
Identification of a Major QTL for Seed Protein Content in Cultivated Peanut (Arachis hypogaea L.) Using QTL-Seq
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
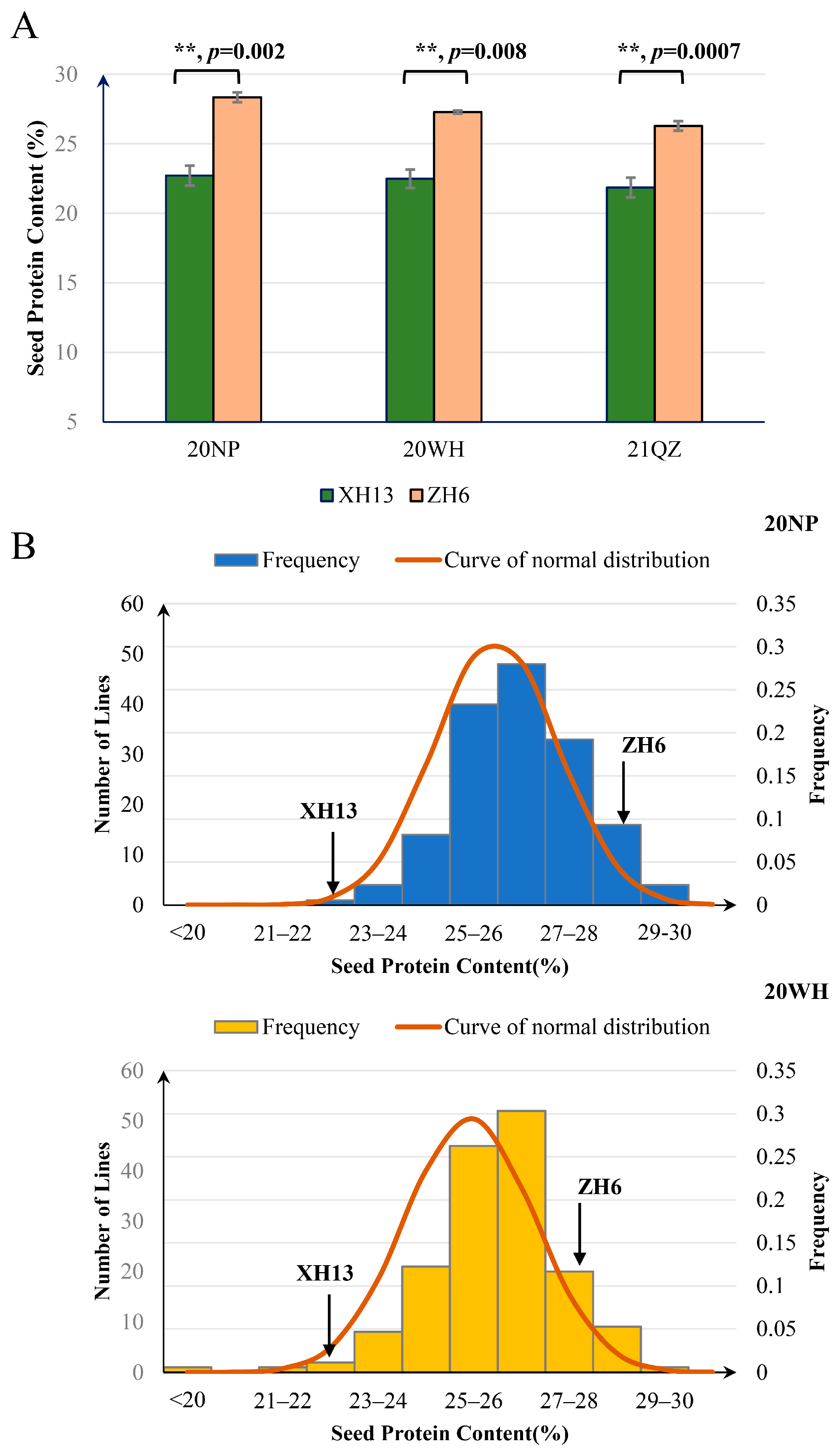
2.1. Phenotypic Variation for SPC in the RIL Population and Its Parental Lines
2.2. QTL-Seq Identification of a QTL Region Controlling SPC
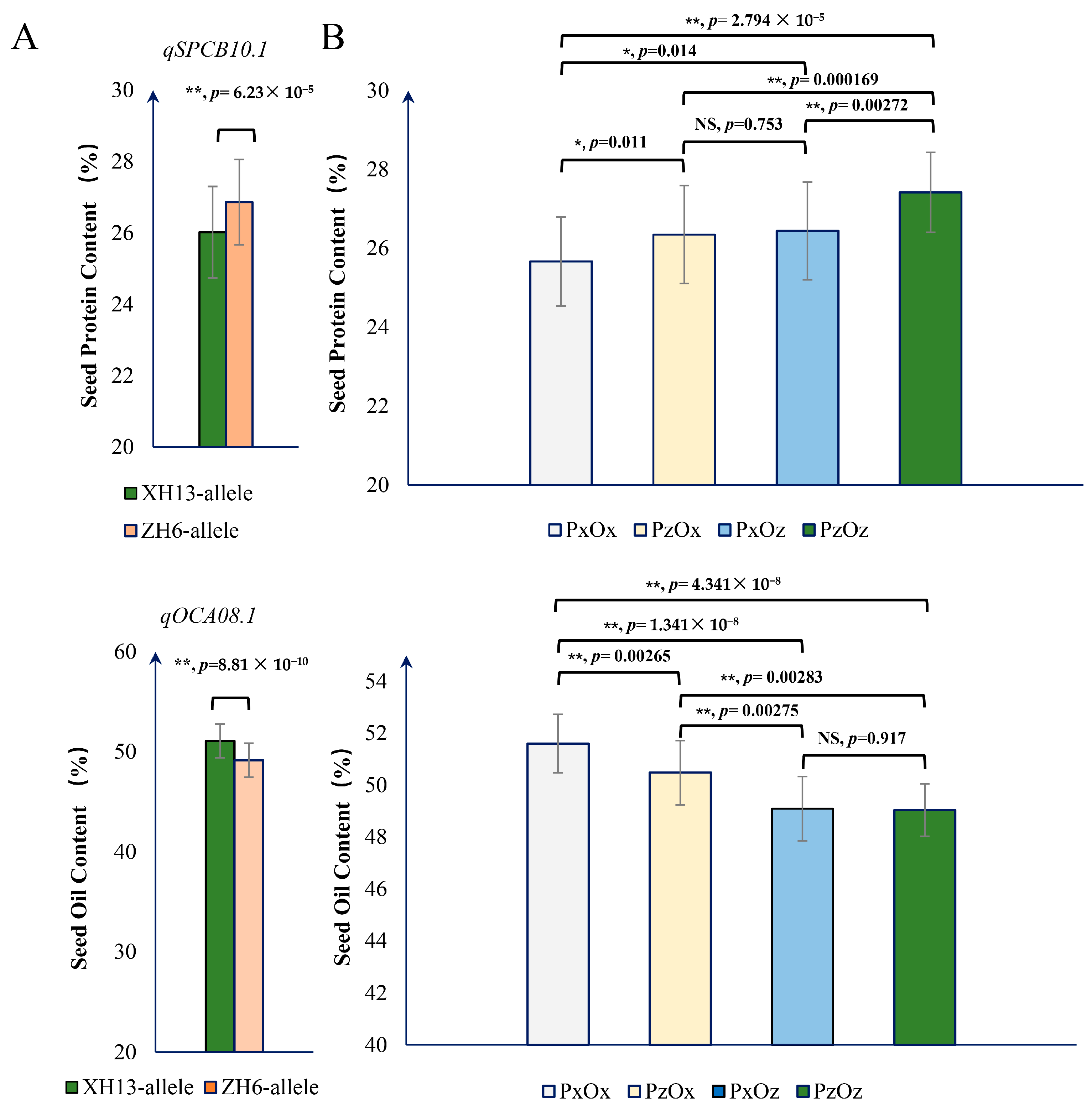
2.3. Gene Candidate Analysis for qSPCB10.1
2.4. Relationship between qSPCB10.1 and qOCA08.1 in SPC and SOC Accumulation
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. Phenotypic Evaluation
4.3. Preparation of DNA Bulks and Illumina Sequencing
4.4. Variant Detection
4.5. BSA-Seq Analysis
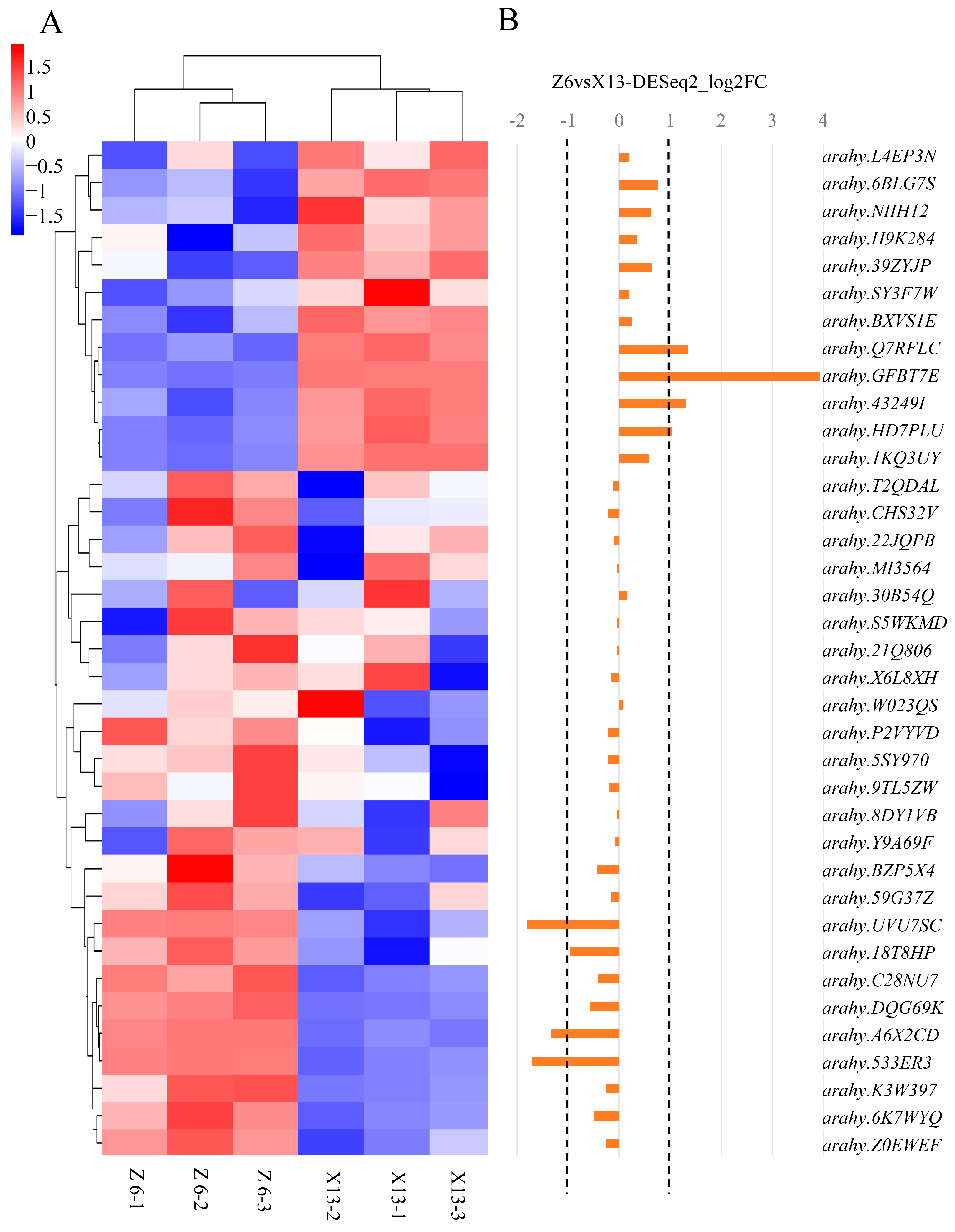
4.6. RNA-Sequencing and Gene Expression Profile Analysis in the Mapped Region
4.7. Marker Development
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Davis, J.P.; Dean, L.L. Peanut composition, flavor and nutrition. In Peanuts; Elsevier: Amsterdam, The Netherlands, 2016; pp. 289–345. [Google Scholar]
- Stephens, A.M.; Dean, L.L.; Davis, J.P.; Osborne, J.A.; Sanders, T.H. Peanuts, peanut oil, and fat free peanut flour reduced cardiovascular disease risk factors and the development of atherosclerosis in Syrian golden hamsters. J. Food Sci. 2010, 75, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Arya, S.S.; Salve, A.R.; Chauhan, S. Peanuts as functional food: A review. J. Food Sci. Technol. 2016, 53, 31–41. [Google Scholar] [CrossRef]
- Settaluri, V.; Kandala, C.; Puppala, N.; Sundaram, J. Peanuts and their nutritional aspects—A review. Food Nutr. Sci. 2012, 3, 25267. [Google Scholar] [CrossRef]
- Javaid, A.; Ghafoor, A.; Anwar, R. Seed storage protein electrophoresis in groundnut for evaluating genetic diversity. Pak. J. Bot. 2004, 36, 25–30. [Google Scholar]
