
An Integrated Strategy for Analyzing the Complete Complex Integrated Structure of Maize MON810 and Identification of an SNP in External Insertion Sequences
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Sampling
2.2. PacBio-HiFi Sequencing
2.3. High-Throughput Sequencing-Based PCR-Free Library
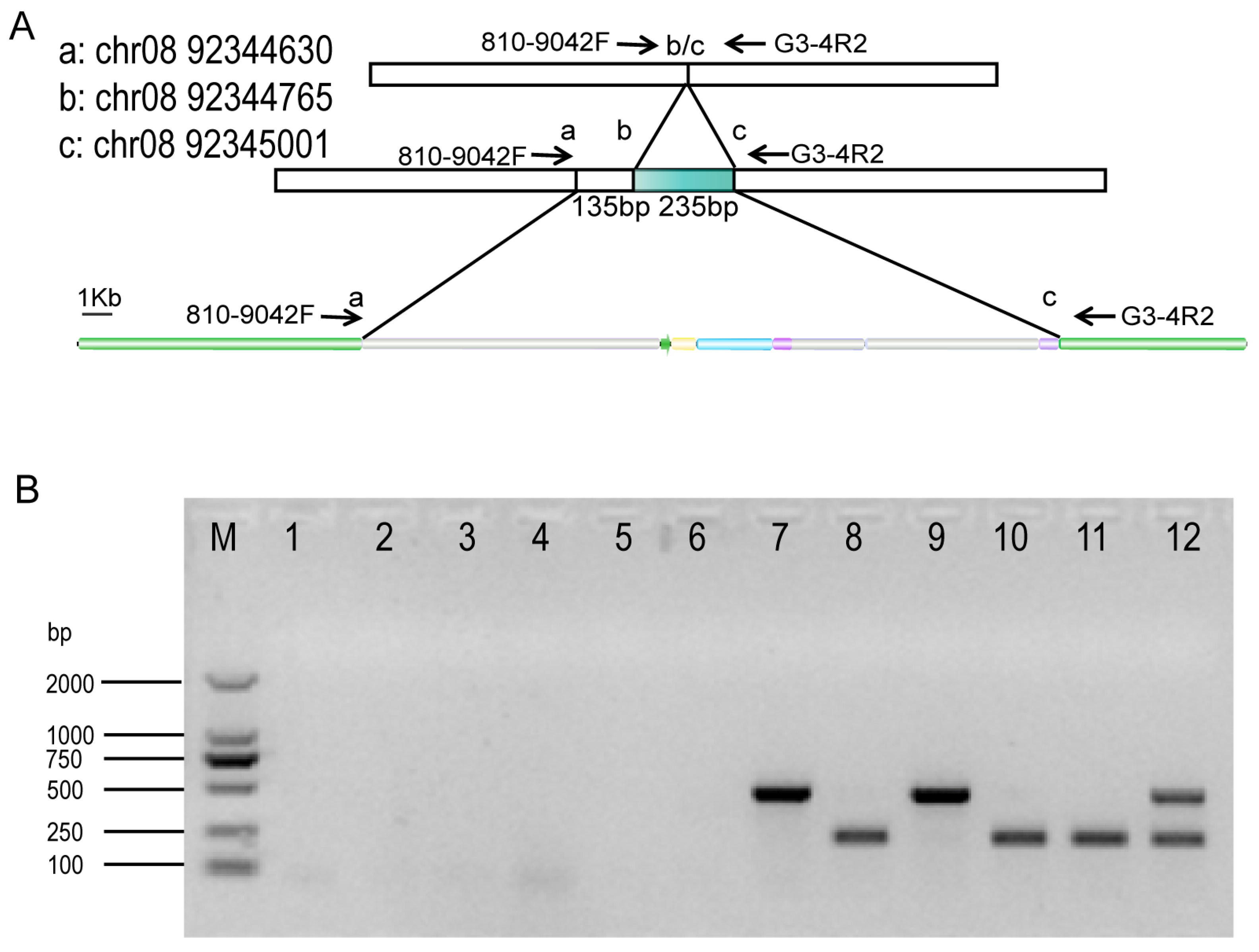
2.4. Design of Diagnostic Assays and Sanger Sequencing
2.5. Detection of Variant Type Using Qualitative AS-PCR
2.6. Detection of Variant Type Using Quantitative AS-PCR-Based Construct-Specific Method
2.7. BDA Method to Detect the Low-Frequency AT Variant
3. Results
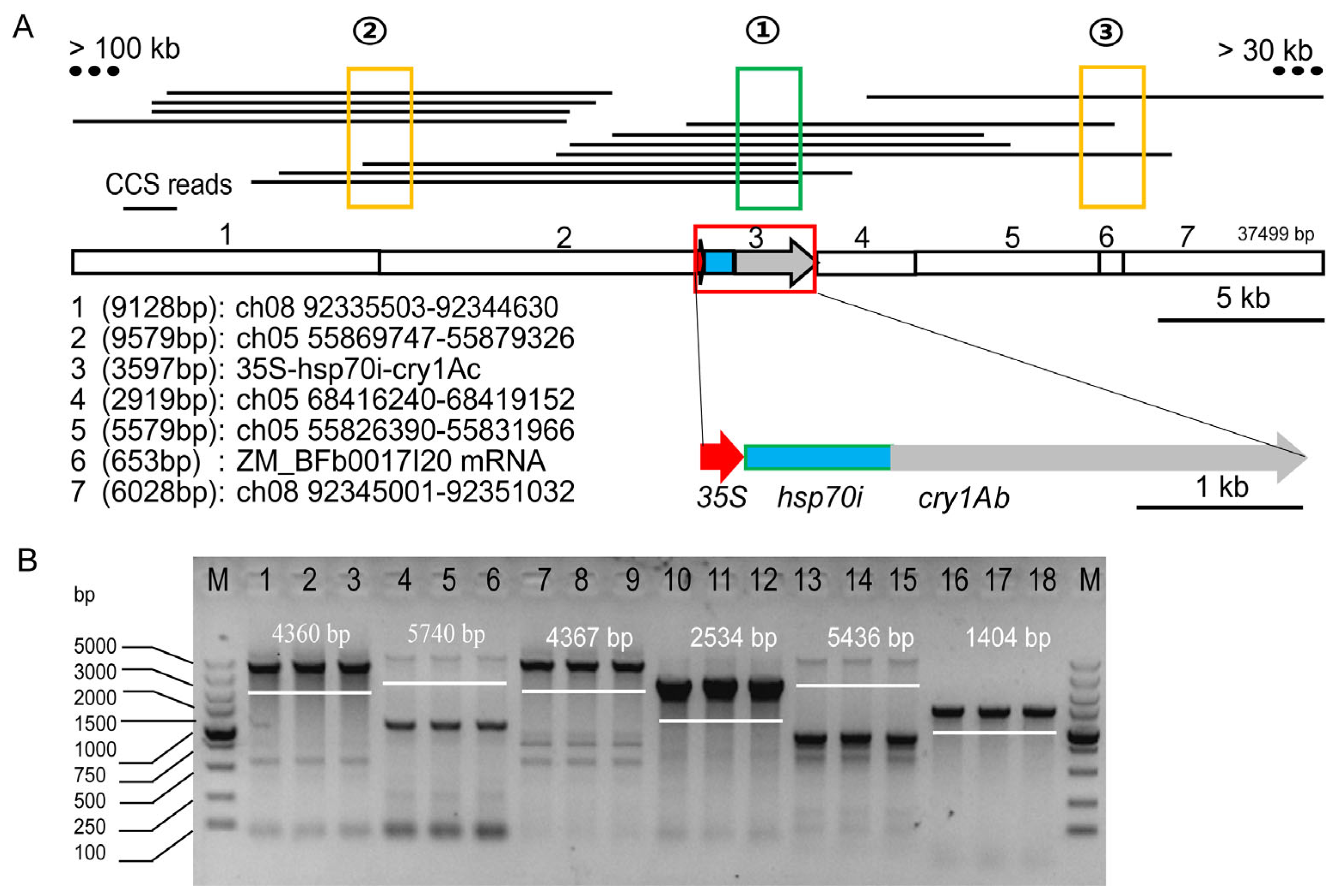
3.1. Pacbio-Hifi Sequencing
3.2. Identification of Sequence Diversity at the Insertion Site
3.3. Next-Generation Sequencing
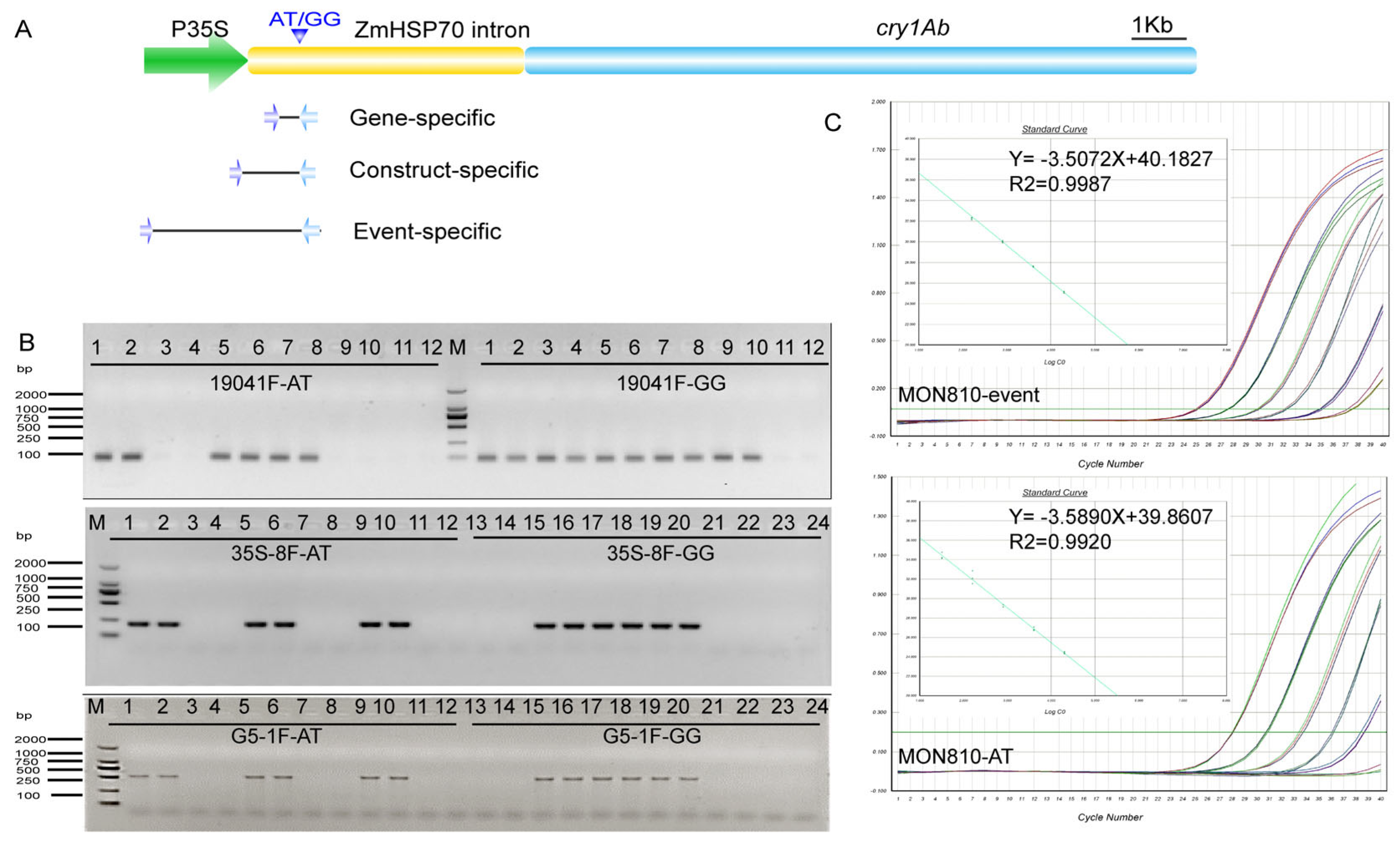
3.4. Detection of the Variant Frequency Using Qualitative AS-PCR
3.5. Detection of the AT Variant Frequency Using Quantitative AS-PCR
3.6. BDA Enrichment to Detect Low Variant Allele Frequency (VAF) of the AT Type
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Li, Y.; Hallerman, E.M.; Wu, K.; Peng, Y. Insect-Resistant Genetically Engineered Crops in China: Development, Application, and Prospects for Use. Annu. Rev. Entomol. 2020, 65, 273–292. [Google Scholar] [CrossRef]
- Zanon Agapito-Tenfen, S.; Guerra, M.P.; Nodari, R.O.; Wikmark, O.G. Untargeted Proteomics-Based Approach to Investigate Unintended Changes in Genetically Modified Maize for Environmental Risk Assessment Purpose. Front. Toxicol. 2021, 3, 655968. [Google Scholar] [CrossRef] [PubMed]
- Tahar, S.B.; Salva, I.; Brants, I.O. Genetic Stability in Two Commercialized Transgenic Lines (MON810). Nat. Biotechnol. 2010, 28, 779. [Google Scholar] [CrossRef]
