
Co-Expression Network Analysis and Introgressive Gene Identification for Fiber Length and Strength Reveal Transcriptional Differences in 15 Cotton Chromosome Substitution Segment Lines and Their Upland and Sea Island Parents

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Phenotypic Evaluation and Statistical Analysis of Cotton Fiber Quality
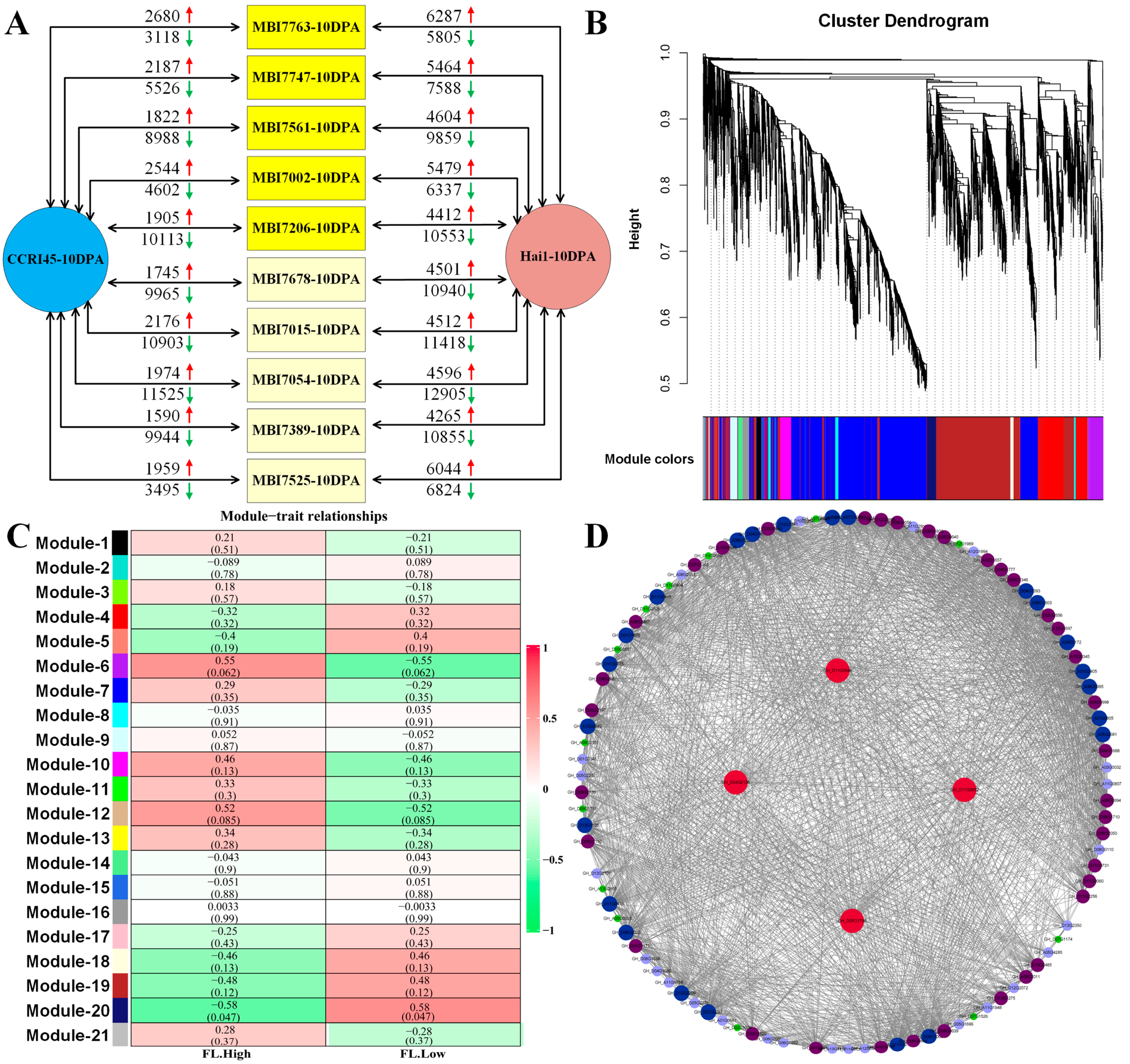
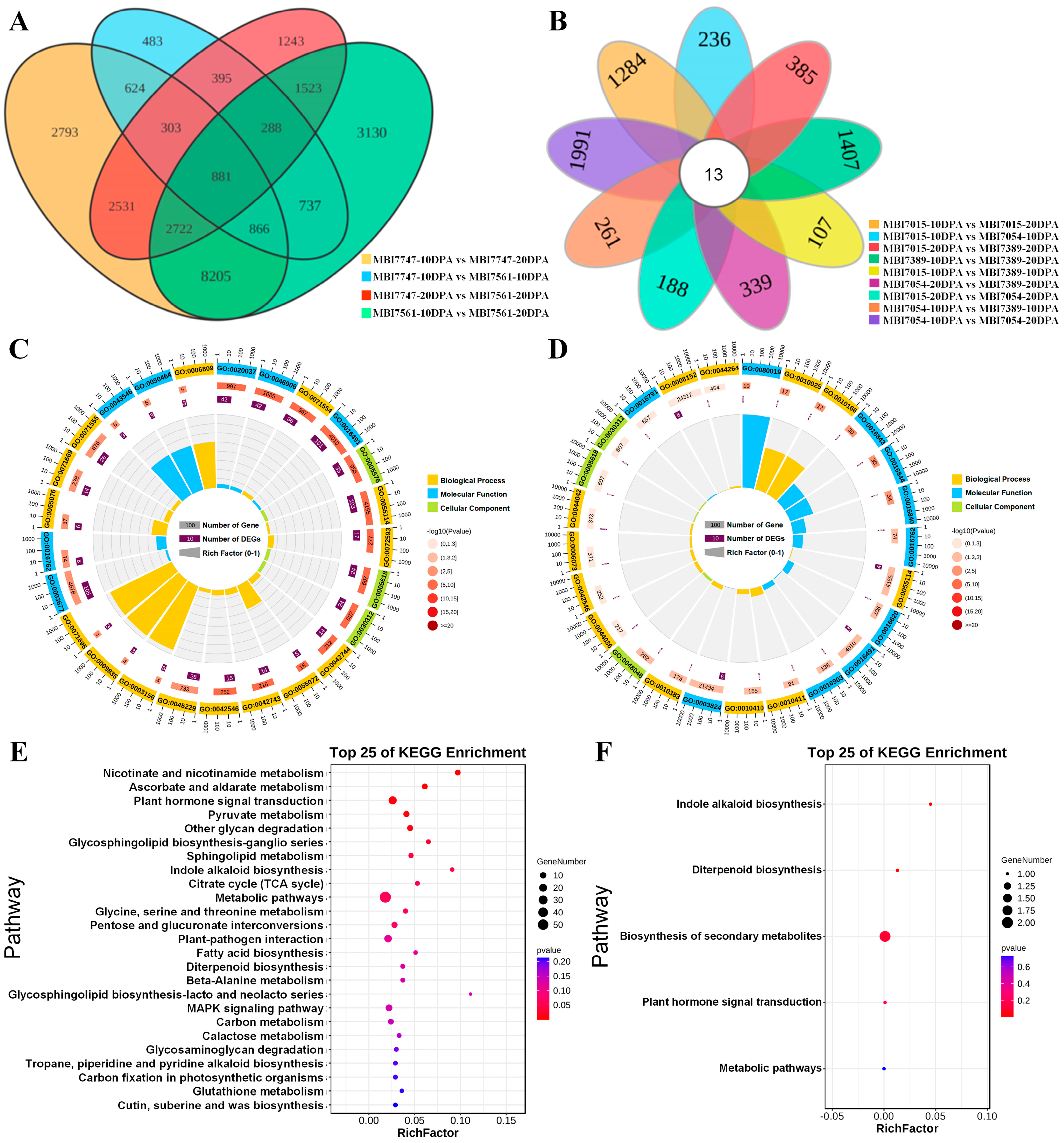
2.2. RNA-Seq, Enrichment Analyses, and WGCNA of FL Related DEGs
2.3. RNA-Seq, Enrichment Analyses, and WGCNA of FS Related DEGs
2.4. RNA-Seq, Enrichment Analyses, and WGCNA of Fiber Quality-Related DEGs
2.5. Prediction and Enrichment Analysis of Introgressed DEGs in CSSLs
2.6. Conjoint Analyses of FQ-Related QTL and Introgressive DEGs
3. Discussion
4. Materials and Methods
4.1. Cotton Materials and Phenotypic Statistics
4.2. Plant Cultivation and Sample Collection
4.3. RNA Extraction and Library Sequencing
4.4. Transcriptome Sequencing and Data Analysis
4.5. Construction of Weight Gene Co-Expressed Network
4.6. Prediction of Introgressive DEGs in CSSLs
4.7. Combination of QTL Intervals and Introgressive DEGs in CSSLs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wen, X.; Chen, Z.; Yang, Z.; Wang, M.; Jin, S.; Wang, G.; Zhang, L.; Wang, L.; Li, J.; Saeed, S.; et al. A comprehensive overview of cotton genomics, biotechnology and molecular biological studies. Sci. China Life Sci. 2023, 66, 2214–2256. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Shang, X.; He, Q.; Zhu, L.; Li, W.; Song, X.; Guo, W. LIPID TRANSFER PROTEIN4 regulates cotton ceramide content and activates fiber cell elongation. Plant Physiol. 2023, 193, 1816–1833. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Guo, H.; Zhang, Q.; Guo, H.; Zhang, L.; Zhang, C.; Gou, Z.; Liu, Y.; Wei, J.; Chen, A.; et al. Arbuscular mycorrhizal fungi (AMF) enhanced the growth, yield, fiber quality and phosphorus regulation in upland cotton (Gossypium hirsutum L.). Sci. Rep. 2020, 10, 2084. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Sun, M.; Li, W.; Xu, M.; Shao, L.; Liu, Y.; Zhao, G.; Liu, Z.; Xu, Z.; You, J.; et al. Single-cell RNA-seq reveals fate determination control of an individual fibre cell initiation in cotton (Gossypium hirsutum). Plant Biotechnol. J. 2022, 20, 2372–2388. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Woodward, A.W.; Chen, Z.J. Gene expression changes and early events in cotton fibre development. Ann. Bot. 2007, 100, 1391–1401. [Google Scholar] [CrossRef] [PubMed]
- Haigler, C.H.; Betancur, L.; Stiff, M.R.; Tuttle, J.R. Cotton fiber: A powerful single-cell model for cell wall and cellulose research. Front. Plant Sci. 2012, 3, 104. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.M.; Zhu, Y.X. How cotton fibers elongate: A tale of linear cell-growth mode. Curr. Opin. Plant Biol. 2011, 14, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Y.L.; Llewellyn, D.J.; Furbank, R.T. The control of single-celled cotton fiber elongation by developmentally reversible gating of plasmodesmata and coordinated expression of sucrose and K+ transporters and expansin. Plant Cell. 2001, 13, 47–60. [Google Scholar] [PubMed]
- Naoumkina, M.; Thyssen, G.N.; Fang, D.D. RNA-seq analysis of short fiber mutants Ligon-lintless-1 (Li 1) and -2 (Li 2) revealed important role of aquaporins in cotton (Gossypium hirsutum L.) fiber elongation. BMC Plant Biol. 2015, 15, 65. [Google Scholar] [CrossRef]
