
Genome-Wide Characterization and Development of Simple Sequence Repeat Markers for Molecular Diversity Analyses in Yellowhorn (Xanthoceras sorbifolium Bunge)

 ,
,
Abstract
1. Introduction
2. Results
2.1. Characterization of SSRs in the Yellowhorn Genome
2.2. Functional Annotation for the SSR-Associated Genes
2.3. Development, Validation and Physical Mapping of Polymorphism SSR Markers
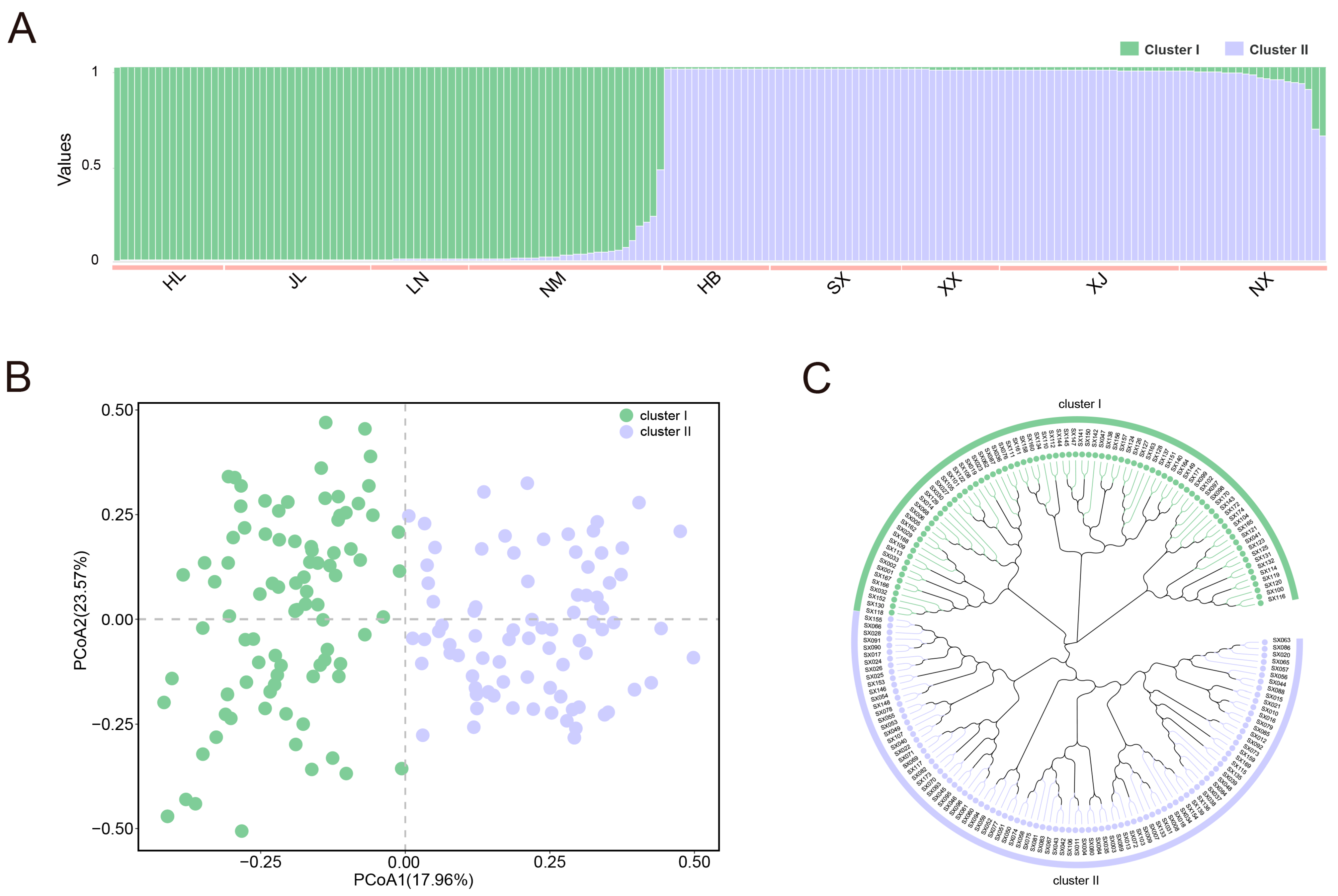
2.4. Analysis of Genetic Diversity and Population Structure
2.5. Establishment of Yellowhorn Germplasm Core Collection
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Characterization of SSR Loci in Yellowhorn Genome
4.3. Functional Annotation of SSR-Containing Genes
4.4. DNA Extraction and PCR Amplification
4.5. Genetic Diversity and Population Structure Analysis
4.6. Identification of Yellowhorn Core Germplasms
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lang, Y.; Sun, Y.; Feng, Y.; Qi, Z.; Yu, M.; Song, K. Recent progress in the molecular investigations of Yellow Horn (Xanthoceras sorbifolia Bunge). Bot. Rev. 2020, 86, 136–148. [Google Scholar] [CrossRef]
- Venegas-Calerón, M.; Ruíz-Méndez, M.V.; Martínez-Force, E.; Garcés, R.; Salas, J.J. Characterization of Xanthoceras sorbifolium Bunge seeds: Lipids, proteins and saponins content. Ind. Crops Prod. 2017, 109, 192–198. [Google Scholar] [CrossRef]
- Chen, X.; Lei, Z.; Cao, J.; Zhang, W.; Wu, R.; Cao, F.; Guo, Q.; Wang, J. Traditional uses, phytochemistry, pharmacology and current uses of underutilized Xanthoceras sorbifolium bunge: A review. J. Ethnopharmacol. 2022, 283, 114747. [Google Scholar] [CrossRef] [PubMed]
- Zang, E.; Qiu, B.; Chen, N.; Li, C.; Liu, Q.; Zhang, M.; Liu, Y.; Li, M. Xanthoceras sorbifolium Bunge: A review on botany, phytochemistry, pharmacology, and applications. Front. Pharmacol. 2021, 12, 70854. [Google Scholar] [CrossRef]
- Wang, M.; Wang, X.; Xu, H.; Liu, X.; Bi, Q.; Wang, L. Integrated transcriptomics and metabolomics analysis to characterize the optimal picking time in yellowhorn (Xanthoceras sorbifolium) flowers. Ind. Crops Prod. 2022, 187, 115389. [Google Scholar] [CrossRef]
- Fu, Y.-B. Understanding crop genetic diversity under modern plant breeding. Theor. Appl. Genet. 2015, 128, 2131–2142. [Google Scholar] [CrossRef]
- Vellend, M.; Geber, M.A. Connections between species diversity and genetic diversity. Ecol. Lett. 2005, 8, 767–781. [Google Scholar] [CrossRef]