- Patil, G.; Mian, R.; Vuong, T.; Pantalone, V.; Song, Q.; Chen, P.; Shannon, G.J.; Carter, T.C.; Nguyen, H.T. Molecular mapping and genomics of soybean seed protein: A review and perspective for the future. Theor. Appl. Genet. 2017, 130, 1975–1991. [Google Scholar] [CrossRef]
- Dwivedi, S.; Jambunathan, R.; Nigam, S.; Raghunath, K.; Shankar, K.R.; Nagabhushanam, G. Relationship of seed mass to oil and protein contents in peanut (Arachis hypogaea L.). Peanut Sci. 1990, 17, 48–52. [Google Scholar] [CrossRef]
- Sarvamangala, C.; Gowda, M.; Varshney, R. Identification of quantitative trait loci for protein content, oil content and oil quality for groundnut (Arachis hypogaea L.). Field Crops Res. 2011, 122, 49–59. [Google Scholar] [CrossRef]
- Sun, Z.; Qi, F.; Liu, H.; Qin, L.; Xu, J.; Shi, L.; Zhang, Z.; Miao, L.; Huang, B.; Dong, W. QTL mapping of quality traits in peanut using whole-genome resequencing. Crop J. 2022, 10, 177–184. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, M.L.; Dang, P.; Jiang, T.; Zhao, S.; Lamb, M.; Chen, C. Identification of potential QTLs and genes associated with seed composition traits in peanut (Arachis hypogaea L.) using GWAS and RNA-Seq analysis. Gene 2021, 769, 145215. [Google Scholar] [CrossRef]
- Liu, N.; Guo, J.; Zhou, X.; Wu, B.; Huang, L.; Luo, H.; Chen, Y.; Chen, W.; Lei, Y.; Huang, Y. High-resolution mapping of a major and consensus quantitative trait locus for oil content to a~ 0.8-Mb region on chromosome A08 in peanut (Arachis hypogaea L.). Theor. Appl. Genet. 2020, 133, 37–49. [Google Scholar] [CrossRef]
- Bonku, R.; Yu, J. Health aspects of peanuts as an outcome of its chemical composition. Food Sci. Hum. Wellness 2020, 9, 21–30. [Google Scholar] [CrossRef]
- Takagi, H.; Abe, A.; Yoshida, K.; Kosugi, S.; Natsume, S.; Mitsuoka, C.; Uemura, A.; Utsushi, H.; Tamiru, M.; Takuno, S. QTL-seq: Rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 2013, 74, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ma, J.; Li, M.; Deng, L.; Li, G.; Xia, H.; Zhao, S.; Hou, L.; Li, P.; Ma, C. Whole-genome