- Olaru, I.; Waigmann, E. Annual Report of the EFSA Scientific Network for Risk Assessment of GMOs for 2017. EFSA Support. Publ. 2018, 15. [Google Scholar] [CrossRef]
- Ben Ali, S.E.; Schamann, A.; Dobrovolny, S.; Indra, A.; Agapito-Tenfen, S.Z.; Hochegger, R.; Haslberger, A.G.; Brandes, C. Genetic and Epigenetic Characterization of the Cry1Ab Coding Region and Its 3′ Flanking Genomic Region in MON810 Maize Using Next-Generation Sequencing. Eur. Food Res. Technol. 2018, 244, 1473–1485. [Google Scholar] [CrossRef]
- Castan, M.; Ben Ali, S.E.; Hochegger, R.; Ruppitsch, W.; Haslberger, A.G.; Brandes, C. Analysis of the Genetic Stability of Event NK603 in Stacked Corn Varieties Using High-Resolution Melting (HRM) Analysis and Sanger Sequencing. Eur. Food Res. Technol. 2017, 243, 353–365. [Google Scholar] [CrossRef]
- Park, D.; Choi, I.Y.; Kim, N.S. Detection of MPing Mobilization in Transgenic Rice Plants. Genes Genom. 2020, 42, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Holck, A.; Va, M.; Didierjean, L.; Rudi, K. 5′-Nuclease PCR for Quantitative Event-Specific Detection of the Genetically Modified Mon810 MaisGard Maize. Eur. Food Res. Technol. 2002, 214, 449–454. [Google Scholar] [CrossRef]
- Holst-Jensen, A.; Bertheau, Y.; de Loose, M.; Grohmann, L.; Hamels, S.; Hougs, L.; Morisset, D.; Pecoraro, S.; Pla, M.; Van den Bulcke, M.; et al. Detecting Un-Authorized Genetically Modified Organisms (GMOs) and Derived Materials. Biotechnol. Adv. 2012, 30, 1318–1335. [Google Scholar] [CrossRef] [PubMed]
- Sim, S.B.; Corpuz, R.L.; Simmonds, T.J.; Geib, S.M. HiFiAdapterFilt, a Memory Efficient Read Processing Pipeline, Prevents Occurrence of Adapter Sequence in PacBio HiFi Reads and Their Negative Impacts on Genome Assembly. BMC Genom. 2022, 23, 1–7. [Google Scholar] [CrossRef]
