
Genetic Diversity Analysis and Prediction of Potential Suitable Areas for the Rare and Endangered Wild Plant Henckelia longisepala
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Data Collection
2.2. Genetic Diversity Assessment
2.2.1. DNA Extraction, Library Construction, and Sequencing
2.2.2. Sequencing Data Quality Control and Filtering
2.2.3. SNP Detection and Screening
2.2.4. Data Analysis
2.3. Suitable Area Prediction for H. longisepala
2.3.1. Data Preprocessing
2.3.2. Correlation Analysis of Climatic Factors
2.3.3. Parameter Selection
2.3.4. Classification and Description of Suitable Areas
3. Results
3.1. ddRAD Sequencing and Data Processing
3.2. SNP Statistics
3.3. Genetic Diversity Analysis
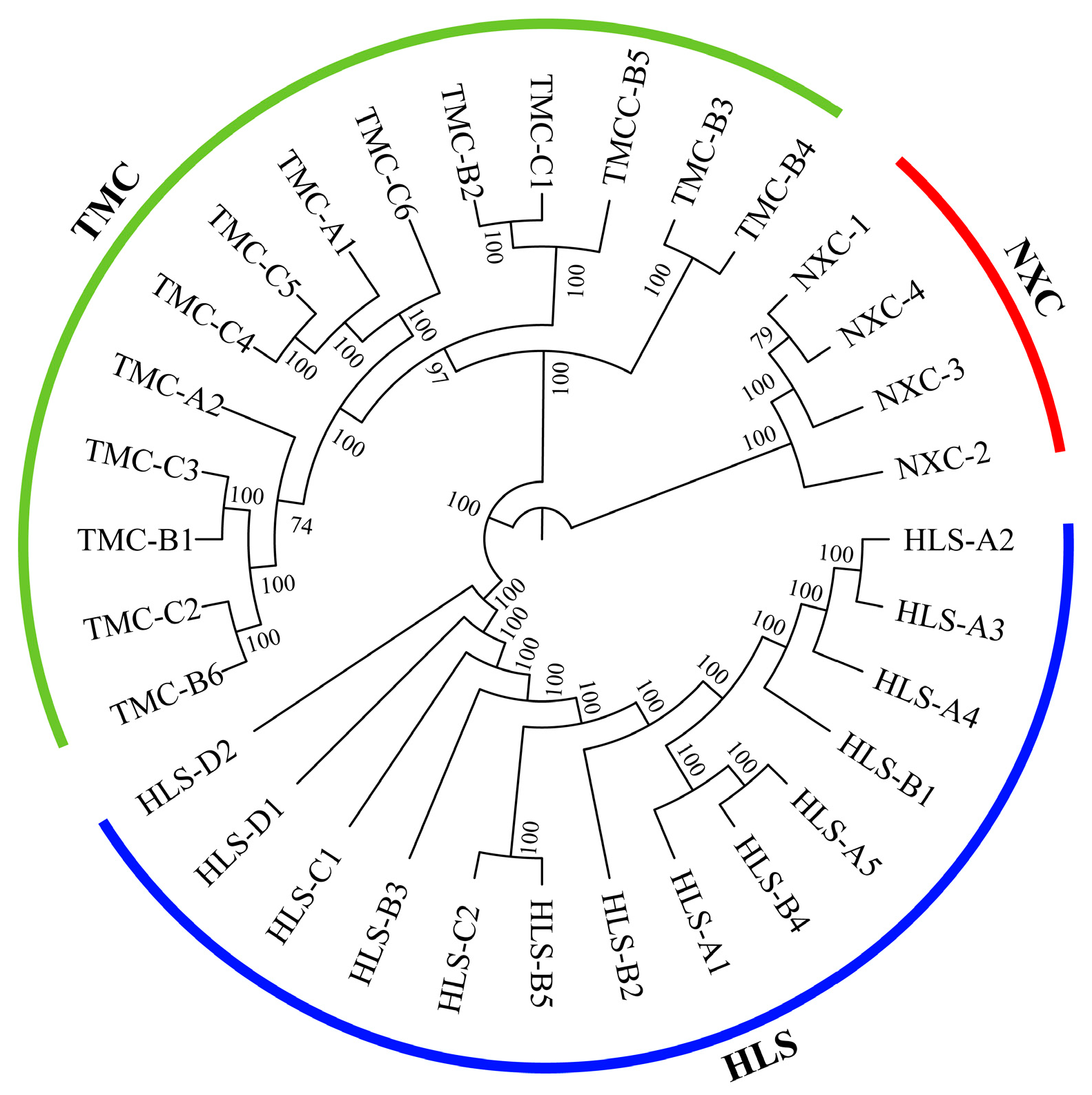
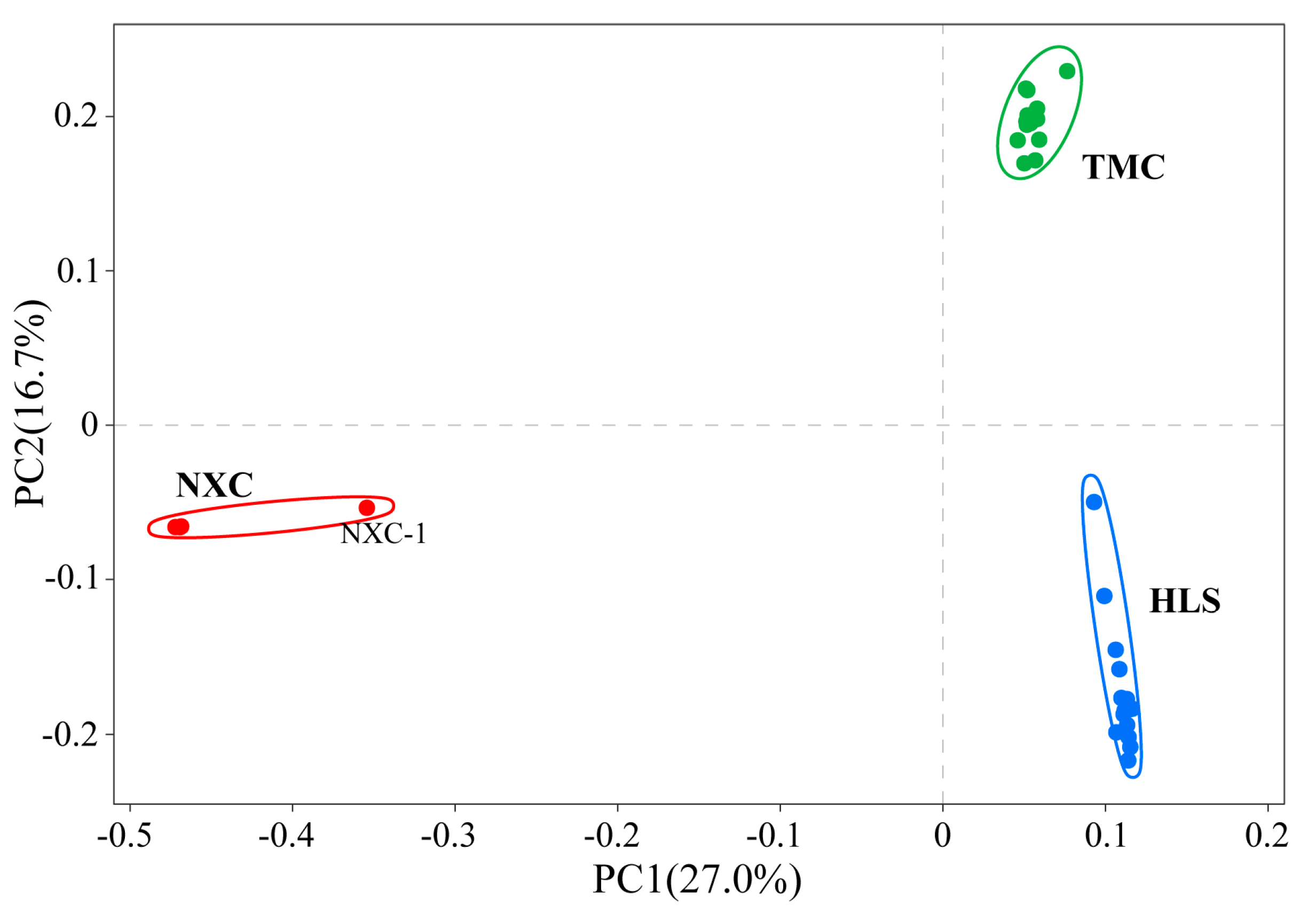
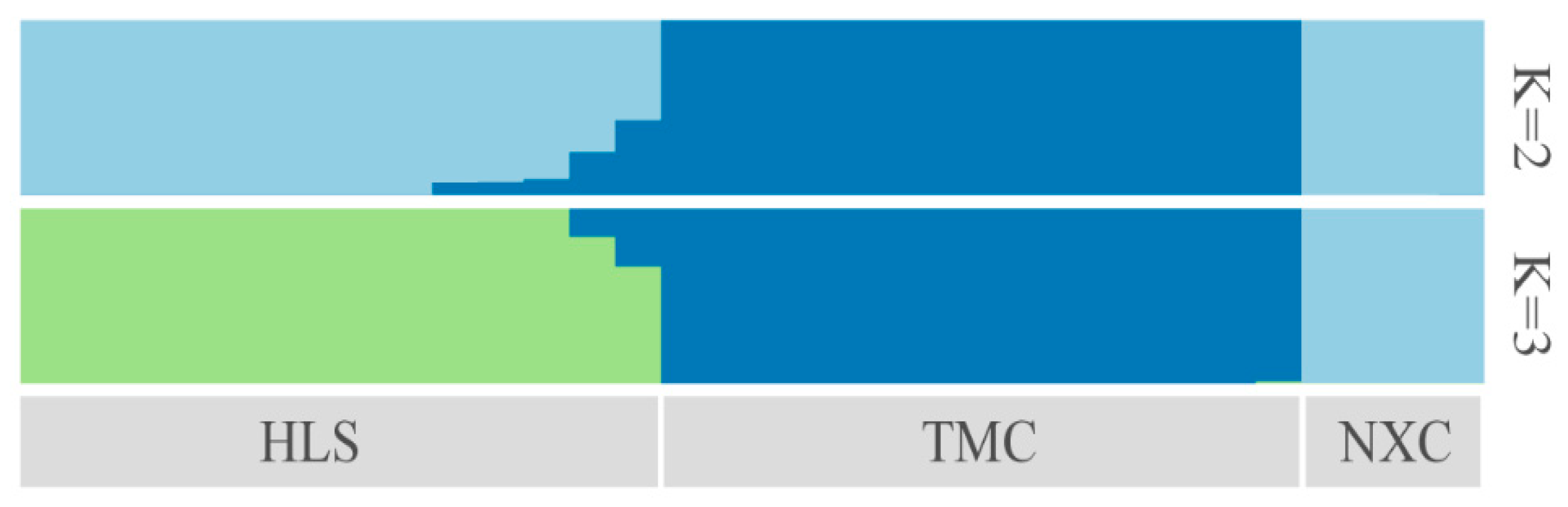
3.4. Genetic Structure Analysis
3.5. MaxEnt Model Prediction and Major Climate Factors
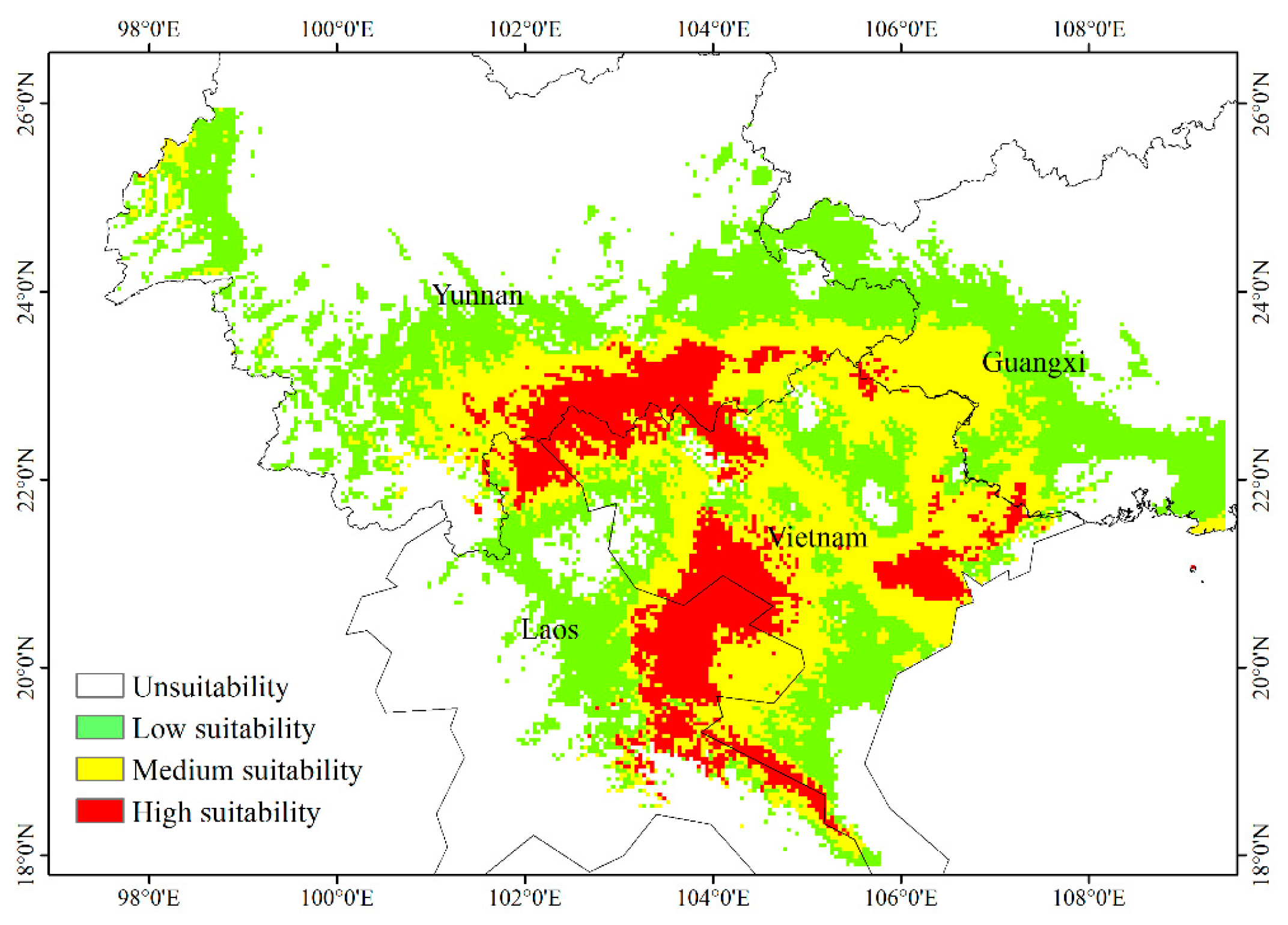
3.6. Current Potential Distribution of H. longisepala
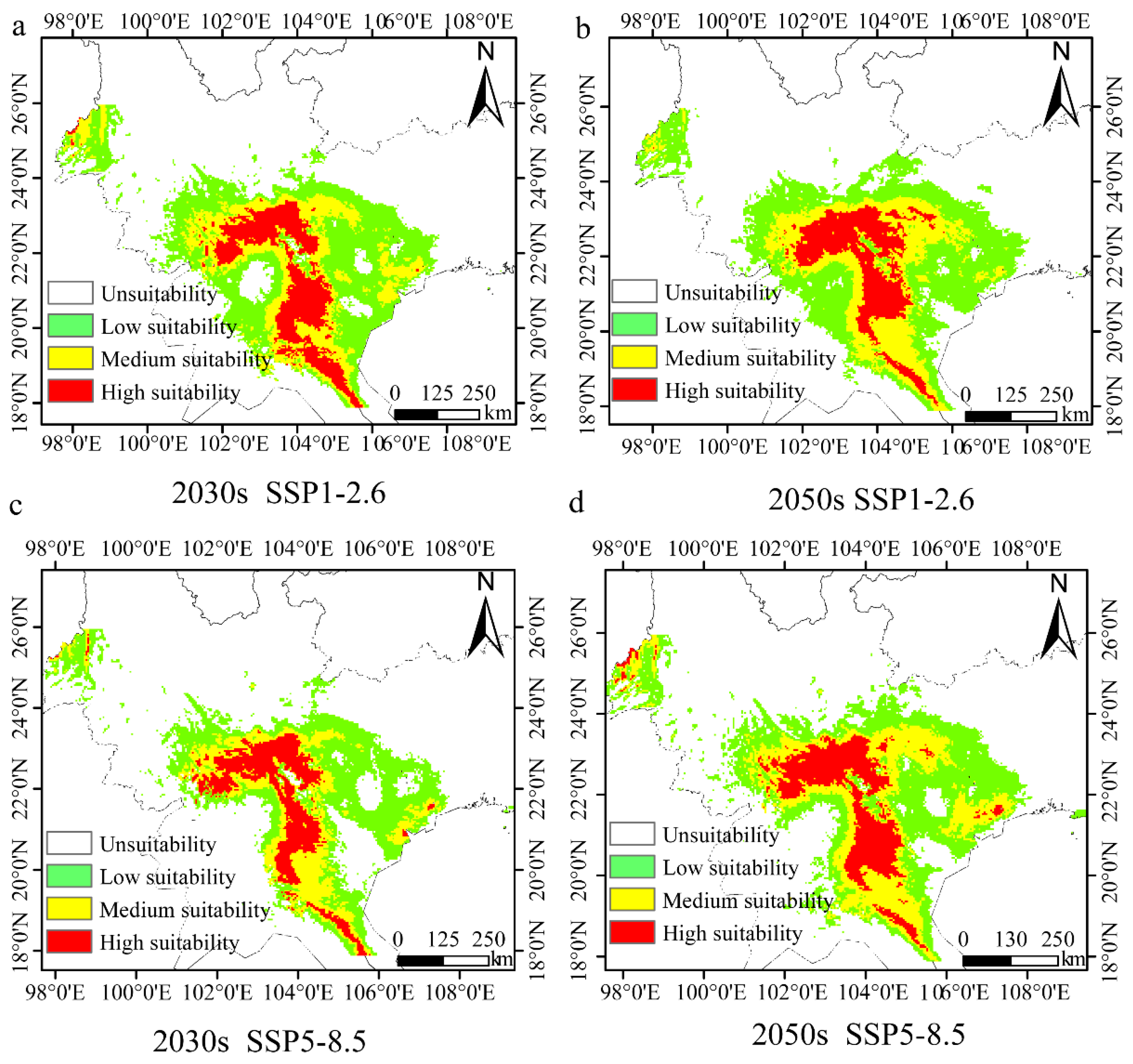
3.7. Potential Future Distribution of H. longisepala
4. Discussion
4.1. Genetic Diversity
4.2. Genetic Structure
4.3. Prediction of Potential Suitable Areas
4.4. Conservation Implications
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tatsuzawa, F.; Hosokawa, M. Flower Colors and Their Anthocyanins in Saintpaulia Cultivars (Gesneriaceae). Hortic. J. 2016, 85, 63–69. [Google Scholar] [CrossRef]
- Jiang, N.; Ning, S.J.; Pan, B.; Shen, X.L.; Huang, S.J. The Resources of Ornamental Plants in Gesneriaceae in Guangxi. Guihaia 2012, 32, 494–500. [Google Scholar]
- Wang, N.; Hu, C.Y.; Xiao, P.G.; Liu, Y.; Bai, Z.F. A Survey of the Traditional Pharmacognosy of Domestic Plants of the Family Gesneriaceae. J. Chin. Med. Mater. 2023, 46, 2975–2979. [Google Scholar]
- Bui, H.Q.; Nuraliev, M.S.; Möller, M.; Kuznetsov, A.N.; Kuznetsova, S.P.; Middleton, D.J.; Wen, F. Henckelia Longisepala (Gesneriaceae), a New Record for Vietnam. Rheedea 2020, 30, 176–186. [Google Scholar] [CrossRef]
- Qin, H.N.; Yang, Y.; Dong, S.Y.; He, Q.; Jia, Y.; Zhao, L.; Yu, S.X.; Liu, H.Y.; Liu, B.; Yan, Y.H.; et al. Threatened Species List of China’s Higher Plants. Biodivers. Sci. 2017, 25, 696–744. [Google Scholar] [CrossRef]
- Zhang, X.J.; Liu, X.F.; Liu, D.T.; Cao, Y.R.; Li, Z.H.; Ma, Y.P.; Ma, H. Genetic Diversity and Structure of Rhododendron Meddianum, a Plant Species with Extremely Small Populations. Plant Divers. 2021, 43, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.Q.; Shen, S.K.; Zhang, X.J.; Wang, Y.H.; Sun, W.B. Genetic Diversity and Population Structure of an Extremely Endangered Species: The World’s Largest Rhododendron. AoB Plants 2015, 7, plu082. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, N.H.; Backes, G.; Stougaard, J.; Andersen, S.U.; Jahoor, A. Genetic Diversity and Population Structure Analysis of European Hexaploid Bread Wheat (Triticum aestivum L.) Varieties. PLoS ONE 2014, 9, e94000. [Google Scholar] [CrossRef] [PubMed]