resequencing-based QTL-seq identified AhTc1 gene encoding a R2R3-MYB transcription factor controlling peanut purple testa colour. Plant Biotechnol. J. 2020, 18, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chen, X.; Xu, R.; Liu, W.; Liu, N.; Huang, L.; Luo, H.; Huai, D.; Lan, X.; Zhang, Y. Fine-mapping and gene candidate analysis for AhRt1, a major dominant locus responsible for testa color in cultivated peanut. Theor. Appl. Genet. 2021, 134, 3721–3730. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Huang, L.; Liu, N.; Chen, Y.; Guo, J.; Yu, B.; Luo, H.; Zhou, X.; Huai, D.; Chen, W. Identification of a stable major sucrose-related QTL and diagnostic marker for flavor improvement in peanut. Theor. Appl. Genet. 2023, 136, 78. [Google Scholar] [CrossRef]
- Clevenger, J.; Chu, Y.; Chavarro, C.; Botton, S.; Culbreath, A.; Isleib, T.G.; Holbrook, C.; Ozias-Akins, P. Mapping late leaf spot resistance in peanut (Arachis hypogaea) using QTL-seq reveals markers for marker-assisted selection. Front. Plant Sci. 2018, 9, 83. [Google Scholar] [CrossRef]
- Pandey, M.K.; Khan, A.W.; Singh, V.K.; Vishwakarma, M.K.; Shasidhar, Y.; Kumar, V.; Garg, V.; Bhat, R.S.; Chitikineni, A.; Janila, P. QTL-seq approach identified genomic regions and diagnostic markers for rust and late leaf spot resistance in groundnut (A rachis hypogaea L.). Plant Biotechnol. J. 2017, 15, 927–941. [Google Scholar] [CrossRef]
- Wang, Z.; Yan, L.; Chen, Y.; Wang, X.; Huai, D.; Kang, Y.; Jiang, H.; Liu, K.; Lei, Y.; Liao, B. Detection of a major QTL and development of KASP markers for seed weight by combining QTL-seq, QTL-mapping and RNA-seq in peanut. Theor. Appl. Genet. 2022, 135, 1779–1795. [Google Scholar] [CrossRef]