- Wenger, A.M.; Peluso, P.; Rowell, W.J.; Chang, P.C.; Hall, R.J.; Concepcion, G.T.; Ebler, J.; Fungtammasan, A.; Kolesnikov, A.; Olson, N.D.; et al. Accurate Circular Consensus Long-Read Sequencing Improves Variant Detection and Assembly of a Human Genome. Nat. Biotechnol. 2019, 37, 1155–1162. [Google Scholar] [CrossRef]
- Zhang, K.; Song, P.; Dai, P.; Zhang, J.X.; Wu, L.R.; Cheng, L.Y.; Pinto, A.; Kwong, L.; Cabrera, K.; Wen, R.; et al. Cost-Efficient Sequence-Based Nonextensible Oligonucleotide in Real-Time PCR and High-Throughput Sequencing. ACS Sens. 2022, 7, 1165–1174. [Google Scholar] [CrossRef]
- Pel, J.; Broemeling, D.; Mai, L.; Poon, H.L.; Tropini, G.; Warren, R.L.; Holt, R.A.; Marziali, A. Nonlinear Electrophoretic Response Yields a Unique Parameter for Separation of Biomolecules. Proc. Natl. Acad. Sci. USA 2009, 106, 14796–14801. [Google Scholar] [CrossRef] [PubMed]
- Kidess, E.; Heirich, K.; Wiggin, M.; Vysotskaia, V.; Visser, B.C.; Marziali, A.; Wiedenmann, B.; Norton, J.A.; Lee, M.; Jeffrey, S.S.; et al. Mutation Profiling of Tumor DNA from Plasma and Tumor Tissue of Colorectal Cancer Patients with a Novel, High-Sensitivity Multiplexed Mutation Detection Platform. Oncotarget 2015, 6, 2549–2561. [Google Scholar] [CrossRef]
- Song, C.; Liu, Y.; Fontana, R.; Makrigiorgos, A.; Mamon, H.; Kulke, M.H.; Makrigiorgos, G.M. Elimination of Unaltered DNA in Mixed Clinical Samples via Nuclease-Assisted Minor-Allele Enrichment. Nucleic Acids Res. 2016, 44, e146. [Google Scholar] [CrossRef]
- Arcila, M.; Lau, C.; Nafa, K.; Ladanyi, M. Detection of KRAS and BRAF Mutations in Colorectal Carcinoma: Roles for High-Sensitivity Locked Nucleic Acid-PCR Sequencing and Broad-Spectrum Mass Spectrometry Genotyping. J. Mol. Diagn. 2011, 13, 64–73. [Google Scholar] [CrossRef]