- Zhu, R.-B.; Wang, Q.; Guan, W.-B.; Mao, Y.; Tian, B.; Cheng, J.-M.; El-Kassaby, Y.A. Conservation of genetic diversity hotspots of the high-valued relic yellowhorn (Xanthoceras sorbifolium) considering climate change predictions. Ecol. Evol. 2019, 9, 3251–3263. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Y. Population genetics and development of a core collection from elite germplasms of Xanthoceras sorbifolium based on genome-Wide SNPs. Forests 2022, 13, 338. [Google Scholar] [CrossRef]
- Taheri, S.; Lee Abdullah, T.; Yusop, M.R.; Hanafi, M.M.; Sahebi, M.; Azizi, P.; Shamshiri, R.R. Mining and development of novel SSR markers using next generation sequencing (NGS) data in plants. Molecules 2018, 23, 399. [Google Scholar] [CrossRef]
- Schulman, A.H. Molecular markers to assess genetic diversity. Euphytica 2007, 158, 313–321. [Google Scholar] [CrossRef]
- Song, X.; Yang, Q.; Bai, Y.; Gong, K.; Wu, T.; Yu, T.; Pei, Q.; Duan, W.; Huang, Z.; Wang, Z.; et al. Comprehensive analysis of SSRs and database construction using all complete gene-coding sequences in major horticultural and representative plants. Hortic. Res. 2021, 8, 122. [Google Scholar] [CrossRef]
- Bi, Q.; Guan, W. Isolation and characterisation of polymorphic genomic SSRs markers for the endangered tree Xanthoceras sorbifolium Bunge. Conserv. Genet. Resour. 2014, 6, 895–898. [Google Scholar] [CrossRef]
- El-Kassaby, Y.A.; Wang, Q.; Wang, T.; Ratcliffe, B.; Bi, Q.-X.; Wang, Z.; Mao, J.-F.; Guan, W. Concept for gene conservation strategy for the endangered Chinese yellowhorn, Xanthoceras sorbifolium, based on simulation of pairwise kinship coefficients. For. Ecol. Manag. 2019, 432, 976–982. [Google Scholar] [CrossRef]
- Liang, Q.; Li, H.; Li, S.; Yuan, F.; Sun, J.; Duan, Q.; Li, Q.; Zhang, R.; Sang, Y.L.; Wang, N.; et al. The genome assembly and annotation of yellowhorn (Xanthoceras sorbifolium Bunge). GigaScience 2019, 8, giz071. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Li, N.; Guo, Y.; Bai, Y.; Wu, T.; Yu, T.; Feng, S.; Zhang, Y.; Wang, Z.; Liu, Z.; et al. Comprehensive identification and characterization of simple sequence repeats based on the whole-genome sequences of 14 forest and fruit trees. For. Res. 2021, 1, 1–10. [Google Scholar] [CrossRef]
- Kalia, R.K.; Rai, M.K.; Kalia, S.; Singh, R.; Dhawan, A.K. Microsatellite markers: An overview of the recent progress in plants. Euphytica 2011, 177, 309–334. [Google Scholar] [CrossRef]