- Luo, H.; Pandey, M.K.; Khan, A.W.; Guo, J.; Wu, B.; Cai, Y.; Huang, L.; Zhou, X.; Chen, Y.; Chen, W. Discovery of genomic regions and candidate genes controlling shelling percentage using QTL-seq approach in cultivated peanut (Arachis hypogaea L.). Plant Biotechnol. J. 2019, 17, 1248–1260. [Google Scholar] [CrossRef]
- Kumar, R.; Janila, P.; Vishwakarma, M.K.; Khan, A.W.; Manohar, S.S.; Gangurde, S.S.; Variath, M.T.; Shasidhar, Y.; Pandey, M.K.; Varshney, R.K. Whole-genome resequencing-based QTL-seq identified candidate genes and molecular markers for fresh seed dormancy in groundnut. Plant Biotechnol. J. 2020, 18, 992–1003. [Google Scholar] [CrossRef]
- Siloto, R.M.; Findlay, K.; Lopez-Villalobos, A.; Yeung, E.C.; Nykiforuk, C.L.; Moloney, M.M. The accumulation of oleosins determines the size of seed oilbodies in Arabidopsis. Plant Cell 2006, 18, 1961–1974. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Yin, Y.; Liu, S.; Guo, Z.; Zhang, K.; Liang, Y.; Zhang, L.; Zhao, W.; Chao, H.; Li, M. Genome-wide identification and functional analysis of oleosin genes in Brassica napus L. BMC Plant Biol. 2019, 19, 294. [Google Scholar] [CrossRef]
- Huang, A.H. Oleosins and oil bodies in seeds and other organs. Plant Physiol. 1996, 110, 1055. [Google Scholar] [CrossRef]
- Huang, M.; Huang, A.H. Bioinformatics reveal five lineages of oleosins and the mechanism of lineage evolution related to structure/function from green algae to seed plants. Plant Physiol. 2015, 169, 453–470. [Google Scholar] [CrossRef]
- Zhang, Y.; Yao, W.; Wang, F.; Su, Y.; Zhang, D.; Hu, S.; Zhang, X. AGC protein kinase AGC1-4 mediates seed size in Arabidopsis. Plant Cell Rep. 2020, 39, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Xing, J.; Liu, X.; Liu, Z.; Yao, Y.; Hu, Z.; Peng, H.; Xin, M.; Zhou, D.X.; Zhang, Y. Histone acetyltransferase general control non-repressed protein 5 (GCN 5) affects the fatty acid composition of Arabidopsis thaliana seeds by acetylating fatty acid desaturase3 (FAD 3). Plant J. 2016, 88, 794–808. [Google Scholar] [CrossRef] [PubMed]