- Guizado, S.J.V.; Nadeem, M.A.; Ali, F.; Barut, M.; Habyarimana, E.; Gómez, T.P.; Santillan, J.A.V.; Canales, E.T.; Gómez, J.C.C.; Chung, G.; et al. Genetic Diversity and Population Structure of Endangered Rosewood from the Peruvian Amazon Using ISSR Markers. Acta Amaz. 2020, 50, 204–212. [Google Scholar] [CrossRef]
- Cao, Y.Y.; Diao, Q.N.; Chen, Y.Y.; Zhang, Y.P. Analysis of Genetic Diversity of Cucumis Melo Based on 2b-RAD Simplified Genome Sequencing. Acta Bot. Boreali-Occident. Sin. 2021, 41, 96–106. [Google Scholar]
- Lavretsky, P.; DaCosta, J.M.; Sorenson, M.D.; McCracken, K.G.; Peters, J.L. ddRAD-seq Data Reveal Significant Genome-wide Population Structure and Divergent Genomic Regions That Distinguish the Mallard and Close Relatives in North America. Mol. Ecol. 2019, 28, 2594–2609. [Google Scholar] [CrossRef] [PubMed]
- Garcia, C.B.; Silva, A.V.D.; Carvalho, I.A.S.D.; Nascimento, W.F.D.; Ramos, S.L.F.; Rodrigues, D.P.; Zucchi, M.I.; Costa, F.M.; Alves-Pereira, A.; Batista, C.E.D.A.; et al. Low Diversity and High Genetic Structure for Platonia Insignis Mart., an Endangered Fruit Tree Species. Plants 2024, 13, 1033. [Google Scholar] [CrossRef] [PubMed]
- Yangkun, W.; Yan, H.; Tianzhen, Z. Current status and perspective of RAD-seq in genomic research. Hereditas 2014, 36, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.Y.; Tong, L.; Sun, M.; Zhu, Y.X.; Wen, J.; Lin, Q.W.; Liu, B. Phylogeny and Divergence Time Estimation of the Walnut Family (Juglandaceae) Based on Nuclear RAD-Seq and Chloroplast Genome Data. Mol. Phylogenet. Evol. 2020, 147, 106802. [Google Scholar] [CrossRef]
- Kajiya-Kanegae, H.; Takanashi, H.; Fujimoto, M.; Ishimori, M.; Ohnishi, N.; Wacera, W.F.; Omollo, E.A.; Kobayashi, M.; Yano, K.; Nakano, M.; et al. RAD-Seq-Based High-Density Linkage Map Construction and QTL Mapping of Biomass-Related Traits in Sorghum Using the Japanese Landrace Takakibi NOG. Plant Cell Physiol. 2020, 61, 1262–1272. [Google Scholar] [CrossRef] [PubMed]
- Peterson, B.K.; Weber, J.N.; Kay, E.H.; Fisher, H.S.; Hoekstra, H.E. Double Digest RADseq: An Inexpensive Method for De Novo SNP Discovery and Genotyping in Model and Non-Model Species. PLoS ONE 2012, 7, e37135. [Google Scholar] [CrossRef] [PubMed]
- Aballay, M.M.; Aguirre, N.C.; Filippi, C.V.; Valentini, G.H.; Sánchez, G. Fine-Tuning the Performance of ddRAD-Seq in the Peach Genome. Sci. Rep. 2021, 11, 6298. [Google Scholar] [CrossRef] [PubMed]
- Li, L.L.; Huang, S.; Hou, J.; Gao, L.Y.; Li, Q.; Li, Y.; Zhu, H.Y.; Yang, L.M.; Hu, J.B. Construction of a High-Density Genetic Map for Melon Using ddRAD-Seq Technology from a Population Derived from Flexuosus and Reticulatus Botanical Groups. Sci. Hortic. 2020, 272, 10953. [Google Scholar] [CrossRef]