- Bi, Q.; Mao, J.-F.; Guan, W. Efficiently developing a large set of polymorphic EST-SSR markers for Xanthoceras sorbifolium by mining raw reads from high-throughput sequencing. Conserv. Genet. Resour. 2015, 7, 423–425. [Google Scholar] [CrossRef]
- Adhikari, S.; Saha, S.; Biswas, A.; Rana, T.S.; Bandyopadhyay, T.K.; Ghosh, P. Application of molecular markers in plant genome analysis: A review. Nucleus 2017, 60, 283–297. [Google Scholar] [CrossRef]
- Lou, W.; Lyu, Y.; Hu, L.; Yang, X.; Zhai, M.; Xuan, J.; Mo, Z. Development and characterization of EST-SSR markers in pecan (Carya illinoinensis). Trees 2023, 37, 297–307. [Google Scholar] [CrossRef]
- Dang, M.; Zhang, T.; Hu, Y.; Zhou, H.; Woeste, K.E.; Zhao, P. De novo assembly and characterization of bud, leaf and flowers transcriptome from Juglans Regia L. for the identification and characterization of new EST-SSRs. Forests 2016, 7, 247. [Google Scholar] [CrossRef]
- Patil, P.G.; Singh, N.V.; Parashuram, S.; Bohra, A.; Sowjanya, R.; Gaikwad, N.; Mundewadikar, D.M.; Sangnure, V.R.; Jamma, S.M.; Injal, A.S.; et al. Genome-wide characterization and development of simple sequence repeat markers for genetic studies in pomegranate (Punica granatum L.). Trees 2020, 34, 987–998. [Google Scholar] [CrossRef]
- Metzgar, D.; Bytof, J.; Wills, C. Selection against frameshift mutations limits microsatellite expansion in coding DNA. Genome Res. 2000, 10, 72–80. [Google Scholar] [PubMed]
- He, X.; Zheng, J.; Zhou, J.; He, K.; Shi, S.; Wang, B. Characterization and comparison of EST-SSRs in Salix, Populus, and Eucalyptus. Tree Genet. Genomes 2014, 11, 820. [Google Scholar] [CrossRef]
- Li, Y.-C.; Korol, A.B.; Fahima, T.; Nevo, E. Microsatellites within genes: Structure, function, and evolution. Mol. Biol. Evol. 2004, 21, 991–1007. [Google Scholar] [CrossRef]
- Singh, A.K.; Chaurasia, S.; Kumar, S.; Singh, R.; Kumari, J.; Yadav, M.C.; Singh, N.; Gaba, S.; Jacob, S.R. Identification, analysis and development of salt responsive candidate gene based SSR markers in wheat. BMC Plant Biol. 2018, 18, 249. [Google Scholar] [CrossRef]
- García-Gómez, B.; Razi, M.; Salazar, J.A.; Prudencio, A.S.; Ruiz, D.; Dondini, L.; Martínez-Gómez, P. Comparative analysis of SSR markers developed in exon, intron, and intergenic regions and distributed in regions controlling fruit quality traits in Prunus species: Genetic diversity and association studies. Plant Mol. Biol. Report. 2018, 36, 23–35. [Google Scholar] [CrossRef]