- Salih, H.; Gong, W.; He, S.; Xia, W.; Odongo, M.R.; Du, X. Long non-coding RNAs and their potential functions in Ligon-lintless-1 mutant cotton during fiber development. BMC Genom. 2019, 20, 661. [Google Scholar] [CrossRef]
- Li, P.T.; Wang, M.; Lu, Q.W.; Ge, Q.; Rashid, M.H.O.; Liu, A.Y.; Gong, J.W.; Shang, H.H.; Gong, W.K.; Li, J.W.; et al. Comparative transcriptome analysis of cotton fiber development of Upland cotton (Gossypium hirsutum) and Chromosome Segment Substitution Lines from G. hirsutum × G. barbadense. BMC Genom. 2017, 18, 705. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Xiao, X.; Gong, J.; Li, P.; Zhao, Y.; Feng, J.; Peng, R.; Shi, Y.; Yuan, Y. Identification of candidate cotton genes associated with fiber length through quantitative trait loci mapping and RNA-sequencing using a chromosome segment substitution line. Front. Plant Sci. 2021, 12, 796722. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.H.; Chen, Y.; Wang, N.N.; Chen, Y.H.; Wei, N.; Lu, R.; Li, Y.; Li, X.B. A basic helix-loop-helix protein (GhFP1) promotes fibre elongation of cotton (Gossypium hirsutum) by modulating brassinosteroid biosynthesis and signalling. New Phytol. 2020, 225, 2439–2452. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Chen, F.; Guo, Y.; Gan, X.; Yang, M.; Zeng, W.; Persson, S.; Li, J.; Xu, W. GhMYB7 promotes secondary wall cellulose deposition in cotton fibres by regulating GhCesA gene expression through three distinct cis-elements. New Phytol. 2021, 232, 1718–1737. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.N.; Li, Y.; Chen, Y.H.; Lu, R.; Zhou, L.; Wang, Y.; Zheng, Y.; Li, X.B. Phosphorylation of WRKY16 by MPK3-1 is essential for its transcriptional activity during fiber initiation and elongation in cotton (Gossypium hirsutum). Plant Cell. 2021, 33, 2736–2752. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y. The actin cytoskeleton and signaling network during pollen tube tip growth. J. Integr. Plant Biol. 2010, 52, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.X.; Chen, Y.; Zeng, W.; Li, Y.; Gao, L.; Li, D.D.; Bacic, A.; Xu, W.L.; Li, X.B. The cotton β-galactosyltransferase 1 (GalT1) that galactosylates arabinogalactan proteins participates in controlling fiber development. Plant J. 2017, 89, 957–971. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Zhao, P.; Zhang, Y. A pivotal role of hormones in regulating cotton fiber development. Front. Plant Sci. 2019, 10, 87. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Liu, Z.; Ge, X.; Lu, L.; Qin, W.; Qanmber, G.; Liu, L.; Wang, Z.; Li, F. Brassinosteroids regulate cotton fiber elongation by modulating very-long-chain fatty acid biosynthesis. Plant Cell. 2023, 35, 2114–2131. [Google Scholar] [CrossRef]
- Niu, E.; Shang, X.; Cheng, C.; Bao, J.; Zeng, Y.; Cai, C.; Du, X.; Guo, W. Comprehensive analysis of the COBRA-like (COBL) gene family in Gossypium identifies two COBLs potentially associated with fiber quality. PLoS ONE 2015, 10, e0145725. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.; He, S.P.; Xu, S.W.; Li, L.; Zheng, Y.; Li, X.B. The transcription factor ERF108 interacts with AUXIN RESPONSE FACTORs to mediate cotton fiber secondary cell wall biosynthesis. Plant Cell. 2023, 35, 4133–4154. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Cao, H.; Xi, J.; Zeng, J.; Huang, J.; Li, B.; Song, S.; Zhao, J.; Pei, Y. Auxin directly upregulates GhRAC13 expression to promote the onset of secondary cell wall deposition in cotton fibers. Front. Plant Sci. 2020, 11, 581983. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Q.; Liu, A.Y.; Kong, L.L.; Gong, J.W.; Li, J.W.; Gong, W.K.; Lu, Q.W.; Li, P.T.; Ge, Q.; Shang, H.H.; et al. QTL mapping and genetic effect of chromosome segment substitution lines with excellent fiber quality from Gossypium hirsutum × Gossypium barbadense. Mol. Genet. Genom. 2019, 294, 1123–1136. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Kong, L.; Xiao, X.; Li, P.; Liu, A.; Li, J.; Gong, J.; Gong, W.; Ge, Q.; Shang, H.; et al. Genome-wide artificial introgressions of Gossypium barbadense into G. hirsutum reveal superior loci for simultaneous improvement of cotton fiber quality and yield traits. J. Adv. Res. 2023, 53, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Liu, A.; Li, J.; Zhang, J.; Li, S.; Zhang, J.; Ma, L.; He, R.; Song, W.; Guo, L.; et al. Examining two sets of introgression lines across multiple environments reveals background-independent and stably expressed quantitative trait loci of fiber quality in cotton. Theor. Appl. Genet. 2020, 133, 2075–2093. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Li, P.; Yang, R.; Xiao, X.; Li, Z.; Wu, Q.; Gong, J.; Ge, Q.; Liu, A.; Du, S.; et al. QTL mapping and candidate gene prediction for fiber yield and quality traits in a high-generation cotton chromosome substitution line with Gossypium barbadense segments. Mol. Genet. Genom. 2022, 297, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, D.; Surapaneni, M.; Mesapogu, S.; Neelamraju, S. Development and use of chromosome segment substitution lines as a genetic resource for crop improvement. Theor. Appl. Genet. 2019, 132, 1–25. [Google Scholar] [CrossRef]
- Liang, P.; Wang, H.; Zhang, Q.; Zhou, K.; Li, M.; Li, R.; Xiang, S.; Zhang, T.; Ling, Y.; Yang, Z.; et al. Identification and pyramiding of QTLs for rice grain size based on short-wide grain CSSL-Z563 and fine-mapping of qGL3-2. Rice 2021, 14, 35. [Google Scholar] [CrossRef]
- Xia, Y.; Wang, B.; Zhu, L.; Wu, W.; Sun, S.; Zhu, Z.; Li, X.; Weng, J.; Duan, C. Identification of a Fusarium ear rot resistance gene in maize by QTL mapping and RNA sequencing. Front. Plant Sci. 2022, 13, 954546. [Google Scholar] [CrossRef]
- Ye, G.; Liang, S.; Wan, J. QTL mapping of protein content in rice using single chromosome segment substitution lines. Theor. Appl. Genet. 2010, 121, 741–750. [Google Scholar] [CrossRef]
- Li, B.; Shi, Y.; Gong, J.; Li, J.; Liu, A.; Shang, H.; Gong, W.; Chen, T.; Ge, Q.; Jia, C.; et al. Genetic effects and heterosis of yield and yield component traits based on Gossypium barbadense chromosome segment substitution lines in two Gossypium hirsutum backgrounds. PLoS ONE 2016, 11, e0157978. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Y.L. Sucrose metabolism: Gateway to diverse carbon use and sugar signaling. Annu. Rev. Plant Biol. 2014, 65, 33–67. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kumar, K.; Pandey, P.; Rajamani, V.; Padmalatha, K.V.; Dhandapani, G.; Kanakachari, M.; Leelavathi, S.; Kumar, P.A.; Reddy, V.S. Glycoproteome of elongating cotton fiber cells. Mol. Cell Proteom. 2013, 12, 3677–3689. [Google Scholar] [CrossRef] [PubMed]
- Maris, A.; Kaewthai, N.; Eklöf, J.M.; Miller, J.G.; Brumer, H.; Fry, S.C.; Verbelen, J.P.; Vissenberg, K. Differences in enzymic properties of five recombinant xyloglucan endotransglucosylase/hydrolase (XTH) proteins of Arabidopsis thaliana. J. Exp. Bot. 2011, 62, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Zhang, X.; Yao, W.; Gao, Y.; Zhao, K.; Guo, Q.; Zhou, B.; Jiang, T. Genome-wide identification and expression analysis of the xyloglucan endotransglucosylase/hydrolase gene family in poplar. BMC Genom. 2021, 22, 804. [Google Scholar] [CrossRef] [PubMed]