- Kobayashi, H.; Haino, Y.; Iwasaki, T.; Tezuka, A.; Nagano, A.J.; Shimada, S. ddRAD-Seq Based Phylogeographic Study of Sargassum Thunbergii (Phaeophyceae, Heterokonta) around Japanese Coast. Mar. Environ. Res. 2018, 140, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Kaviriri, D.K.; Zhang, Q.; Zhang, X.; Jiang, L.; Zhang, J.; Wang, J.; Khasa, D.P.; You, X.; Zhao, X. Phenotypic Variability and Genetic Diversity in a Pinus Koraiensis Clonal Trial in Northeastern China. Genes 2020, 11, 673. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.X.; Zhou, M.Y.; Yang, G.Q.; Zhang, Y.X.; Ma, P.F.; Guo, C.; Vorontsova, M.S.; Li, D.Z. ddRAD Analyses Reveal a Credible Phylogenetic Relationship of the Four Main Genera of Bambusa-Dendrocalamus-Gigantochloa Complex (Poaceae: Bambusoideae). Mol. Phylogenet. Evol. 2020, 146, 106758. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.Q.; Chen, Y.M.; Wang, J.P.; Guo, C.; Zhao, L.; Wang, X.-Y.; Guo, Y.; Li, L.; Li, D.-Z.; Guo, Z.-H. Development of a Universal and Simplified ddRAD Library Preparation Approach for SNP Discovery and Genotyping in Angiosperm Plants. Plant Methods 2016, 12, 39. [Google Scholar] [CrossRef] [PubMed]
- García, V.; Castro, P.; Die, J.V.; Millán, T.; Gil, J.; Moreno, R. QTL Analysis of Morpho-Agronomic Traits in Garden Asparagus (Asparagus Officinalis L.). Horticulturae 2022, 9, 41. [Google Scholar] [CrossRef]
- Feng, J.Y.; Zhao, S.; Li, M.; Zhang, C.; Qu, H.J.; Li, Q.; Li, J.W.; Lin, Y.; Pu, Z.G. Genome-Wide Genetic Diversity Detection and Population Structure Analysis in Sweetpotato (Ipomoea Batatas) Using RAD-Seq. Genomics 2020, 112, 1978–1987. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.D.; Yin, Q.; Sang, Z.Y.; Zhu, Z.L.; Jia, Z.K.; Ma, L.Y. Prediction of Potentially Suitable Areas for the Introduction of Magnolia Wufengensis under Climate Change. Ecol. Indic. 2021, 127, 107762. [Google Scholar] [CrossRef]
- Elith, J.; Leathwick, J.R. Species Distribution Models: Ecological Explanation and Prediction Across Space and Time. Annu. Rev. Ecol. Evol. Syst. 2009, 40, 677–697. [Google Scholar] [CrossRef]
- Liu, Y.; Tian, B.; Ou, G.L. Displacement Distribution and Climate Explanation on Humid and Semi-Humid Ever green Broadleaved Forests Using Niche Model of Cyclobalanopsis Glauca and C. Glaucoides in China. Guihaia 2022, 42, 460–469. [Google Scholar] [CrossRef]
- Merow, C.; Smith, M.J.; Silander, J.A. A Practical Guide to MaxEnt for Modeling Species’ Distributions: What It Does, and Why Inputs and Settings Matter. Ecography 2013, 36, 1058–1069. [Google Scholar] [CrossRef]
- Hernandez, P.A.; Graham, C.H.; Master, L.L.; Albert, D.L. The Effect of Sample Size and Species Characteristics on Performance of Different Species Distribution Modeling Methods. Ecography 2006, 29, 773–785. [Google Scholar] [CrossRef]
- Yu, Y.L.; Wang, H.C.; Yu, Z.X.; Schinnerl, J.; Tang, R.; Geng, Y.P.; Chen, G. Genetic Diversity and Structure of the Endemic and Endangered Species Aristolochia Delavayi Growing along the Jinsha River. Plant Divers. 2021, 43, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.M.; Cai, L.; Dao, Z.L.; Sun, W.B. Genomic Data Reveals Population Genetic and Demographic History of Magnolia Fistulosa (Magnoliaceae), a Plant Species With Extremely Small Populations in Yunnan Province, China. Front. Plant Sci. 2022, 13, 811312. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Catchen, J.; Bassham, S.; Wilson, T.; Currey, M.; O’Brien, C.; Yeates, Q.; Cresko, W.A. The Population Structure and Recent Colonization History of Oregon Threespine Stickleback Determined Using RAD-Seq. Mol. Ecol. 2013, 22, 2864–2883. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-Generation PLINK: Rising to the Challenge of Larger and Richer Datasets. GigaScience 2015, 4, s13742-015-0047-8. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A Tool for Genome-Wide Complex Trait Analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Pfaff, C.L.; Parra, E.J.; Bonilla, C.; Hiester, K.; McKeigue, P.M.; Kamboh, M.I.; Hutchinson, R.G.; Ferrell, R.E.; Boerwinkle, E.; Shriver, M.D. Population Structure in Admixed Populations: Effect of Admixture Dynamics on the Pattern of Linkage Disequilibrium. Am. J. Hum. Genet. 2001, 68, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.R.; Hodgkin, T. Genetic Diversity and Conservation and Utilization of Plant Genetic Resources. Plant Cell Tissue Organ. Cult. 2002, 68, 1–19. [Google Scholar] [CrossRef]
- Gao, R.; Liu, L.; Zhao, L.; Cui, S. Potentially Suitable Geographical Area for Monochamus Alternatus under Current and Future Climatic Scenarios Based on Optimized MaxEnt Model. Insects 2023, 14, 182. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.N.; Liu, Z.G.; Yu, T.; Liao, H.; Zhou, J.Y. Prediction of Potential Distribution of Prunus Mume Based on MaxEnt Model. Chin. Wild Plant Resour. 2024, 43, 107–113+126. [Google Scholar]
- Wu, T.; Lu, Y.; Fang, Y.; Xin, X.; Li, L.; Li, W.; Jie, W.; Zhang, J.; Liu, Y.; Zhang, L.; et al. The Beijing Climate Center Climate System Model (BCC-CSM): The Main Progress from CMIP5 to CMIP6. Geosci. Model Dev. 2019, 12, 1573–1600. [Google Scholar] [CrossRef]
- Zhang, R.L.; Liu, W.Y.; Zhang, Y.X.; Tu, D.D.; Zhu, L.Y.; Zhang, W.J. Genetic Diversity Analysis of Melocalamus Arrectus Based on Reduced-Representation Genome Sequencing. Mol. Plant Breed. 2021. [Google Scholar]
- Willoughby, J.R.; Sundaram, M.; Wijayawardena, B.K.; Kimble, S.J.A.; Ji, Y.; Fernandez, N.B.; Antonides, J.D.; Lamb, M.C.; Marra, N.J.; DeWoody, J.A. The Reduction of Genetic Diversity in Threatened Vertebrates and New Recommendations Regarding IUCN Conservation Rankings. Biol. Conserv. 2015, 191, 495–503. [Google Scholar] [CrossRef]
- Jorde, L.B. Linkage Disequilibrium and the Search for Complex Disease Genes. Genome Res. 2000, 10, 1435–1444. [Google Scholar] [CrossRef] [PubMed]
- Tikendra, L.; Potshangbam, A.M.; Amom, T.; Dey, A.; Nongdam, P. Understanding the Genetic Diversity and Population Structure of Dendrobium Chrysotoxum Lindl.-An Endangered Medicinal Orchid and Implication for Its Conservation. S. Afr. J. Bot. 2021, 138, 364–376. [Google Scholar] [CrossRef]
- Miller, M.R.; Dunham, J.P.; Amores, A.; Cresko, W.A.; Johnson, E.A. Rapid and Cost-Effective Polymorphism Identification and Genotyping Using Restriction Site Associated DNA (RAD) Markers. Genome Res. 2007, 17, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Berihulay, H.; Li, Y.; Liu, X.; Gebreselassie, G.; Islam, R.; Liu, W.; Jiang, L.; Ma, Y. Genetic Diversity and Population Structure in Multiple Chinese Goat Populations Using a SNP Panel. Anim. Genet. 2019, 50, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Marrano, A.; Birolo, G.; Prazzoli, M.L.; Lorenzi, S.; Valle, G.; Grando, M.S. SNP-Discovery by RAD-Sequencing in a Germplasm Collection of Wild and Cultivated Grapevines (V. Vinifera L.). PLoS ONE 2017, 12, e0170655. [Google Scholar] [CrossRef] [PubMed]
