
Comparative Analysis of Transposable Elements in the Genomes of Citrus and Citrus-Related Genera



Abstract
:1. Introduction
2. Results
2.1. Construction of the Pan-Genome TE Library
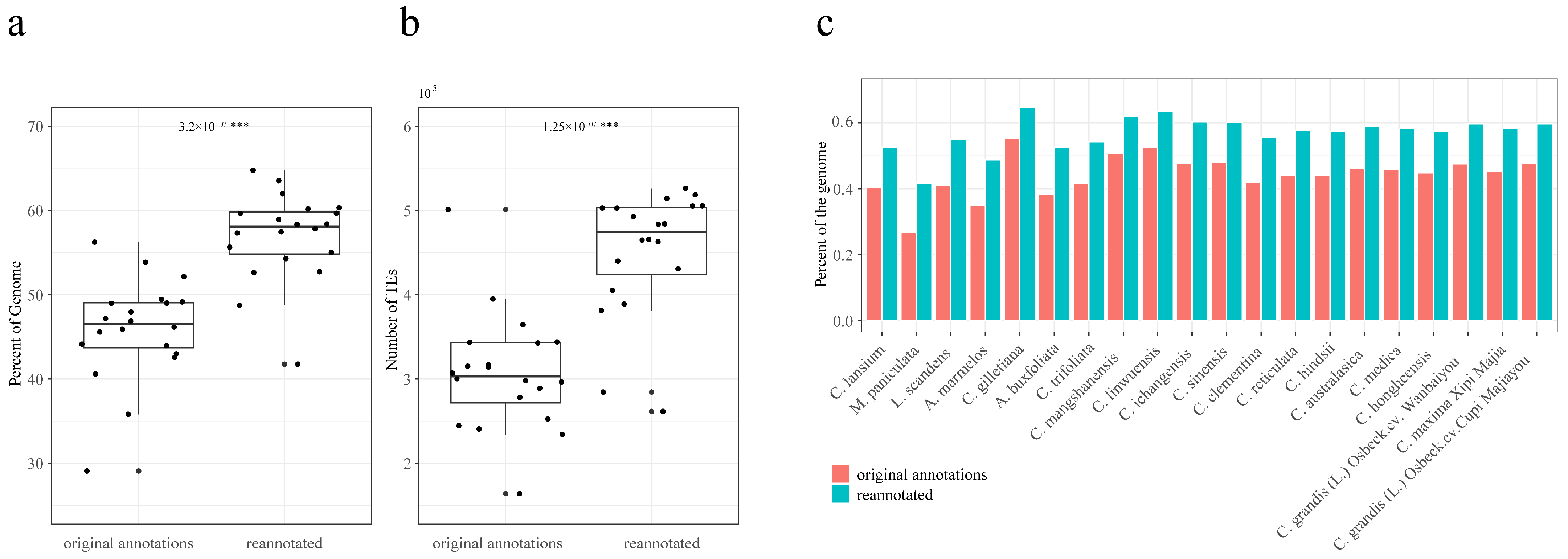
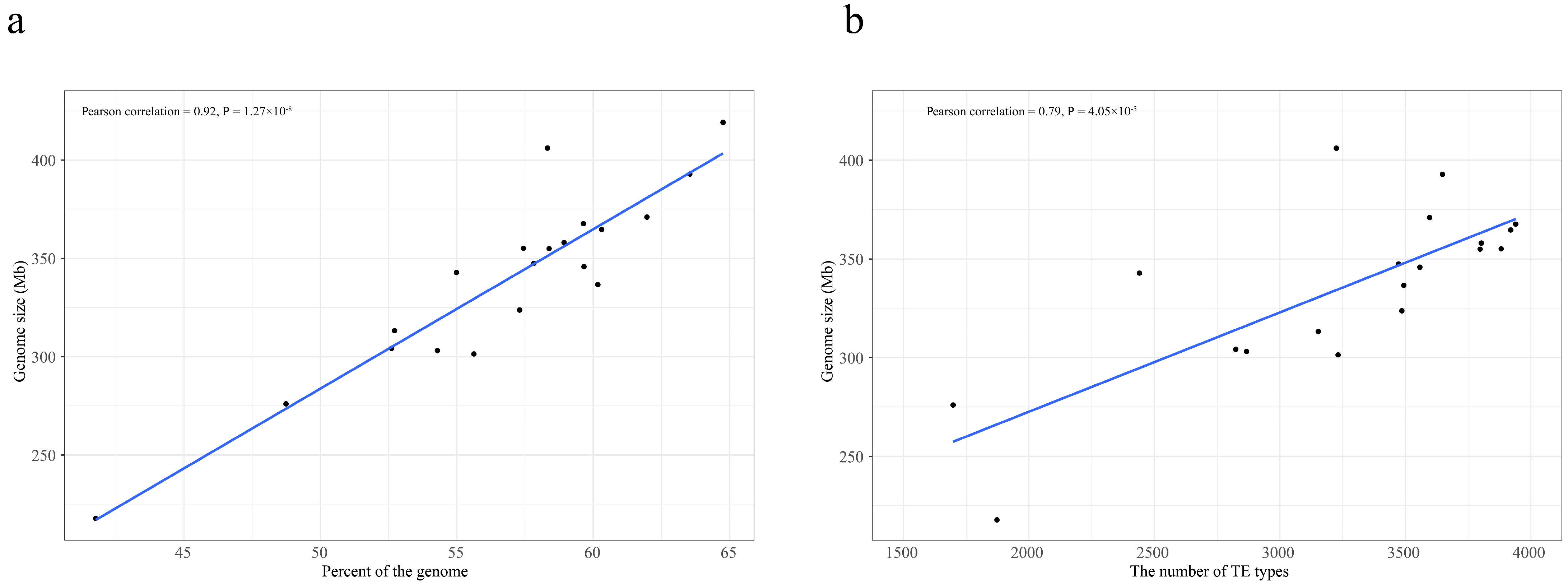
2.2. Summary of TEs in Reannotated Genomes
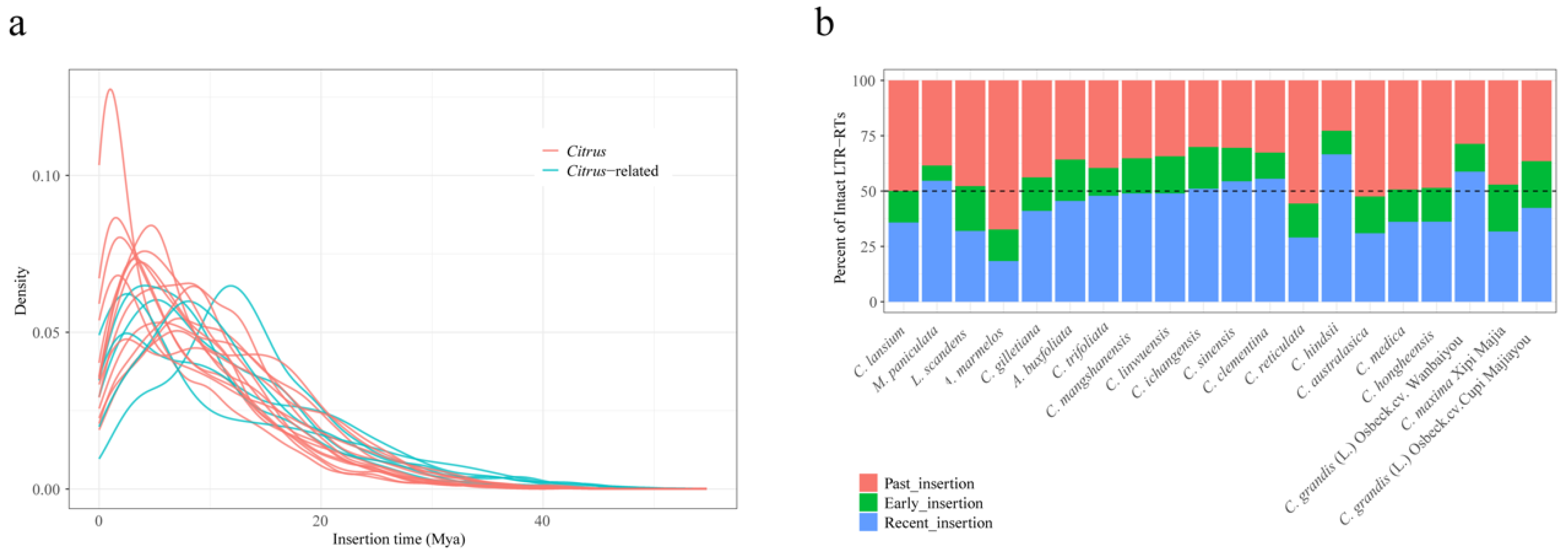
2.3. TE Evolutionary History in Citrus and Citrus-Related Genera
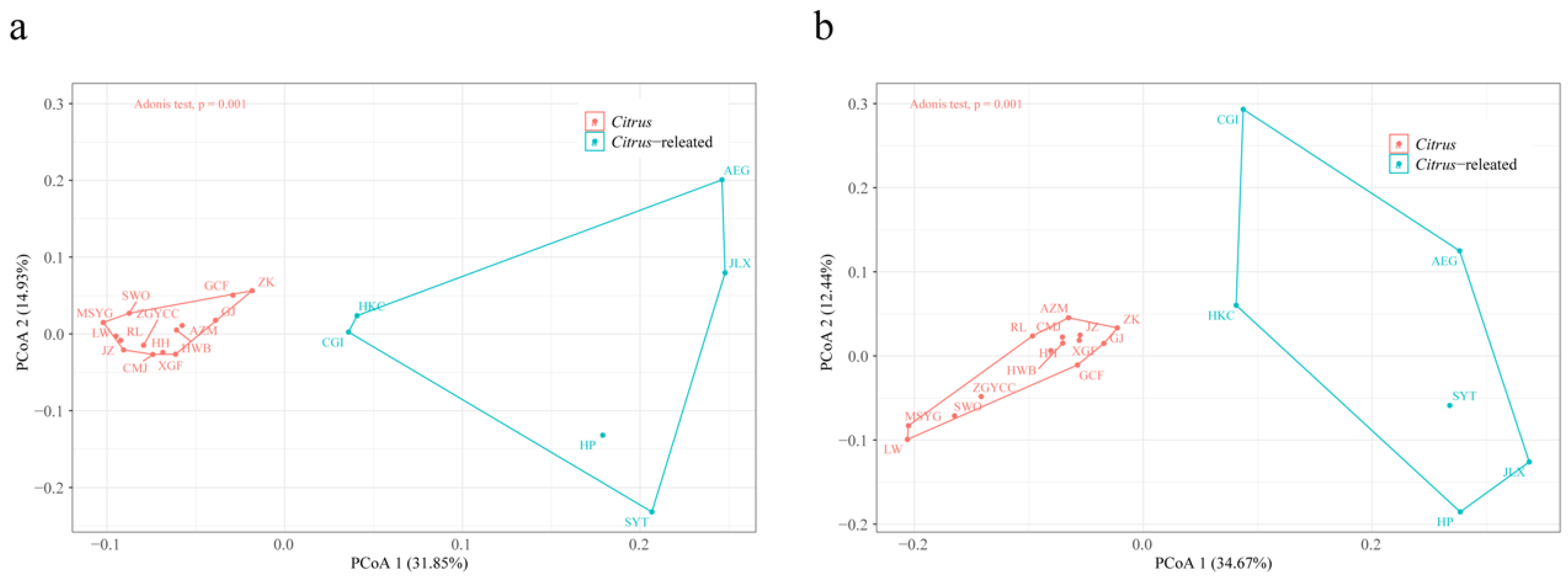
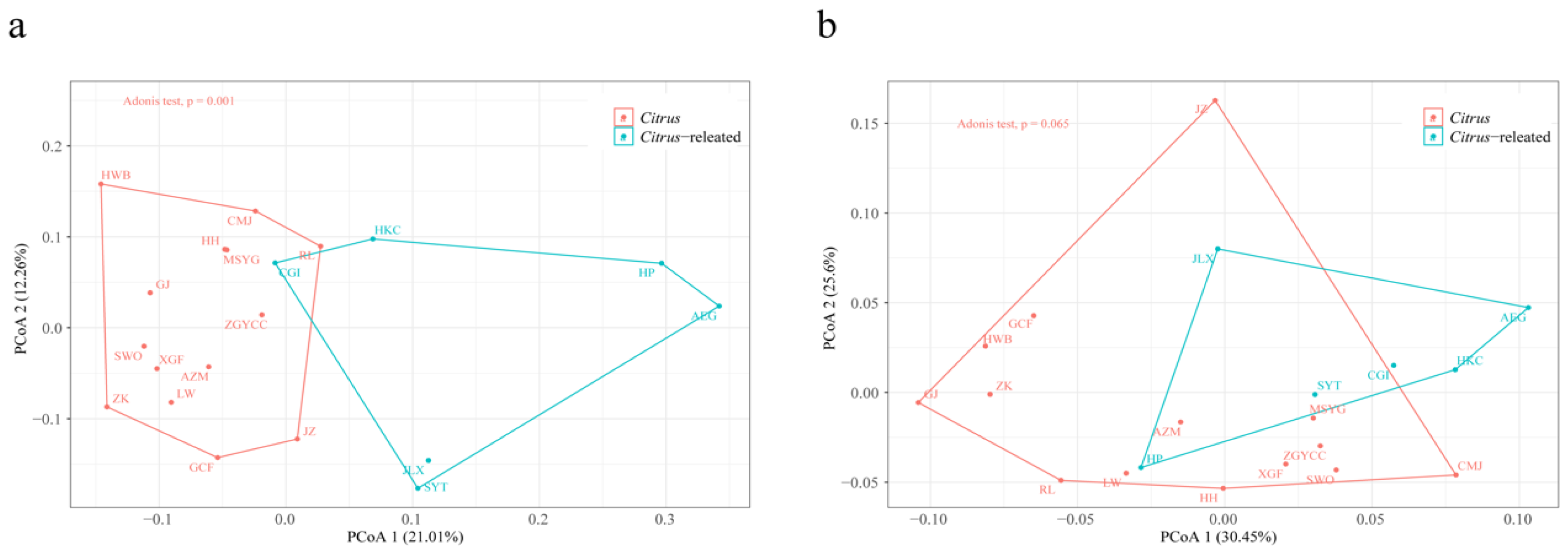
2.4. TE Variation in Citrus and Citrus-Related Genera
2.5. Important Implications of TEs for Citrus Bitterness
3. Discussion
4. Materials and Methods
4.1. Genome Dataset Collection
4.2. Construction of the Pan-Genome TE Library
4.3. Genome Reannotation with the Pan-Genome TE Library
4.4. Intact TEs and Citrus Bitterness
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kazazian, H.H., Jr. Mobile elements: Drivers of genome evolution. Science 2004, 303, 1626–1632. [Google Scholar] [CrossRef] [PubMed]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvák, Z.; Levin, H.L.; Macfarlan, T.S. Ten things you should know about transposable elements. Genome Biol. 2018, 19, 199. [Google Scholar] [CrossRef] [PubMed]
- Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 2000, 408, 796–815. [Google Scholar] [CrossRef]
- International Rice Genome Sequencing Project; Sasaki, T. The map-based sequence of the rice genome. Nature 2005, 436, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.A.; Terol, J.; Ibanez, V.; Lopez-Garcia, A.; Perez-Roman, E.; Borreda, C.; Domingo, C.; Tadeo, F.R.; Carbonell-Caballero, J.; Alonso, R.; et al. Genomics of the origin and evolution of Citrus. Nature 2018, 554, 311–316. [Google Scholar] [CrossRef]