- Zhang, H.; Li, R.; Guo, Y.; Zhang, Y.; Zhang, D.; Yang, L. LIFE-Seq: A Universal Large Integrated DNA Fragment Enrichment Sequencing Strategy for Deciphering the Transgene Integration of Genetically Modified Organisms. Plant Biotechnol. J. 2022, 20, 964–976. [Google Scholar] [CrossRef]
- Zhou, X.; Zhou, L.; Qian, F.; Chen, J.; Zhang, Y.; Yu, Z.; Zhang, J.; Yang, Y.; Li, Y.; Song, C.; et al. TFTG: A Comprehensive Database for Human Transcription Factors and Their Targets. Comput. Struct. Biotechnol. J. 2024, 23, 1877–1885. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, Z.; Tan, K.; Huang, W.; Shi, J.; Li, T.; Hu, J.; Wang, K.; Wang, C.; Xin, B.; et al. A Complete Telomere-to-Telomere Assembly of the Maize Genome. Nat. Genet. 2023, 55, 1221–1231. [Google Scholar] [CrossRef]
- Nurk, S.; Walenz, B.P.; Rhie, A.; Vollger, M.R.; Logsdon, G.A.; Grothe, R.; Miga, K.H.; Eichler, E.E.; Phillippy, A.M.; Koren, S. HiCanu: Accurate Assembly of Segmental Duplications, Satellites, and Allelic Variants from High-Fidelity Long Reads. Genome Res. 2020, 30, 1291–1305. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.N.; Gonzalez, C. Minimap: An Interactive Dynamic Decision Making Game for Search and Rescue Missions. Behav. Res. Methods 2024, 56, 2311–2332. [Google Scholar] [CrossRef]
- Delcher, A.L.; Salzberg, S.L.; Phillippy, A.M. Using MUMmer to Identify Similar Regions in Large Sequence Sets. Curr. Protoc. Bioinform. 2003, 1–18. [Google Scholar] [CrossRef]
- Cock, J.M.; Sterck, L.; Rouzé, P.; Scornet, D.; Allen, A.E.; Amoutzias, G.; Anthouard, V.; Artiguenave, F.; Aury, J.M.; Badger, J.H.; et al. The Ectocarpus Genome and the Independent Evolution of Multicellularity in Brown Algae. Nature 2010, 465, 617–621. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Wu, Z.; Wang, T.; Chen, J.; Zhang, Y.; Lv, G. Sweet Corn Association Panel and Genome-Wide Association Analysis Reveal Loci for Chilling-Tolerant Germination. Sci. Rep. 2024, 14, 10791. [Google Scholar] [CrossRef]