- Wang, L.; Ruan, C.; Bao, A.; Li, H. Small RNA profiling for identification of microRNAs involved in regulation of seed development and lipid biosynthesis in yellowhorn. BMC Plant Biol. 2021, 21, 464. [Google Scholar] [CrossRef]
- Ruan, C.J.; Yan, R.; Wang, B.X.; Mopper, S.; Guan, W.K.; Zhang, J. The importance of yellow horn (Xanthoceras sorbifolia) for restoration of arid habitats and production of bioactive seed oils. Ecol. Eng. 2017, 99, 504–512. [Google Scholar] [CrossRef]
- Shen, Z.; Zhang, K.; Ma, L.; Duan, J.; Ao, Y. Analysis of the genetic relationships and diversity among 11 populations of Xanthoceras sorbifolia using phenotypic and microsatellite marker data. Electron. J. Biotechnol. 2017, 26, 33–39. [Google Scholar] [CrossRef]
- Grueber, C.E.; Wallis, G.P.; Jamieson, I.G. Heterozygosity–fitness correlations and their relevance to studies on inbreeding depression in threatened species. Mol. Ecol. 2008, 17, 3978–3984. [Google Scholar] [CrossRef]
- Ellegren, H.; Galtier, N. Determinants of genetic diversity. Nat. Rev. Genet. 2016, 17, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Di Nunzio, A.; Tinto, N.; Iafusco, F.; Zibetti, A.; Ricci, P.; Di Nunzio, C. Null allele can bring to interpretative problems in a deficitary paternity case. Forensic Sci. Int. Genet. Suppl. Ser. 2022, 8, 115–116. [Google Scholar] [CrossRef]
- Shen, Z.; Duan, J.; Ma, L. Genetic diversity of Xanthoceras sorbifolium bunge germplasm using morphological traits and microsatellite molecular markers. PLoS ONE 2017, 12, e0177577. [Google Scholar] [CrossRef]
- Eckert, C.G.; Samis, K.E.; Lougheed, S.C. Genetic variation across species’ geographical ranges: The central–marginal hypothesis and beyond. Mol. Ecol. 2008, 17, 1170–1188. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, T.; Li, D.; Zhang, X.; Yu, W.; Cai, J.; Wang, G.; Guo, Q.; Yang, X.; Cao, F. The genetic diversity and population structure of Sophora alopecuroides (Faboideae) as determined by microsatellite markers developed from transcriptome. PLoS ONE 2019, 14, e0226100. [Google Scholar] [CrossRef]
- Du, Q.; Wang, B.; Wei, Z.; Zhang, D.; Li, B. Genetic diversity and population structure of Chinese white poplar (Populus tomentosa) revealed by SSR markers. J. Hered. 2012, 103, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Odong, T.L.; Jansen, J.; van Eeuwijk, F.A.; van Hintum, T.J.L. Quality of core collections for effective utilisation of genetic resources review, discussion and interpretation. Theor. Appl. Genet. 2013, 126, 289–305. [Google Scholar] [CrossRef]