- SanMiguel, P.; Tikhonov, A.; Jin, Y.-K.; Motchoulskaia, N.; Zakharov, D.; Melake-Berhan, A.; Springer, P.S.; Edwards, K.J.; Lee, M.; Avramova, Z. Nested retrotransposons in the intergenic regions of the maize genome. Science 1996, 274, 765–768. [Google Scholar] [CrossRef]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef]
- Pritham, E.J.; Putliwala, T.; Feschotte, C. Mavericks, a novel class of giant transposable elements widespread in eukaryotes and related to DNA viruses. Gene 2007, 390, 3–17. [Google Scholar] [CrossRef]
- Goodwin, T.J.; Butler, M.I.; Poulter, R.T. Cryptons: A group of tyrosine-recombinase-encoding DNA transposons from pathogenic fungi. Microbiology 2003, 149, 3099–3109. [Google Scholar] [CrossRef]
- Cai, X.; Lin, R.; Liang, J.; King, G.J.; Wu, J.; Wang, X. Transposable element insertion: A hidden major source of domesticated phenotypic variation in Brassica rapa. Plant Biotechnol. J. 2022, 20, 1298–1310. [Google Scholar] [CrossRef]
- Bergman, C.M.; Quesneville, H.; Anxolabéhère, D.; Ashburner, M. Recurrent insertion and duplication generate networks of transposable element sequences in the Drosophila melanogaster genome. Genome Biol. 2006, 7, R112. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Shukla, H.; Lee, Y.C.G. Species-specific chromatin landscape determines how transposable elements shape genome evolution. Elife 2022, 11, e81567. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; He, J.; Xu, Y.; Zheng, W.; Wang, S.; Chen, P.; Zeng, B.; Yang, S.; Jiang, X.; Liu, Z.; et al. Pangenome analysis provides insight into the evolution of the orange subfamily and a key gene for citric acid accumulation in citrus fruits. Nat. Genet. 2023, 55, 1964–1975. [Google Scholar] [CrossRef]
- McClintock, B. The origin and behavior of mutable loci in maize. Proc. Natl. Acad. Sci. USA 1950, 36, 344–355. [Google Scholar] [CrossRef] [PubMed]
- Finnegan, D.J. Transposable elements. Curr. Opin. Genet. Dev. 1992, 2, 861–867. [Google Scholar] [CrossRef]