- Arpaia, S.; Christiaens, O.; Giddings, K.; Jones, H.; Mezzetti, B.; Moronta-Barrios, F.; Perry, J.N.; Sweet, J.B.; Taning, C.N.T.; Smagghe, G.; et al. Biosafety of GM Crop Plants Expressing DsRNA: Data Requirements and EU Regulatory Considerations. Front. Plant Sci. 2020, 11, 940. [Google Scholar] [CrossRef] [PubMed]
- Neumann, G.; Brandes, C.; Joachimsthaler, A.; Hochegger, R. Assessment of the Genetic Stability of GMOs with a Detailed Examination of MON810 Using Scorpion Probes. Eur. Food Res. Technol. 2011, 233, 19–30. [Google Scholar] [CrossRef]
- Rosati, A.; Bogani, P.; Santarlasci, A.; Buiatti, M. Characterisation of 3′ Transgene Insertion Site and Derived MRNAs in MON810 YieldGard® Maize. Plant Mol. Biol. 2008, 67, 271–281. [Google Scholar] [CrossRef]
- Wu, L.R.; Chen, S.X.; Wu, Y.; Patel, A.A.; Zhang, D.Y. Multiplexed Enrichment of Rare DNA Variants via Sequence-Selective and Temperature-Robust Amplification. Nat. Biomed. Eng. 2017, 1, 714–723. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assay | Target | Primer | Sequence (5′–3′) | Product (bp) |
|---|---|---|---|---|
| MON810 full length | The full length of MON810 | MON810_1F | ATGACCAGGGGTACGTTCGATA | 5015 |
| MON810_1R | CGTTGAGCAATCAAAGCGTGAG | |||
| MON810_2F | TCTGCGACTTCTTCAGCTGTTC | 5107 | ||
| MON810_2R | AGTCACTAGGTGGTTGGAGTGA | |||
| MON810_3F | GAGCAACGTCTACTTCGCGTAA | 4935 | ||
| MON810_3R | GACTACACAATCACTTGGCCGT | |||
| MON810_4F | TACGGAGTCCAAAAGTTGCCG | 4929 | ||
| MON810_4R | TTTCGGGCGAAGGTTATGAAGG | |||
| MON810_5F | CGGCTTCTGAAGGTCCTCAAAA | 5169 | ||
| MON810_5R | ATACTTCCCGGCGGATACTGAT | |||