- Dubreuil, M.; Riba, M.; Mayol, M. Genetic Structure and Diversity in Ramonda Myconi (Gesneriaceae): Effects of Historical Climate Change on a Preglacial Relict Species. Am. J. Bot. 2008, 95, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Lu, G.H.; Fu, Y.H.; Wang, Y.Q. Genetic Differentiation between Two Varieties of Oreocharis Benthamii (Gesneriaceae) in Sympatric and Allopatric Regions. Ecol. Evol. 2020, 10, 7792–7805. [Google Scholar] [CrossRef] [PubMed]
- Ni, X.W.; Huang, Y.L.; Wu, L.; Zhou, R.C.; Deng, S.; Wu, D.R.; Wang, B.S.; Su, G.H.; Tang, T.; Shi, S.H. Genetic Diversity of the Endangered Chinese Endemic Herb Primulina Tabacum (Gesneriaceae) Revealed by Amplified Fragment Length Polymorphism (AFLP). Genetica 2006, 127, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Zou, R.; Tang, J.; Deng, L.; Wei, X. Genetic Diversity Variation during the Natural Regeneration of Vatica Guangxiensis, an Endangered Tree Species with Extremely Small Populations. Glob. Ecol. Conserv. 2023, 42, e02400. [Google Scholar] [CrossRef]
- Xu, G.; Liang, Y.; Jiang, Y.; Liu, X.S.; Hu, S.L.; Xiao, Y.F.; Hao, B.B. Genetic diversity and population structure of Bretschneidera sinensis, an endangered species: Genetic diversity and population structure of Bretschneidera sinensis, an endangered species. Biodivers. Sci. 2014, 21, 723–731. [Google Scholar]
- Mao, C.L.; Zhang, F.L.; Li, X.Q.; Yang, T.; Zhao, Q.; Hu, Y.H.; Wu, Y. Genetic Diversity of Horsfieldia Pandurifolia Based on AFLP Markers. J. Trop. Subtrop. Bot. 2020, 28, 271–276. [Google Scholar] [CrossRef]
- Pan, Y.Z.; Wang, X.Q.; Sun, G.L.; Li, F.S.; Gong, X. Application of RAD Sequencing for Evaluating the Genetic Diversity of Domesticated Panax Notoginseng (Araliaceae). PLoS ONE 2016, 11, e0166419. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.P.; Liu, G.Q.; Bu, W.J.; Gao, Y. Ecological niche modeling and its applications in biodiversity conservation: Ecological niche modeling and its applications in biodiversity conservation. Biodivers. Sci. 2013, 21, 90–98. [Google Scholar]
- Garzón, M.B.; Blazek, R.; Neteler, M.; Dios, R.S.D.; Ollero, H.S.; Furlanello, C. Predicting Habitat Suitability with Machine Learning Models: The Potential Area of Pinus Sylvestris L. in the Iberian Peninsula. Ecol. Model. 2006, 197, 383–393. [Google Scholar] [CrossRef]
- Manthey, M.; Box, E.O. Realized Climatic Niches of Deciduous Trees: Comparing Western Eurasia and Eastern North America. J. Biogeogr. 2007, 34, 1028–1040. [Google Scholar] [CrossRef]
- Kramer-Schadt, S.; Niedballa, J.; Pilgrim, J.D.; Schröder, B.; Lindenborn, J.; Reinfelder, V.; Stillfried, M.; Heckmann, I.; Scharf, A.K.; Augeri, D.M.; et al. The Importance of Correcting for Sampling Bias in MaxEnt Species Distribution Models. Divers. Distrib. 2013, 19, 1366–1379. [Google Scholar] [CrossRef]