- Bennetzen, J.L. Transposable elements, gene creation and genome rearrangement in flowering plants. Curr. Opin. Genet. Dev. 2005, 15, 621–627. [Google Scholar] [CrossRef]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory activities of transposable elements: From conflicts to benefits. Nat. Rev. Genet. 2017, 18, 71–86. [Google Scholar] [CrossRef]
- Tian, Y.; Thrimawithana, A.; Ding, T.; Guo, J.; Gleave, A.; Chagne, D.; Ampomah-Dwamena, C.; Ireland, H.S.; Schaffer, R.J.; Luo, Z.; et al. Transposon insertions regulate genome-wide allele-specific expression and underpin flower colour variations in apple (Malus spp.). Plant Biotechnol. J. 2022, 20, 1285–1297. [Google Scholar] [CrossRef]
- Fiol, A.; García, S.; Dujak, C.; Pacheco, I.; Infante, R.; Aranzana, M.J. An LTR retrotransposon in the promoter of a PsMYB10. 2 gene associated with the regulation of fruit flesh color in Japanese plum. Hort. Res. 2022, 9, uhac206. [Google Scholar] [CrossRef]
- Zhang, L.; Hu, J.; Han, X.; Li, J.; Gao, Y.; Richards, C.M.; Zhang, C.; Tian, Y.; Liu, G.; Gul, H. A high-quality apple genome assembly reveals the association of a retrotransposon and red fruit colour. Nat. Commun. 2019, 10, 1494. [Google Scholar] [CrossRef]
- Domínguez, M.; Dugas, E.; Benchouaia, M.; Leduque, B.; Jiménez-Gómez, J.M.; Colot, V.; Quadrana, L. The impact of transposable elements on tomato diversity. Nat. Commun. 2020, 11, 4058. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.; Collins, T.; Qiu, Y.; Seetharam, A.S.; Menard, C.C.; Manchanda, N.; Gent, J.I.; Schatz, M.C.; Anderson, S.N.; Hufford, M.B. Differences in activity and stability drive transposable element variation in tropical and temperate maize. bioRxiv 2022. [Google Scholar] [CrossRef]
- Lyu, K.; Xiao, J.; Lyu, S.; Liu, R. Comparative analysis of transposable elements in strawberry genomes of different ploidy levels. Int. J. Mol. Sci. 2023, 24, 16935. [Google Scholar] [CrossRef] [PubMed]