- Bajwa, K.S.; Shahid, A.A.; Rao, A.Q.; Bashir, A.; Aftab, A.; Husnain, T. Stable transformation and expression of GhEXPA8 fiber expansin gene to improve fiber length and micronaire value in cotton. Front. Plant Sci. 2015, 6, 838. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Xiao, S.; Zuo, D.; Cheng, H.; Zhang, Y.; Wang, Q.; Lv, L.; Song, G. Genome-wide analysis elucidates the roles of GhHMA genes in different abiotic stresses and fiber development in upland cotton. Plant Physiol. Biochem. 2023, 194, 281–301. [Google Scholar] [CrossRef] [PubMed]
- Xue, T.; Liu, L.; Zhang, X.; Li, Z.; Sheng, M.; Ge, X.; Xu, W.; Su, Z. Genome-wide investigation and co-expression network analysis of SBT family gene in Gossypium. Int. J. Mol. Sci. 2023, 24, 5760. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Zheng, C.; Feng, W.; Shao, J.; Pang, C.; Li, P.; Dong, H. Low soil available phosphorus level reduces cotton fiber length via osmoregulation. Front. Plant Sci. 2023, 14, 1254103. [Google Scholar] [CrossRef]
- Yu, D.; Li, X.; Li, Y.; Ali, F.; Li, F.; Wang, Z. Dynamic roles and intricate mechanisms of ethylene in epidermal hair development in Arabidopsis and cotton. New Phytol. 2022, 234, 375–391. [Google Scholar] [CrossRef]
- Li, S.; Geng, S.; Pang, B.; Zhao, J.; Huang, Y.; Rui, C.; Cui, J.; Jiao, Y.; Zhang, R.; Gao, W. Revealing genetic differences in fiber elongation between the offspring of sea island cotton and upland cotton backcross populations based on transcriptome and weighted gene coexpression networks. Genes 2022, 13, 954. [Google Scholar] [CrossRef]
- Guo, H.; Guo, H.; Zhang, L.; Tang, Z.; Yu, X.; Wu, J.; Zeng, F. Metabolome and transcriptome association analysis reveals dynamic regulation of purine metabolism and flavonoid synthesis in transdifferentiation during somatic embryogenesis in cotton. Int. J. Mol. Sci. 2019, 20, 2070. [Google Scholar] [CrossRef]
- Baillo, E.H.; Kimotho, R.N.; Zhang, Z.; Xu, P. Transcription factors associated with abiotic and biotic stress tolerance and their potential for crops improvement. Genes 2019, 10, 771. [Google Scholar] [CrossRef]
- Wang, L.; Guo, D.; Zhao, G.; Wang, J.; Zhang, S.; Wang, C.; Guo, X. Group IIc WRKY transcription factors regulate cotton resistance to Fusarium oxysporum by promoting GhMKK2-mediated flavonoid biosynthesis. New Phytol. 2022, 236, 249–265. [Google Scholar] [CrossRef]
- Ding, M.; Chen, J.; Jiang, Y.; Lin, L.; Cao, Y.; Wang, M.; Zhang, Y.; Rong, J.; Ye, W. Genome-wide investigation and transcriptome analysis of the WRKY gene family in Gossypium. Mol. Genet. Genom. 2015, 290, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, B.; Qin, G.; Zhang, Z.; Tian, S. Identification and functional characterization of a tonoplast dicarboxylate transporter in tomato (Solanum lycopersicum). Front. Plant Sci. 2017, 8, 186. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Guo, L.; Lu, X.; Malik, W.A.; Zhang, Y.; Wang, J.; Chen, X.; Wang, S.; Wang, J.; Wang, D.; et al. Structure and character analysis of cotton response regulator genes family reveals that GhRR7 responses to draught stress. Biol. Res. 2022, 55, 27. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhao, W.; Yao, W.; Wang, Y.; Jiang, T.; Liu, H. Genome-wide analysis of strictosidine synthase-like gene family revealed their response to biotic/abiotic stress in poplar. Int. J. Mol. Sci. 2023, 24, 10117. [Google Scholar] [CrossRef]