- Coelho, N.; Gonçalves, S.; Romano, A. Endemic Plant Species Conservation: Biotechnological Approaches. Plants 2020, 9, 345. [Google Scholar] [CrossRef] [PubMed]
- Desai, P.; Gajera, B.; Mankad, M.; Shah, S.; Patel, A.; Patil, G.; Narayanan, S.; Kumar, N. Comparative Assessment of Genetic Diversity among Indian Bamboo Genotypes Using RAPD and ISSR Markers. Mol. Biol. Rep. 2015, 42, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Lozano, M.; Sánchez-González, A.; Octavio-Aguilar, P.; Galván-Hernández, D.M.; Vázquez-García, J.A. Genetic Diversity and Structure of Magnolia Mexicana (Magnoliacea): A Threatened Species in Eastern Mexico. Silvae Genet. 2023, 72, 132–142. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pop | Longitude (° E) | Latitude (° N) | Altitude (m) | Locality | Number of Samples |
|---|---|---|---|---|---|
| HLS | 102.3494 | 22.6457 | 1009 | Yunnan Lüchun | 14 |
| NXC | 103.8897 | 22.6095 | 462 | Yunnan Hekou | 4 |
| TMC | 103.0264 | 22.6250 | 947 | Yunnan Jinping | 14 |
| Climate Variable | Description | Climate Variable | Description |
|---|---|---|---|
| bio1 | Annual mean temperature | bio11 | Mean temperature of coldest quarter |
| bio2 | Mean diurnal range | bio12 | Annual precipitation |
| bio3 | Isothermality | bio13 | Precipitation of wettest month |
| bio4 | Temperature seasonality | bio14 | Precipitation of driest month |
| bio5 | Max temperature of warmest month | bio15 | Precipitation seasonality |
| bio6 | Min temperature of coldest month | bio16 | Precipitation of wettest quarter |
| bio7 | Temperature annual range | bio17 | Precipitation of driest quarter |
| bio8 | Mean temperature of wettest quarter | bio18 | Precipitation of warmest quarter |
| bio9 | Mean temperature of driest quarter | bio19 | Precipitation of coldest quarter |
| bio10 | Mean temperature of warmest quarter |
| Sample_Name | HQ Reads | Total_Base (bp) | HQ Bases (bp) | HQ Bases % | GC (%) | Q30 (%) |
|---|---|---|---|---|---|---|
| HLS_A1 | 12,508,536 | 1,847,065,824 | 1,781,995,529 | 96.48 | 40.97 | 94.94 |
| HLS_A2 | 14,332,744 | 2,120,032,512 | 2,041,843,119 | 96.31 | 43.12 | 94.92 |
| HLS_A3 | 36,818,088 | 5,410,729,440 | 5,257,449,583 | 97.17 | 40.22 | 96.41 |
| HLS_A4 | 34,107,414 | 5,009,117,184 | 4,872,148,968 | 97.27 | 39.26 | 96.19 |
| HLS_A5 | 30,046,774 | 4,413,880,800 | 4,292,836,294 | 97.26 | 38.49 | 96.10 |
| HLS_B1 | 32,220,506 | 4,733,449,056 | 4,602,590,836 | 97.24 | 39.03 | 96.29 |
| HLS_B2 | 32,318,314 | 4,827,110,569 | 4,675,400,724 | 96.86 | 38.35 | 96.19 |
| HLS_B3 | 32,792,914 | 4,906,151,916 | 4,745,839,024 | 96.73 | 38.04 | 96.00 |
| HLS_B4 | 14,576,362 | 2,136,863,520 | 2,087,739,170 | 97.70 | 38.95 | 97.89 |
| HLS_B5 | 15,292,504 | 2,244,107,808 | 2,189,839,488 | 97.58 | 39.19 | 97.82 |
| HLS_C1 | 15,307,248 | 2,253,815,136 | 2,191,488,136 | 97.23 | 42.68 | 97.67 |
| HLS_C2 | 18,084,322 | 2,656,833,696 | 2,589,175,360 | 97.45 | 42.18 | 97.71 |
| HLS_D1 | 16,292,676 | 2,398,851,360 | 2,331,478,275 | 97.19 | 41.94 | 97.71 |
| HLS_D2 | 16,406,160 | 2,414,157,408 | 2,347,565,196 | 97.24 | 43.57 | 97.66 |
