress and prospects on naturally-colored cotton. J. Ethnobiol. Ethnomed. 2004, 4, 249–254. [Google Scholar] [CrossRef]
- Zhang, S.; Yu, X.; Chen, M.; Chang, C.; Zhu, J.; Zhao, H. Comparative Transcriptome and Metabolome Profiling Reveal Mechanisms of Red Leaf Color Fading in Populus × euramericana cv. ‘Zhonghuahongye’. Plants 2023, 12, 3511. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Chen, J.; Guo, X.; Li, X.; Shen, Q.; Fu, X.; Tang, D. Metabolic Patterns of Flavonoid and Its Key Gene Expression Characteristics of Five Cultivars of Tulipa gesneriana during Flower Development. Plants 2024, 13, 459. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Burks, D.J.; Azad, R.K. RNA-Seq Data Analysis Pipeline for Plants: Transcriptome Assembly, Alignment, and Differential Expression Analysis. In Plant Metabolic Engineering. Methods in Molecular Biology; Humana: New York, NY, USA, 2022; pp. 47–60. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Liang, C.; Meng, Z.; Sun, G.; Meng, Z.; Guo, S.; Zhang, R. CottonFGD: An integrated functional genomics database for cotton. BMC Plant Biol. 2017, 17, 101. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Tian, F.; Yang, D.C.; Meng, Y.Q.; Jin, J.; Gao, G. PlantRegMap: Charting functional regulatory maps in plants. Nucleic Acids Res. 2019, 48, D1104–D1113. [Google Scholar] [CrossRef]
- Chen, C.; Wu, Y.; Li, J.; Wang, X.; Zeng, Z.; Xu, J.; Liu, Y.; Feng, J.; Chen, H.; He, Y.; et al. TBtools-II: A “one for all, all for one” bioinformatics platform for biological big-data mining. Mol. Plant 2023, 16, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Bai, C.; Chen, Y.; Yu, L.; Wu, W.; Hu, K. Comparing univariate filtration preceding and succeeding PLS-DA analysis on the differential variables/metabolites identified from untargeted LC-MS metabolomics data. Anal. Chim. Acta 2024, 1287, 342103. [Google Scholar] [CrossRef]
- Mishra, P.; Singh, U.; Pandey, C.; Mishra, P.; Pandey, G. Application of student’s t-test, analysis of variance, and covariance. Ann. Card. Anaesth. 2019, 22, 407–411. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, S.; Tang, R.; Hao, X.; Tang, S.; Chen, W.; Jiang, C.; Long, M.; Chen, K.; Hu, X.; Xie, Q.; et al. Integrative Transcriptomic and Metabolic Analyses Reveal That Flavonoid Biosynthesis Is the Key Pathway Regulating Pigment Deposition in Naturally Brown Cotton Fibers. Plants 2024, 13, 2028. https://doi.org/10.3390/plants13152028
Shi S, Tang R, Hao X, Tang S, Chen W, Jiang C, Long M, Chen K, Hu X, Xie Q, et al. Integrative Transcriptomic and Metabolic Analyses Reveal That Flavonoid Biosynthesis Is the Key Pathway Regulating Pigment Deposition in Naturally Brown Cotton Fibers. Plants. 2024; 13(15):2028. https://doi.org/10.3390/plants13152028
Chicago/Turabian StyleShi, Shandang, Rui Tang, Xiaoyun Hao, Shouwu Tang, Wengang Chen, Chao Jiang, Mengqian Long, Kailu Chen, Xiangxiang Hu, Quanliang Xie, and et al. 2024. "Integrative Transcriptomic and Metabolic Analyses Reveal That Flavonoid Biosynthesis Is the Key Pathway Regulating Pigment Deposition in Naturally Brown Cotton Fibers" Plants 13, no. 15: 2028. https://doi.org/10.3390/plants13152028
APA StyleShi, S., Tang, R., Hao, X., Tang, S., Chen, W., Jiang, C., Long, M., Chen, K., Hu, X., Xie, Q., Xie, S., Meng, Z., Ismayil, A., Jin, X., Wang, F., Liu, H., & Li, H. (2024). Integrative Transcriptomic and Metabolic Analyses Reveal That Flavonoid Biosynthesis Is the Key Pathway Regulating Pigment Deposition in Naturally Brown Cotton Fibers. Plants, 13(15), 2028. https://doi.org/10.3390/plants13152028