- Yang, Y.; Lai, W.; Long, L.; Gao, W.; Xu, F.; Li, P.; Zhou, S.; Ding, Y.; Hu, H. Comparative proteomic analysis identified proteins and the phenylpropanoid biosynthesis pathway involved in the response to ABA treatment in cotton fiber development. Sci. Rep. 2023, 13, 1488. [Google Scholar] [CrossRef]
- Zhang, J.; Mei, H.; Lu, H.; Chen, R.; Hu, Y.; Zhang, T. Transcriptome time-course analysis in the whole period of cotton fiber development. Front. Plant Sci. 2022, 13, 864529. [Google Scholar] [CrossRef]
- Matsuba, Y.; Zi, J.; Jones, A.D.; Peters, R.J.; Pichersky, E. Biosynthesis of the diterpenoid lycosantalonol via nerylneryl diphosphate in Solanum lycopersicum. PLoS ONE 2015, 10, e0119302. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Tian, R.; Chen, J.; Wang, S.; Li, X.; Wang, P.; Zhang, T. Transcriptomic analysis of fiber strength in upland cotton chromosome introgression lines carrying different Gossypium barbadense chromosomal segments. PLoS ONE 2014, 9, e94642. [Google Scholar] [CrossRef] [PubMed]
- Padmalatha, K.V.; Dhandapani, G.; Kanakachari, M.; Kumar, S.; Dass, A.; Patil, D.P.; Rajamani, V.; Kumar, K.; Pathak, R.; Rawat, B.; et al. Genome-wide transcriptomic analysis of cotton under drought stress reveal significant down-regulation of genes and pathways involved in fibre elongation and up-regulation of defense responsive genes. Plant Mol. Biol. 2012, 78, 223–246. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Xiao, Y.; Li, X.; Lu, X.; Deng, W.; Li, D.; Hou, L.; Hu, M.; Li, Y.; Pei, Y. GhDET2, a steroid 5alpha-reductase, plays an important role in cotton fiber cell initiation and elongation. Plant J. 2007, 51, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.H.; Saha, S.; Jenkins, J.N.; Ma, D.P. Characterization and promoter analysis of a cotton RING-type ubiquitin ligase (E3) gene. Mol. Biotechnol. 2010, 46, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Khodaeiaminjan, M.; Gomes, C.; Pagano, A.; Kruszka, D.; Sulima, P.; Przyborowski, J.A.; Krajewski, P.; Paiva, J.A.P. Impacts of in-vitro zebularine treatment on genome-wide DNA methylation and transcriptomic profiles in Salix purpurea L. Physiol. Plant. 2024, 176, e14403. [Google Scholar] [CrossRef] [PubMed]
- Kurth, E.G.; Peremyslov, V.V.; Turner, H.L.; Makarova, K.S.; Iranzo, J.; Mekhedov, S.L.; Koonin, E.V.; Dolja, V.V. Myosin-driven transport network in plants. Proc. Natl. Acad. Sci. USA 2017, 114, E1385–E1394. [Google Scholar] [CrossRef]
- Xu, Y.; Magwanga, R.O.; Cai, X.; Zhou, Z.; Wang, X.; Wang, Y.; Zhang, Z.; Jin, D.; Guo, X.; Wei, Y.; et al. Deep Transcriptome analysis reveals reactive oxygen species (ROS) network evolution, response to abiotic stress, and regulation of fiber development in cotton. Int. J. Mol. Sci. 2019, 20, 1863. [Google Scholar] [CrossRef]
- Wei, Z.; Li, Y.; Ali, F.; Wang, Y.; Liu, J.; Yang, Z.; Wang, Z.; Xing, Y.; Li, F. Transcriptomic analysis reveals the key role of histone deacetylation via mediating different phytohormone signalings in fiber initiation of cotton. Cell Biosci. 2022, 12, 107. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, J.Y. Cotton cytosolic pyruvate kinase GhPK6 participates in fast fiber elongation regulation in a ROS-mediated manner. Planta 2016, 244, 915–926. [Google Scholar] [CrossRef]
- Andleeb, A.; Dikici, S.; Waris, T.S.; Bashir, M.M.; Akhter, S.; Chaudhry, A.A.; MacNeil, S.; Yar, M. Developing affordable and accessible pro-angiogenic wound dressings; incorporation of 2 deoxy D-ribose (2dDR) into cotton fibres and wax-coated cotton fibres. J. Tissue Eng. Regen. Med. 2020, 14, 973–988. [Google Scholar] [CrossRef]