| NX_1 | 15,163,360 | 2,221,348,320 | 2,170,983,575 | 97.73 | 38.85 | 97.84 |
| NX_2 | 17,780,208 | 2,614,287,456 | 2,545,830,403 | 97.38 | 39.43 | 97.73 |
| NX_3 | 15,545,240 | 2,282,696,928 | 2,225,675,817 | 97.50 | 39.43 | 97.73 |
| NX_4 | 14,758,408 | 2,167,298,496 | 2,112,253,626 | 97.46 | 38.15 | 97.72 |
| TM_A1 | 14,494,752 | 2,130,803,712 | 2,076,078,685 | 97.43 | 43.20 | 97.82 |
| TM_A2 | 16,837,792 | 2,481,160,320 | 2,410,517,148 | 97.15 | 43.16 | 97.59 |
| TM_B1 | 14,951,876 | 2,195,641,152 | 2,140,724,678 | 97.50 | 39.91 | 97.74 |
| TM_B2 | 14,226,138 | 2,093,088,960 | 2,037,690,222 | 97.35 | 42.23 | 97.68 |
| TM_B3 | 14,467,912 | 2,133,347,904 | 2,071,285,801 | 97.09 | 41.71 | 97.65 |
| TM_B4 | 14,766,900 | 2,175,391,872 | 2,115,346,007 | 97.24 | 41.65 | 97.70 |
| TM_B5 | 13,956,478 | 2,053,735,200 | 1,999,096,791 | 97.34 | 40.38 | 97.66 |
| TM_B6 | 15,690,434 | 2,297,819,808 | 2,247,479,371 | 97.81 | 39.11 | 97.97 |
| TM_C1 | 15,504,368 | 2,272,298,112 | 2,218,737,176 | 97.64 | 38.23 | 97.86 |
| TM_C2 | 17,817,304 | 2,612,644,992 | 2,550,693,042 | 97.63 | 39.69 | 97.88 |
| TM_C3 | 16,814,138 | 2,466,775,008 | 2,407,381,170 | 97.59 | 40.25 | 97.84 |
| TM_C4 | 17,449,764 | 2,558,971,008 | 2,495,422,887 | 97.52 | 41.10 | 97.80 |
| TM_C5 | 18,474,068 | 2,713,303,584 | 2,644,282,676 | 97.46 | 39.40 | 97.75 |
| TM_C6 | 17,334,360 | 2,539,707,264 | 2,482,303,852 | 97.74 | 38.78 | 97.87 |
| Sample | SNP Number | Transitions | Transversions | Number of Heterozygous SNPs | Heterozygosity (%) | Number of Homozygous SNPs | Homozygosity (%) | Ts/Tv |
|---|---|---|---|---|---|---|---|---|
| HLS_A1 | 37,028 | 21,251 | 15,777 | 19,185 | 51.81 | 17,843 | 48.19 | 1.35 |
| HLS_A2 | 37,915 | 21,857 | 16,058 | 21,569 | 56.89 | 16,346 | 43.11 | 1.36 |
| HLS_A3 | 50,957 | 29,202 | 21,755 | 29,499 | 57.89 | 21,458 | 42.11 | 1.34 |
| HLS_A4 | 49,246 | 28,245 | 21,001 | 28,974 | 58.84 | 20,272 | 41.16 | 1.34 |
| HLS_A5 | 46,992 | 26,996 | 19,996 | 26,903 | 57.25 | 20,089 | 42.75 | 1.35 |
| HLS_B1 | 48,142 | 27,603 | 20,539 | 27,442 | 57.00 | 20,700 | 43.00 | 1.34 |
| HLS_B2 | 52,104 | 29,696 | 22,408 | 29,235 | 56.11 | 22,869 | 43.89 | 1.33 |
| HLS_B3 | 42,699 | 24,250 | 18,449 | 23,372 | 54.74 | 19,327 | 45.26 | 1.31 |
| HLS_B4 | 48,884 | 27,799 | 21,085 | 27,867 | 57.01 | 21,017 | 42.99 | 1.32 |
| HLS_B5 | 48,644 | 27,821 | 20,823 | 23,862 | 49.05 | 24,782 | 50.95 | 1.34 |
| HLS_C1 | 44,796 | 25,954 | 18,842 | 24,137 | 53.88 | 20,659 | 46.12 | 1.38 |
| HLS_C2 | 50,281 | 28,952 | 21,329 | 29,745 | 59.16 | 20,536 | 40.84 | 1.36 |
| HLS_D1 | 49,453 | 28,730 | 20,723 | 31,828 | 64.36 | 17,625 | 35.64 | 1.39 |
| HLS_D2 | 43,263 | 25,069 | 18,194 | 20,529 | 47.45 | 22,734 | 52.55 | 1.38 |
| NX_1 | 78,996 | 46,842 | 32,154 | 27,571 | 34.90 | 51,425 | 65.10 | 1.46 |
| NX_2 | 65,403 | 39,064 | 26,339 | 17,770 | 27.17 | 47,633 | 72.83 | 1.48 |
| NX_3 | 83,268 | 49,298 | 33,970 | 29,080 | 34.92 | 54,188 | 65.08 | 1.45 |
| NX_4 | 79,208 | 46,794 | 32,414 | 27,658 | 34.92 | 51,550 | 65.08 | 1.44 |
| TM_A1 | 40,827 | 23,647 | 17,180 | 22,512 | 55.14 | 18,315 | 44.86 | 1.38 |