| MON810_6F | AAGTCAGACGAGACCCTCCAAT | 5039 | ||
| MON810_6R | GATTCAATCCCAGGCGTTAGCT | |||
| MON810_7F | TGTTGAGGACCGCTCTTTCAAG | 5201 | ||
| MON810_7R | CCCTGCGATAAAGTTAGCCCAT | |||
| MON810_8F | AGCTAAGGGGGTTAAACAACTTGT | 4930 | ||
| MON810_8R | AGGTCTGAGTTGGCGTGAGATA | |||
| Insertion DL | Detection for Insertion site | G3-4R2 | CTTTACCACAAGAGATAAGG | 235/370 |
| 810-9042F | AGAGACGAATAAGCAAGTTAGC | |||
| Gene specific | SNP for AT type | 810-19041F | CTTCGGTACGCGCTCACTCC | GG 88/AT 92 |
| Construct specific | 35S-8F | TAAGGGATGACGCACAATCC | GG 200/AT 204 | |
| Event specific | G5-1F | TATGTCCTTCATAACCTTCG | GG 513/AT 517 | |
| AT type | 810-AT | GCTAAACCACTCTCAGCAATCAAT | / | |
| GG type | 810-GG | AACCACTCTCAGCAATCACC | ||
| BDA | BDA method | 810-18907F | TGATGTGATATCTCCACTGACG | / |
| 810-19133R | AGCTAAACCACTCTCAGCAATCA | 227 | ||
| 810B-19120R | TCAGCAATCACCACACAAGAGAGCAAAAA | 214 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, C.; Zhang, Y.; Yu, H.; Chen, X.; Xie, J. An Integrated Strategy for Analyzing the Complete Complex Integrated Structure of Maize MON810 and Identification of an SNP in External Insertion Sequences. Plants 2024, 13, 2276. https://doi.org/10.3390/plants13162276
Huang C, Zhang Y, Yu H, Chen X, Xie J. An Integrated Strategy for Analyzing the Complete Complex Integrated Structure of Maize MON810 and Identification of an SNP in External Insertion Sequences. Plants. 2024; 13(16):2276. https://doi.org/10.3390/plants13162276
Chicago/Turabian StyleHuang, Chunmeng, Yongjun Zhang, Huilin Yu, Xiuping Chen, and Jiajian Xie. 2024. "An Integrated Strategy for Analyzing the Complete Complex Integrated Structure of Maize MON810 and Identification of an SNP in External Insertion Sequences" Plants 13, no. 16: 2276. https://doi.org/10.3390/plants13162276