- Chen, B.; Sun, Y.; Tian, Z.; Fu, G.; Pei, X.; Pan, Z.; Nazir, M.F.; Song, S.; Li, H.; Wang, X.; et al. GhGASA10-1 promotes the cell elongation in fiber development through the phytohormones IAA-induced. BMC Plant Biol. 2021, 21, 448. [Google Scholar] [CrossRef]
- Tian, Y.; Zhang, T. MIXTAs and phytohormones orchestrate cotton fiber development. Curr. Opin. Plant Biol. 2021, 59, 101975. [Google Scholar] [CrossRef]
- Tang, W.; Tu, L.; Yang, X.; Tan, J.; Deng, F.; Hao, J.; Guo, K.; Lindsey, K.; Zhang, X. The calcium sensor GhCaM7 promotes cotton fiber elongation by modulating reactive oxygen species (ROS) production. New Phytol. 2014, 202, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Lian, B.; Hao, P.; Fu, X.; Zhang, M.; Lu, J.; Ma, L.; Yu, S.; Wei, H.; Wang, H. GhMYB30-GhMUR3 affects fiber elongation and secondary wall thickening in cotton. Plant J. 2024, 117, 694–712. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Mo, H.; Xu, R. Arginine decarboxylase gene ADC2 regulates fiber elongation in cotton. Genes 2022, 13, 784. [Google Scholar] [CrossRef]
- Pang, C.Y.; Wang, H.; Pang, Y.; Xu, C.; Jiao, Y.; Qin, Y.M.; Western, T.L.; Yu, S.X.; Zhu, Y.X. Comparative proteomics indicates that biosynthesis of pectic precursors is important for cotton fiber and Arabidopsis root hair elongation. Mol. Cell Proteom. 2010, 9, 2019–2033. [Google Scholar] [CrossRef]
- Zhu, H.; Hu, W.; Li, Y.; Zou, J.; He, J.; Wang, Y.; Meng, Y.; Chen, B.; Zhao, W.; Wang, S.; et al. Drought decreases cotton fiber strength by altering sucrose flow route. J. Exp. Bot. 2024, 75, 3579–3595. [Google Scholar] [CrossRef]
- Wang, L.; Cheng, H.; Xiong, F.; Ma, S.; Zheng, L.; Song, Y.; Deng, K.; Wu, H.; Li, F.; Yang, Z. Comparative phosphoproteomic analysis of BR-defective mutant reveals a key role of GhSK13 in regulating cotton fiber development. Sci. China Life Sci. 2020, 63, 1905–1917. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wen, J.; Lease, K.A.; Doke, J.T.; Tax, F.E.; Walker, J.C. BAK1, an Arabidopsis LRR receptor-like protein kinase, interacts with BRI1 and modulates brassinosteroid signaling. Cell 2002, 110, 213–222. [Google Scholar] [CrossRef]
- Yang, L.; Wang, X.; Zhao, F.; Zhang, X.; Li, W.; Huang, J.; Pei, X.; Ren, X.; Liu, Y.; He, K.; et al. Roles of S-adenosylmethionine and its derivatives in salt tolerance of cotton. Int. J. Mol. Sci. 2023, 24, 9517. [Google Scholar] [CrossRef]
- Shi, Z.; Chen, X.; Xue, H.; Jia, T.; Meng, F.; Liu, Y.; Luo, X.; Xiao, G.; Zhu, S. GhBZR3 suppresses cotton fiber elongation by inhibiting very-long-chain fatty acid biosynthesis. Plant J. 2022, 111, 785–799. [Google Scholar] [CrossRef]
- Liu, L.; Chen, G.; Li, S.; Gu, Y.; Lu, L.; Qanmber, G.; Mendu, V.; Liu, Z.; Li, F.; Yang, Z. A brassinosteroid transcriptional regulatory network participates in regulating fiber elongation in cotton. Plant Physiol. 2023, 191, 1985–2000. [Google Scholar] [CrossRef]
- Lu, R.; Li, Y.; Zhang, J.; Wang, Y.; Zhang, J.; Li, Y.; Zheng, Y.; Li, X.B. The bHLH/HLH transcription factors GhFP2 and GhACE1 antagonistically regulate fiber elongation in cotton. Plant Physiol. 2022, 189, 628–643. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, C.; Yang, X.; Liu, K.; Wu, Z.; Zhang, X.; Zheng, W.; Xun, Q.; Liu, C.; Lu, L.; et al. PAG1, a cotton brassinosteroid catabolism gene, modulates fiber elongation. New Phytol. 2014, 203, 437–448. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, H.; Zhu, J.; Wang, X.; Jiang, B.; Hou, L.; Xiao, G. A conserved brassinosteroid-mediated BES1-CERP-EXPA3 signaling cascade controls plant cell elongation. Cell Rep. 2023, 42, 112301. [Google Scholar] [CrossRef]