| TM_A2 | 42,953 | 24,843 | 18,110 | 24,407 | 56.82 | 18,546 | 43.18 | 1.37 |
| TM_B1 | 37,417 | 21,613 | 15,804 | 22,603 | 60.41 | 14,814 | 39.59 | 1.37 |
| TM_B2 | 40,854 | 23,637 | 17,217 | 22,332 | 54.66 | 18,522 | 45.34 | 1.37 |
| TM_B3 | 40,333 | 23,211 | 17,122 | 23,883 | 59.21 | 16,450 | 40.79 | 1.36 |
| TM_B4 | 43,117 | 24,863 | 18,254 | 25,114 | 58.25 | 18,003 | 41.75 | 1.36 |
| TM_B5 | 38,974 | 22,396 | 16,578 | 21,086 | 54.10 | 17,888 | 45.90 | 1.35 |
| TM_B6 | 47,327 | 27,388 | 19,939 | 27,724 | 58.58 | 19,603 | 41.42 | 1.37 |
| TM_C1 | 40,341 | 23,226 | 17,115 | 18,166 | 45.03 | 22,175 | 54.97 | 1.36 |
| TM_C2 | 48,286 | 27,923 | 20,363 | 29,165 | 60.40 | 19,121 | 39.60 | 1.37 |
| TM_C3 | 48,601 | 27,859 | 20,742 | 30,902 | 63.58 | 17,699 | 36.42 | 1.34 |
| TM_C4 | 43,142 | 24,796 | 18,346 | 24,096 | 55.85 | 19,046 | 44.15 | 1.35 |
| TM_C5 | 48,577 | 27,991 | 20,586 | 29,012 | 59.72 | 19,565 | 40.28 | 1.36 |
| TM_C6 | 95,535 | 55,927 | 39,608 | 35,017 | 36.65 | 60,518 | 63.35 | 1.41 |
| Pop | Obs Het | Obs Hom | Exp Het | Exp Hom | Pi | FIS |
|---|---|---|---|---|---|---|
| HLS | 0.1256 | 0.8744 | 0.1883 | 0.8117 | 0.1990 | 0.2079 |
| TMC | 0.1182 | 0.8818 | 0.1572 | 0.8428 | 0.1795 | 0.1868 |
| NXC | 0.1210 | 0.8790 | 0.1151 | 0.8849 | 0.1408 | 0.0420 |
| Pop | HLS | TMC | NXC |
|---|---|---|---|
| HLS | 0.1924 | 0.3680 | |
| TMC | 0.4071 |
| HLS | TMC | |
|---|---|---|
| TMC | 0.4524 | |
| NXC | 0.8276 | 0.7672 |
| Environment Variable | Percent Contribution | Permutation Importance |
|---|---|---|
| bio18 | 36.7 | 43.9 |
| bio7 | 25.4 | 33.9 |
| bio16 | 17.5 | 16.3 |
| bio9 | 11.9 | 2.5 |
| bio17 | 3.2 | 2.3 |
| bio12 | 3.1 | 0.6 |
| bio3 | 2.2 | 0.5 |
| Scenario | Period | High Suitability | Ratio % | Medium Suitability | Ratio % | Low Suitability | Ratio % | Total | Ratio % |
|---|---|---|---|---|---|---|---|---|---|
| Current | — | 5.50 | — | 11.25 | — | 15.67 | — | 32.42 | — |
| ssp1-2.6 | 2030s | 5.24 | −4.73 | 7.26 | −35.47 | 14.12 | −9.89 | 26.63 | −17.86 |
| 2050s | 5.10 | −7.27 | 7.76 | −31.02 | 14.93 | −4.72 | 27.79 | −14.28 | |
| ssp5-8.5 | 2030s | 4.03 | −26.73 | 4.99 | −55.64 | 10.76 | −31.33 | 19.78 | −38.99 |
| 2050s | 4.98 | −9.45 | 7.66 | −31.91 | 12.63 | −19.40 | 25.27 | −22.05 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, R.; Huang, N.; Zhang, Z.; Luo, W.; Xiang, J.; Xu, Y.; Wang, Y. Genetic Diversity Analysis and Prediction of Potential Suitable Areas for the Rare and Endangered Wild Plant Henckelia longisepala. Plants 2024, 13, 2093. https://doi.org/10.3390/plants13152093
Zhao R, Huang N, Zhang Z, Luo W, Xiang J, Xu Y, Wang Y. Genetic Diversity Analysis and Prediction of Potential Suitable Areas for the Rare and Endangered Wild Plant Henckelia longisepala. Plants. 2024; 13(15):2093. https://doi.org/10.3390/plants13152093
Chicago/Turabian StyleZhao, Renfen, Nian Huang, Zhiyan Zhang, Wei Luo, Jianying Xiang, Yuanjie Xu, and Yizhi Wang. 2024. "Genetic Diversity Analysis and Prediction of Potential Suitable Areas for the Rare and Endangered Wild Plant Henckelia longisepala" Plants 13, no. 15: 2093. https://doi.org/10.3390/plants13152093