- Parthibane, V.; Iyappan, R.; Vijayakumar, A.; Venkateshwari, V.; Rajasekharan, R. Serine/threonine/tyrosine protein kinase phosphorylates oleosin, a regulator of lipid metabolic functions. Plant Physiol. 2012, 159, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.; Zhang, M.; Zhang, Z.; Liang, S.; Fan, L.; Yang, X.; Yuan, Y.; Pan, Y.; Zhou, G.; Liu, S. Natural allelic variation of GmST05 controlling seed size and quality in soybean. Plant Biotechnol. J. 2022, 20, 1807–1818. [Google Scholar] [CrossRef]
- Goettel, W.; Zhang, H.; Li, Y.; Qiao, Z.; Jiang, H.; Hou, D.; Song, Q.; Pantalone, V.R.; Song, B.-H.; Yu, D. POWR1 is a domestication gene pleiotropically regulating seed quality and yield in soybean. Nat. Commun. 2022, 13, 3051. [Google Scholar] [CrossRef]
- Wang, S.; Liu, S.; Wang, J.; Yokosho, K.; Zhou, B.; Yu, Y.-C.; Liu, Z.; Frommer, W.B.; Ma, J.F.; Chen, L.-Q. Simultaneous changes in seed size, oil content and protein content driven by selection of SWEET homologues during soybean domestication. Natl. Sci. Rev. 2020, 7, 1776–1786. [Google Scholar] [CrossRef]
- Duan, Z.; Li, Q.; Wang, H.; He, X.; Zhang, M. Genetic regulatory networks of soybean seed size, oil and protein contents. Front. Plant Sci. 2023, 14, 1160418. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, H.; Zhu, Y.; Huang, X.; Li, S.; Wu, X.; Zhao, Y.; Bao, Z.; Qin, L.; Jin, Y. THP9 enhances seed protein content and nitrogen-use efficiency in maize. Nature 2022, 612, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Pan, T.; Zhao, Y.; Su, B.; Ren, Y.; Qiu, L. The small GTPase Rab5a and its guanine nucleotide exchange factors are involved in post-Golgi trafficking of storage proteins in developing soybean cotyledon. J. Exp. Bot. 2020, 71, 808–822. [Google Scholar] [CrossRef] [PubMed]
- Carter, E.; Rowland, D.; Tillman, B.; Erickson, J.; Grey, T.; Gillett-Kaufman, J.; Clark, M. Pod maturity in the shelling process. Peanut Sci. 2017, 44, 26–34. [Google Scholar] [CrossRef]