- Niu, E.; Cai, C.; Zheng, Y.; Shang, X.; Fang, L.; Guo, W. Genome-wide analysis of CrRLK1L gene family in Gossypium and identification of candidate CrRLK1L genes related to fiber development. Mol. Genet. Genom. 2016, 291, 1137–1154. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Gao, Y.; Zhang, Z.; Chen, T.; Guo, W.; Zhang, T. A receptor-like kinase gene (GbRLK) from Gossypium barbadense enhances salinity and drought-stress tolerance in Arabidopsis. BMC Plant Biol. 2013, 13, 110. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Gao, J.; Cao, Z.; Chee, P.W.; Guo, Q.; Xu, Z.; Paterson, A.H.; Zhang, X.; Shen, X. Fine mapping and candidate gene analysis of qFL-chr1, a fiber length QTL in cotton. Theor. Appl. Genet. 2017, 130, 1309–1319. [Google Scholar] [CrossRef]
- Xiao, X.; Liu, R.; Gong, J.; Li, P.; Li, Z.; Gong, W.; Liu, A.; Ge, Q.; Deng, X.; Li, S.; et al. Fine mapping and candidate gene analysis of qFL-A12-5: A fiber length-related QTL introgressed from Gossypium barbadense into Gossypium hirsutum. Theor. Appl. Genet. 2023, 136, 48. [Google Scholar] [CrossRef] [PubMed]
- Li, P.T.; Rashid, M.H.O.; Chen, T.T.; Lu, Q.W.; Ge, Q.; Gong, W.K.; Liu, A.Y.; Gong, J.W.; Shang, H.H.; Deng, X.Y.; et al. Transcriptomic and biochemical analysis of upland cotton (Gossypium hirsutum) and a chromosome segment substitution line from G. hirsutum × G. barbadense in response to Verticillium dahliae infection. BMC Plant Biol. 2019, 19, 19. [Google Scholar]
- Shi, Y.; Zhang, B.; Liu, A.; Li, W.; Li, J.; Lu, Q.; Zhang, Z.; Li, S.; Gong, W.; Shang, H.; et al. Quantitative trait loci analysis of Verticillium wilt resistance in interspecific backcross populations of Gossypium hirsutum × Gossypium barbadense. BMC Genom. 2016, 17, 877. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Chen, J.; Fang, L.; Zhang, Z.; Ma, W.; Niu, Y.; Ju, L.; Deng, J.; Zhao, T.; Lian, J.; et al. Gossypium barbadense and Gossypium hirsutum genomes provide insights into the origin and evolution of allotetraploid cotton. Nat. Genet. 2019, 51, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Li, P.; Cheng, S.; Zhao, Z.; Liu, Y.; Wei, Y.; Lu, Q.; Han, J.; Cai, X.; Zhou, Z.; et al. Protoplast dissociation and transcriptome analysis provides insights to salt stress response in cotton. Int. J. Mol. Sci. 2022, 23, 2845. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Long, Y.; Xu, K.; Zhao, F.; Zhao, J.; Li, S.; Geng, S.; Gao, W.; Sun, P.; Deng, X.; et al. Weighted gene co-expression network analysis reveals hub genes for fuzz development in Gossypium hirsutum. Genes 2023, 14, 208. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | QTL | G. hirsutum ID | G. barbadense ID | Non-Synonymous/Synonymous Mutation or Same Sequences |
|---|---|---|---|---|
| Fiber Length | qFL-C7-3 | GH_A07G1073 | GB_A07G1063 | Non-synonymous |
| GH_A07G1074 | GB_A07G1064 | Non-synonymous | ||
| qFL-C12-5 | GH_A12G1295 | GB_A12G1365 | Non-synonymous | |
| qFL-C13-1 | GH_A13G0163 | GB_A13G0168 | Same sequences | |
| qFL-C13-2 | GH_A13G0454 | GB_A13G0460 | Non-synonymous | |
| GH_A13G0460 | GB_A13G0468 | Non-synonymous | ||
| qFL-C14-2 | GH_D02G0665 | GB_D02G0693 | Non-synonymous | |
| qFL-C14-5 | GH_D02G1983 | GB_D02G2045 | Non-synonymous | |
| qFL-C15-2 | GH_D01G0849 | GB_D01G0882 | Non-synonymous | |
