
Genetic Diversity within a Collection of Italian Maize Inbred Lines: A Resource for Maize Genomics and Breeding
 ,
,  ,
,
Abstract
:1. Introduction
2. Results
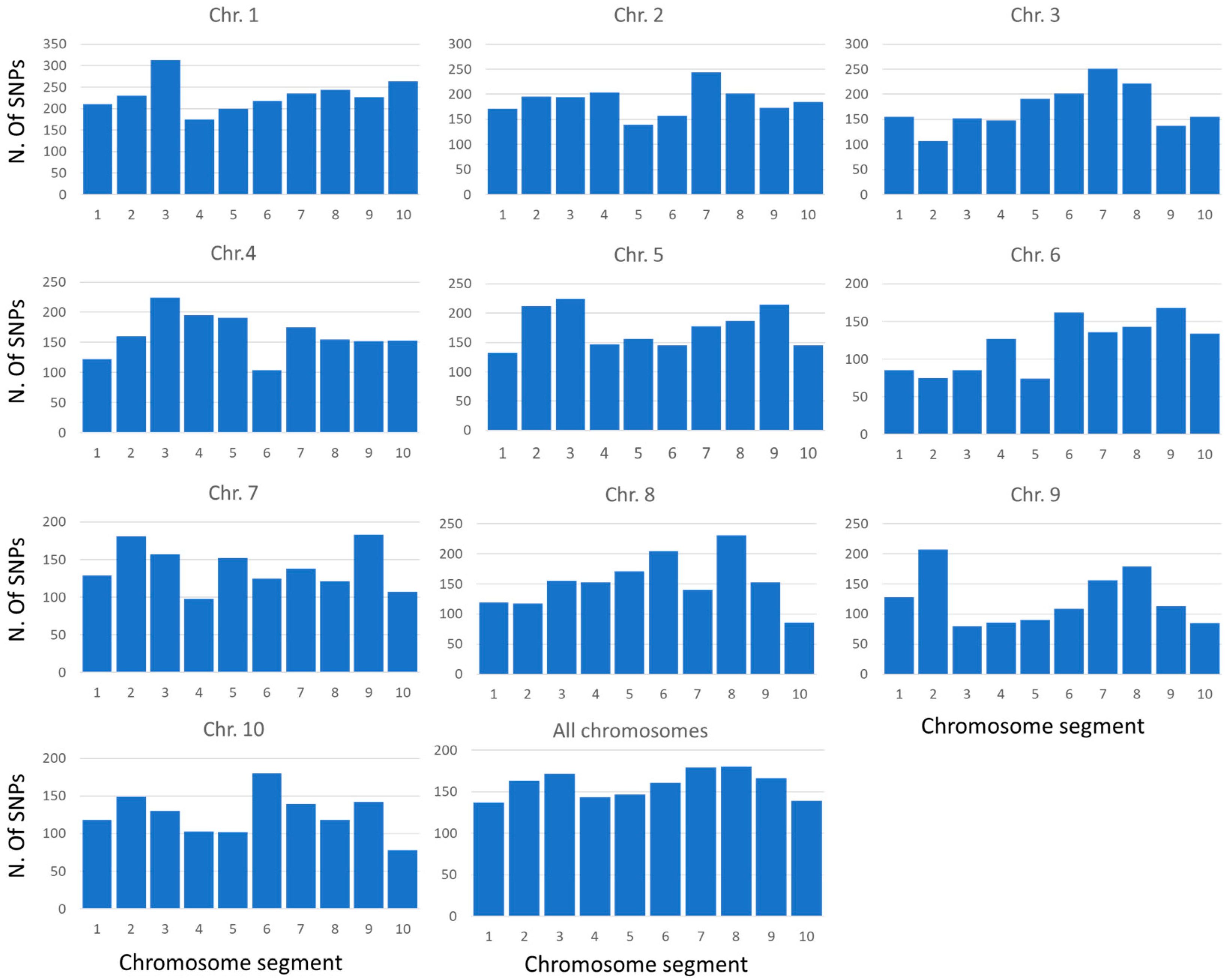
2.1. Genotyping of the Maize Collection
2.2. Linkage Disequilibrium Analysis
2.3. Stratification Analysis of the Maize Collection
2.4. Genetic Diversity Analysis
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. Genotyping and Data Processing
4.3. LD Decay
4.4. Population Structure and Cluster Analysis
4.5. Phylogenetic Tree
4.6. Analysis of Genetic Diversity within the Maize Collection
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- ISTAT. 2023. Available online: https://dati.istat.it/Index.aspx?DataSetCode=DCSP_COLTIVAZIONI (accessed on 9 October 2023).