- Caruso, M.; Smith, M.W.; Froelicher, Y.; Russo, G.; Gmitter, F.G., Jr. Traditional breeding. In The Genus Citrus; Elsevier: Amsterdam, The Netherlands, 2020; pp. 129–148. [Google Scholar]
- Mitani, N.; Matsumoto, R.; Yoshioka, T.; Kuniga, T. Citrus hybrid seedlings reduce initial time to flower when grafted onto shiikuwasha rootstock. Sci. Hortic. 2008, 116, 452–455. [Google Scholar] [CrossRef]
- Xu, Q.; Chen, L.L.; Ruan, X.; Chen, D.; Zhu, A.; Chen, C.; Bertrand, D.; Jiao, W.B.; Hao, B.H.; Lyon, M.P.; et al. The draft genome of sweet orange (Citrus sinensis). Nat. Genet. 2013, 45, 59–66. [Google Scholar] [CrossRef]
- Butelli, E.; Licciardello, C.; Zhang, Y.; Liu, J.; Mackay, S.; Bailey, P.; Reforgiato-Recupero, G.; Martin, C. Retrotransposons control fruit-specific, cold-dependent accumulation of anthocyanins in blood oranges. Plant Cell. 2012, 24, 1242–1255. [Google Scholar] [CrossRef]
- Hu, J.; Liu, C.; Du, Z.; Guo, F.; Song, D.; Wang, N.; Wei, Z.; Jiang, J.; Cao, Z.; Shi, C. Transposable elements cause the loss of self-incompatibility in citrus. Plant Biotechnol. J. 2024, 22, 1113–1131. [Google Scholar] [CrossRef]
- Wang, X.; Xu, Y.; Zhang, S.; Cao, L.; Huang, Y.; Cheng, J.; Wu, G.; Tian, S.; Chen, C.; Liu, Y. Genomic analyses of primitive, wild and cultivated citrus provide insights into asexual reproduction. Nat. Genet. 2017, 49, 765–772. [Google Scholar] [CrossRef]
- Wang, L.; Huang, Y.; Liu, Z.; He, J.; Jiang, X.; He, F.; Lu, Z.; Yang, S.; Chen, P.; Yu, H. Somatic variations led to the selection of acidic and acidless orange cultivars. Nat. Plants 2021, 7, 954–965. [Google Scholar] [CrossRef]
- Liu, H.; Wang, X.; Liu, S.; Huang, Y.; Guo, Y.-X.; Xie, W.-Z.; Liu, H.; ul Qamar, M.T.; Xu, Q.; Chen, L.-L. Citrus Pan-Genome to Breeding Database (CPBD): A comprehensive genome database for citrus breeding. Mol. Plant 2022, 15, 1503–1505. [Google Scholar] [CrossRef]