| qFL-C17-2 | GH_D03G0331 | GB_D03G0325 | Non-synonymous | |
| qFL-C21-1 | GH_D11G0612 | GB_D11G0613 | Non-synonymous | |
| qFL-C21-2 | GH_D11G0693 | GB_D11G0698 | Non-synonymous | |
| GH_D11G0711 | GB_D11G0717 | Synonymous | ||
| qFL-C21-4 | GH_D11G2888 | GB_D11G2913 | Non-synonymous | |
| Fiber Strength | qFS-C2-2 | GH_A02G1378 | GB_A02G1399 | Same sequences |
| qFS-C7-4 | GH_A07G1076 | GB_A07G1066 | Non-synonymous | |
| GH_A07G1078 | GB_A07G1068 | Non-synonymous | ||
| qFS-C15-1 | GH_D01G0056 | GB_D01G0054 | Non-synonymous | |
| GH_D01G0067 | GB_D01G0064 | Non-synonymous | ||
| GH_D01G0071 | GB_D01G0068 | Non-synonymous | ||
| qFS-C15-4 | GH_D01G0480 | GB_D01G0498 | Non-synonymous | |
| qFS-C19-1 | GH_D05G0262 | GB_D05G0265 | Non-synonymous | |
| GH_D05G0264 | GB_D05G0267 | Non-synonymous | ||
| qFS-C21-1 | GH_D11G0616 | GB_D11G0417 | Non-synonymous | |
| qFS-C21-5 | GH_D11G2944 | GB_D11G2973 | Non-synonymous | |
| qFS-C25-1 | GH_D06G0090 | GB_D06G0111 | Non-synonymous | |
| GH_D06G0098 | GB_D06G0118 | Synonymous | ||
| Fiber Quality | qFL-C2-5 | GH_A02G1372 | GB_A02G1393 | Same sequences |
| qFS-C7-1 | GH_A07G0378 | GB_A07G0371 | Same sequences | |
| GH_A07G0380 | GB_A07G0373 | Same sequences | ||
| qFS-C7-2 | GH_A07G0464 | GB_A07G0458 | Same sequences | |
| qFS-C9-1 | GH_A09G1695 | GB_A09G1819 | Non-synonymous | |
| qFL-C12-5 | GH_A12G1294 | GB_A12G1364 | Same sequence | |
| qFS-C13-1 | GH_A13G0148 | GB_D13G0149 | Non-synonymous | |
| GH_A13G0165 | GB_A13G0170 | Non-synonymous | ||
| qFL-C13-2 | GH_A13G0456 | GB_A13G0463 | Same sequences | |
| qFL-C15-2 | GH_D01G0853 | GB_D01G0886 | Non-synonymous | |
| qFS-C14-3 | GH_D02G1927 | GB_D02G1995 | Non-synonymous | |
| qFL-C14-5 | GH_D02G1984 | GB_D02G2046 | Non-synonymous | |
| qFS-C19-1 | GH_D05G0268 | GB_D05G0271 | Non-synonymous | |
| qFL-C19-2 | GH_D05G1491 | GB_D05G1497 | Non-synonymous | |
| GH_D05G1492 | GB_D05G1498 | Synonymous | ||
| qFL-C21-2 | GH_D11G0692 | GB_D11G0697 | Non-synonymous | |
| GH_D11G0696 | GB_D11G0701 | Synonymous | ||
| GH_D11G0701 | GB_D11G0706 | Synonymous | ||
| GH_D11G0711 | GB_D11G0717 | Synonymous | ||
| qFS-C21-5 | GH_D11G2942 | GB_D11G2971 | Non-synonymous | |
| qFS-C25-1 | GH_D06G0090 | GB_D06G0111 | Non-synonymous |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, P.; Chen, Y.; Yang, R.; Sun, Z.; Ge, Q.; Xiao, X.; Yang, S.; Li, Y.; Liu, Q.; Zhang, A.; et al. Co-Expression Network Analysis and Introgressive Gene Identification for Fiber Length and Strength Reveal Transcriptional Differences in 15 Cotton Chromosome Substitution Segment Lines and Their Upland and Sea Island Parents. Plants 2024, 13, 2308. https://doi.org/10.3390/plants13162308
Li P, Chen Y, Yang R, Sun Z, Ge Q, Xiao X, Yang S, Li Y, Liu Q, Zhang A, et al. Co-Expression Network Analysis and Introgressive Gene Identification for Fiber Length and Strength Reveal Transcriptional Differences in 15 Cotton Chromosome Substitution Segment Lines and Their Upland and Sea Island Parents. Plants. 2024; 13(16):2308. https://doi.org/10.3390/plants13162308
Chicago/Turabian StyleLi, Pengtao, Yu Chen, Rui Yang, Zhihao Sun, Qun Ge, Xianghui Xiao, Shuhan Yang, Yanfang Li, Qiankun Liu, Aiming Zhang, and et al. 2024. "Co-Expression Network Analysis and Introgressive Gene Identification for Fiber Length and Strength Reveal Transcriptional Differences in 15 Cotton Chromosome Substitution Segment Lines and Their Upland and Sea Island Parents" Plants 13, no. 16: 2308. https://doi.org/10.3390/plants13162308