- Boccacci, P.; Aramini, M.; Ordidge, M.; van Hintum, T.J.L.; Marinoni, D.T.; Valentini, N.; Sarraquigne, J.-P.; Solar, A.; Rovira, M.; Bacchetta, L.; et al. Comparison of selection methods for the establishment of a core collection using SSR markers for hazelnut (Corylus avellana L.) accessions from European germplasm repositories. Tree Genet. Genomes 2021, 17, 48. [Google Scholar] [CrossRef]
- Wang, D.; Zhou, Q.; Le, L.; Fu, F.; Wang, G.; Cao, F.; Yang, X. Molecular characterization and genetic diversity of ginkgo (Ginkgo biloba L.) based on insertions and deletions (InDel) markers. Plants 2023, 12, 2567. [Google Scholar] [CrossRef]
- Du, L.; Zhang, C.; Liu, Q.; Zhang, X.; Yue, B. Krait: An ultrafast tool for genome-wide survey of microsatellites and primer design. Bioinformatics 2018, 34, 681–683. [Google Scholar] [CrossRef]
- Wang, Y.; Jia, L.; Tian, G.; Dong, Y.; Zhang, X.; Zhou, Z.; Luo, X.; Li, Y.; Yao, W. shinyCircos-V2.0: Leveraging the creation of Circos plot with enhanced usability and advanced features. iMeta 2023, 2, e109. [Google Scholar] [CrossRef]
- Paysan-Lafosse, T.; Blum, M.; Chuguransky, S.; Grego, T.; Pinto, B.L.; Salazar, G.A.; Bileschi, M.L.; Bork, P.; Bridge, A.; Colwell, L.; et al. InterPro in 2022. Nucleic Acids Res. 2023, 51, D418–D427. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Voorrips, R.E. MapChart: Software for the graphical presentation of linkage maps and QTLs. J. Hered. 2002, 93, 77–78. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef]
- Rosenberg, N.A. distruct: A program for the graphical display of population structure. Mol. Ecol. Notes 2004, 4, 137–138. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus | Na | Ne | Ho | He | I | PIC |
|---|---|---|---|---|---|---|
| XsSSR01 | 9 | 3.039 | 0.516 | 0.651 | 0.623 | 0.552 |
| XsSSR02 | 8 | 5.124 | 0.469 | 0.528 | 0.421 | 0.506 |
| XsSSR03 | 10 | 6.712 | 0.594 | 0.721 | 1.201 | 0.734 |
| XsSSR04 | 10 | 5.212 | 0.392 | 0.586 | 0.854 | 0.512 |
| XsSSR05 | 9 | 5.562 | 0.452 | 0.523 | 0.875 | 0.525 |
| XsSSR06 | 10 | 5.254 | 0.698 | 0.754 | 1.035 | 0.584 |
| XsSSR07 | 9 | 4.589 | 0.582 | 0.685 | 0.784 | 0.574 |
| XsSSR08 | 11 | 6.575 | 0.587 | 0.758 | 0.895 | 0.852 |
| XsSSR09 | 7 | 4.258 | 0.457 | 0.762 | 1.324 | 0.721 |
| XsSSR10 | 8 | 4.521 | 0.458 | 0.757 | 1.356 | 0.712 |
| XsSSR11 | 8 | 5.311 | 0.412 | 0.623 | 1.023 | 0.582 |
| XsSSR12 | 9 | 6.563 | 0.667 | 0.854 | 1.896 | 0.756 |