- Ou, S.; Su, W.; Liao, Y.; Chougule, K.; Hufford, M.B. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 2019, 20, 275. [Google Scholar] [CrossRef] [PubMed]
- Bennetzen, J.L. Transposable element contributions to plant gene and genome evolution. Plant Mol. Biol. 2000, 42, 251–269. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Borges-Monroy, R.; Viswanadham, V.V.; Lee, S.; Li, H.; Lee, E.A.; Park, P.J. Comprehensive identification of transposable element insertions using multiple sequencing technologies. Nat. Commun. 2021, 12, 3836. [Google Scholar] [CrossRef]
- Gong, Z.; Wu, Y.; Koblížková, A.; Torres, G.A.; Wang, K.; Iovene, M.; Neumann, P.; Zhang, W.; Novák, P.; Buell, C.R.; et al. Repeatless and Repeat-Based Centromeres in Potato: Implications for Centromere Evolution. Plant Cell 2012, 24, 3559–3574. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Huang, Y.; Brachi, B.; Yun, Q.Z.; Zhang, W.; Lu, W.; Li, H.N.; Li, W.Q.; Sun, X.D.; Wang, G.Y.; et al. Genome-wide analysis of Cushion willow provides insights into alpine plant divergence in a biodiversity hotspot. Nat. Commun. 2019, 10, 5230. [Google Scholar] [CrossRef]
- Jiang, N.; Wessler, S.R. Insertion preference of maize and rice miniature inverted repeat transposable elements as revealed by the analysis of nested elements. Plant Cell 2001, 13, 2553–2564. [Google Scholar]
- Adrion, J.R.; Song, M.J.; Schrider, D.R.; Hahn, M.W.; Schaack, S. Genome-wide estimates of transposable element insertion and deletion rates in Drosophila melanogaster. Genome Biol. Evol. 2017, 9, 1329–1340. [Google Scholar] [CrossRef]
- Dugo, G.; Verzera, A.; Stagno d’Alcontres, I.; Cotroneo, A.; Ficarra, R. On the genuineness of citrus essential oils. Part XLI. Italian bitter orange essential oil: Composition and detection of contamination and additions of oils and terpenes of sweet orange and of lemon. Flavour Fragr J. 1993, 8, 25–33. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, Y.; Gao, M.; Wang, Y. Alcohol dehydrogenases regulated by a MYB44 transcription factor underlie Lauraceae citral biosynthesis. Plant Physiol. 2024, 194, 1674–1691. [Google Scholar] [CrossRef]