- Chen, H.; Yang, X.; Xu, R.; Chen, X.; Zhong, H.; Liu, N.; Huang, L.; Luo, H.; Huai, D.; Liu, W. Genetic mapping of AhVt1, a novel genetic locus that confers the variegated testa color in cultivated peanut (Arachis hypogaea L.) and its utilization for marker-assisted selection. Front. Plant Sci. 2023, 14, 1145098. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Magwene, P.M.; Willis, J.H.; Kelly, J.K. The statistics of bulk segregant analysis using next generation sequencing. PLoS Comput. Biol. 2011, 7, e1002255. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Mansfeld, B.N.; Grumet, R. QTLseqr: An R package for bulk segregant analysis with next-generation sequencing. Plant Genome 2018, 11, 180006. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Campbell, N.R.; Harmon, S.A.; Narum, S.R. Genotyping-in-Thousands by sequencing (GT-seq): A cost effective SNP genotyping method based on custom amplicon sequencing. Mol. Ecol. Resour. 2015, 15, 855–867. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, H.; Liu, N.; Huang, L.; Huai, D.; Xu, R.; Chen, X.; Guo, S.; Chen, J.; Jiang, H. Identification of a Major QTL for Seed Protein Content in Cultivated Peanut (Arachis hypogaea L.) Using QTL-Seq. Plants 2024, 13, 2368. https://doi.org/10.3390/plants13172368
Chen H, Liu N, Huang L, Huai D, Xu R, Chen X, Guo S, Chen J, Jiang H. Identification of a Major QTL for Seed Protein Content in Cultivated Peanut (Arachis hypogaea L.) Using QTL-Seq. Plants. 2024; 13(17):2368. https://doi.org/10.3390/plants13172368
Chicago/Turabian StyleChen, Hao, Nian Liu, Li Huang, Dongxin Huai, Rirong Xu, Xiangyu Chen, Shengyao Guo, Jianhong Chen, and Huifang Jiang. 2024. "Identification of a Major QTL for Seed Protein Content in Cultivated Peanut (Arachis hypogaea L.) Using QTL-Seq" Plants 13, no. 17: 2368. https://doi.org/10.3390/plants13172368