| XsSSR13 | 7 | 4.268 | 0.428 | 0.665 | 1.101 | 0.683 |
| XsSSR14 | 6 | 3.254 | 0.319 | 0.621 | 0.825 | 0.488 |
| XsSSR15 | 7 | 4.332 | 0.682 | 0.768 | 1.524 | 0.635 |
| XsSSR16 | 7 | 3.085 | 0.601 | 0.607 | 1.425 | 0.587 |
| XsSSR17 | 6 | 2.092 | 0.582 | 0.520 | 1.058 | 0.487 |
| XsSSR18 | 7 | 4.756 | 0.452 | 0.789 | 1.526 | 0.758 |
| XsSSR19 | 5 | 2.636 | 0.467 | 0.622 | 1.223 | 0.478 |
| XsSSR20 | 5 | 3.345 | 0.501 | 0.782 | 1.354 | 0.654 |
| XsSSR21 | 9 | 5.369 | 0.745 | 0.827 | 1.582 | 0.752 |
| XsSSR22 | 10 | 5.458 | 0.704 | 0.812 | 1.586 | 0.761 |
| XsSSR23 | 7 | 4.528 | 0.751 | 0.778 | 1.526 | 0.723 |
| XsSSR24 | 6 | 3.485 | 0.712 | 0.859 | 1.452 | 0.631 |
| XsSSR25 | 8 | 6.012 | 0.776 | 0.842 | 1.821 | 0.758 |
| XsSSR26 | 8 | 5.125 | 0.648 | 0.768 | 1.752 | 0.658 |
| XsSSR27 | 9 | 4.231 | 0.425 | 0.605 | 1.205 | 0.561 |
| XsSSR28 | 7 | 4.526 | 0.623 | 0.785 | 1.523 | 0.598 |
| XsSSR29 | 7 | 5.127 | 0.705 | 0.759 | 1.652 | 0.658 |
| XsSSR30 | 5 | 2.852 | 0.498 | 0.587 | 1.058 | 0.532 |
| Mean | 7.8 | 4.573 | 0.563 | 0.689 | 1.249 | 0.634 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, X.; Wang, Y.; Yang, Y.; Shareng, T.; Xing, Y.; Bai, G.; Xing, Z.; Ji, Y.; Liu, L.; Cao, G. Genome-Wide Characterization and Development of Simple Sequence Repeat Markers for Molecular Diversity Analyses in Yellowhorn (Xanthoceras sorbifolium Bunge). Plants 2024, 13, 2794. https://doi.org/10.3390/plants13192794
Yang X, Wang Y, Yang Y, Shareng T, Xing Y, Bai G, Xing Z, Ji Y, Liu L, Cao G. Genome-Wide Characterization and Development of Simple Sequence Repeat Markers for Molecular Diversity Analyses in Yellowhorn (Xanthoceras sorbifolium Bunge). Plants. 2024; 13(19):2794. https://doi.org/10.3390/plants13192794
Chicago/Turabian StyleYang, Xiaoming, Yuan Wang, Yuewen Yang, Tuya Shareng, Yukun Xing, Gaowa Bai, Zhongyu Xing, Yuanyuan Ji, Liling Liu, and Gongxiang Cao. 2024. "Genome-Wide Characterization and Development of Simple Sequence Repeat Markers for Molecular Diversity Analyses in Yellowhorn (Xanthoceras sorbifolium Bunge)" Plants 13, no. 19: 2794. https://doi.org/10.3390/plants13192794
APA StyleYang, X., Wang, Y., Yang, Y., Shareng, T., Xing, Y., Bai, G., Xing, Z., Ji, Y., Liu, L., & Cao, G. (2024). Genome-Wide Characterization and Development of Simple Sequence Repeat Markers for Molecular Diversity Analyses in Yellowhorn (Xanthoceras sorbifolium Bunge). Plants, 13(19), 2794. https://doi.org/10.3390/plants13192794