- Yan, H.; Bombarely, A.; Li, S. DeepTE: A computational method for de novo classification of transposons with convolutional neural network. Bioinformatics 2020, 36, 4269–4275. [Google Scholar] [CrossRef]
- Stritt, C.; Wyler, M.; Gimmi, E.L.; Pippel, M.; Roulin, A.C. Diversity, dynamics and effects of long terminal repeat retrotransposons in the model grass Brachypodium distachyon. New Phytol. 2020, 227, 1736–1748. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2. Wiley Interdiscip. Rev. Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017, 45, D200–D203. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Latin Name | Code Name | Bitterness | Version | Assembly Level | Burst Time | Half-Life Ratio |
|---|---|---|---|---|---|---|
| A. marmelos | AEG | v1 | Contig | 11.86 | 9.51 | |
| C. australasica | AZM | 1 | v1 | Contig | 7.15 | 7.97 |
| C. gilletiana | CGI | v1 | Contig | 5.19 | 7.11 | |
| C. grandis (L.) Osbeck. cv. Cupi Majiayou | CMJ | 2 | v1 | Chromosome | 8.09 | 6.28 |
| C. clementina | GCF | 1 | v1 | Scaffold | 1.90 | 5.59 |
| C. hindsii | GJ | 1 | v1 | Chromosome | 0.99 | 4.20 |
| C. hongheensis | HH | 4 | v1 | Contig | 5.43 | 7.56 |
| A. buxfoliata | HKC | v2 | Chromosome | 4.09 | 6.59 | |
| C. lansium | HP | 1 | v1 | Contig | 2.48 | 8.18 |
| C. grandis (L.) Osbeck. cv. Wanbaiyou | HWB | 2 | v1 | Chromosome | 1.51 | 5.15 |
| M. paniculata | JLX | v1 | Scaffold | 2.55 | 7.17 | |
| C. reticulata | JZ | 1 | v1 | Scaffold | 7.01 | 8.35 |
| C. linwuensis | LW | 1 | v1 | Contig | 3.79 | 6.16 |
| C. mangshanensis | MSYG | 3 | v1 | Contig | 3.21 | 6.19 |
| C. medica | RL | 2 | v1 | Scaffold | 2.44 | 7.84 |
| C. sinensis | SWO | 1 | v3 | Chromosome | 4.68 | 5.76 |
| L. scandens | SYT | v1 | Contig | 8.03 | 8.37 | |
| C. maxima Xipi Majia | XGF | 2 | v1 | Contig | 8.54 | 7.28 |
| C. ichangensis | ZGYCC | 3 | v2 | Contig | 4.07 | 5.57 |
| C. trifoliata | ZK | 3 | v1 | Chromosome | 1.66 | 6.71 |
| Class | Order | Superfamily | Count |
|---|---|---|---|
| Class I | DIRS | DIRS | 2 |
| Class I | LINE | I | 4 |
| Class I | LINE | L1 | 36 |
| Class I | LINE | unknown | 196 |
| Class I | LTR | Copia | 4072 |
| Class I | LTR | Gypsy | 3427 |
| Class I | LTR | Retrovirus | 54 |
| Class I | LTR | unknown | 116 |
| Class I | PLE | Penelope | 38 |
| Class I | nonLTR | unknown | 10 |
| Class I | SINE | SINE | 57 |
| Class II | Helitron | Helitron | 125 |
| Class II | Unknown | 724 | |
| Class II | TIR | CACTA | 1170 |
| Class II | TIR | Mutator | 3258 |
| Class II | TIR | PIF_Harbinger | 753 |
| Class II | TIR | Tc1_Mariner | 1382 |
| Class II | TIR | Transib | 2 |
| Class II | TIR | hAT | 4866 |
| Class II | TIR | unknown | 1388 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.; Wang, F.; Lyu, K.; Liu, R. Comparative Analysis of Transposable Elements in the Genomes of Citrus and Citrus-Related Genera. Plants 2024, 13, 2462. https://doi.org/10.3390/plants13172462
Wu Y, Wang F, Lyu K, Liu R. Comparative Analysis of Transposable Elements in the Genomes of Citrus and Citrus-Related Genera. Plants. 2024; 13(17):2462. https://doi.org/10.3390/plants13172462
Chicago/Turabian StyleWu, Yilei, Fusheng Wang, Keliang Lyu, and Renyi Liu. 2024. "Comparative Analysis of Transposable Elements in the Genomes of Citrus and Citrus-Related Genera" Plants 13, no. 17: 2462. https://doi.org/10.3390/plants13172462
APA StyleWu, Y., Wang, F., Lyu, K., & Liu, R. (2024). Comparative Analysis of Transposable Elements in the Genomes of Citrus and Citrus-Related Genera. Plants, 13(17), 2462. https://doi.org/10.3390/plants13172462