- Ardenghi, N.M.G.; Rossi, G.; Guzzon, F. Back to beaked: Zea mays subsp. mays Rostrata Group in northern Italy, refugia and revival of open-pollinated maize landraces in an intensive cropping system. Peer J. 2018, 6, e5123. [Google Scholar] [CrossRef] [PubMed]
- Brandolini, A.; Brandolini, A. Maize introduction, evolution and diffusion in Italy. Maydica 2009, 54, 233–242. [Google Scholar]
- Levi, G. The Diffusion of Maize in Italy: From Resistance to the Peasants’ Defeat. In Global Goods and the Spanish Empire, 1492–1824: Circulation, Resistance and Diversity; Palgrave Macmillan UK: London, UK, 2014; pp. 100–115. [Google Scholar]
- Brandolini, A. Il germoplasma del mais e la sua conservazione. Maydica 1958, 3, 4–14. [Google Scholar]
- Evers, T.; Millar, S. Cereal Grain Structure and Development: Some Implications for Quality. J. Cereal Sci. 2002, 36, 261–284. [Google Scholar] [CrossRef]
- Gouesnard, B.; Negro, S.; Laffray, A.; Glaubitz, J.; Melchinger, A.; Revilla, P.; Moreno-Gonzalez, J.; Madur, D.; Combes, V.; Tollon-Cordet, C.; et al. Genotyping-by-sequencing highlights original diversity patterns within a European collection of 1191 maize flint lines, as compared to the maize USDA genebank. Theor. Appl. Genet. 2017, 130, 2165–2189. [Google Scholar] [CrossRef]
- Ganal, M.W.; Durstewitz, G.; Polley, A.; Bérard, A.; Buckler, E.S.; Charcosset, A.; Clarke, J.D.; Graner, E.-M.; Hansen, M.; Joets, J.; et al. A large maize (Zea mays L.) SNP genotyping array: Development and germplasm genotyping, and genetic mapping to compare with the B73 reference genome. PLoS ONE 2011, 6, e28334. [Google Scholar] [CrossRef]
- Zhu, X.-M.; Shao, X.-Y.; Pei, Y.-H.; Guo, X.-M.; Li, J.; Song, X.-Y.; Zhao, M.-A. Genetic diversity and genome-wide association study of major ear quantitative traits using high-density SNPs in maize. Front. Plant Sci. 2018, 9, 966. [Google Scholar] [CrossRef]
- Negro, S.S.; Millet, E.J.; Madur, D.; Bauland, C.; Combes, V.; Welcker, C.; Tardieu, F.; Charcosset, A.; Nicolas, S.D. Genotyping-by-sequencing and SNP-arrays are complementary for detecting quantitative trait loci by tagging different haplotypes in association studies. BMC Plant Biol. 2019, 19, 318. [Google Scholar] [CrossRef]
- Wu, X.; Wang, A.; Guo, X.; Liu, P.; Zhu, Y.; Li, X.; Chen, Z. Genetic characterization of maize germplasm derived from Suwan population and temperate resources. Hereditas 2019, 156, 2. [Google Scholar] [CrossRef]
- Hu, H.; Schrag, T.A.; Peis, R.; Unterseer, S.; Schipprack, W.; Chen, S.; Lai, J.; Yan, J.; Prasanna, B.M.; Nair, S.K.; et al. The genetic basis of haploid induction in maize identified with a novel genome-wide association method. Genetics 2016, 202, 1267–1276. [Google Scholar] [CrossRef]
- Millet, E.J.; Welcker, C.; Kruijer, W.; Negro, S.; Coupel-Ledru, A.; Nicolas, S.D.; Laborde, J.; Bauland, C.; Praud, S.; Ranc, N.; et al. Genome-wide analysis of yield in Europe: Allelic effects vary with drought and heat scenarios. Plant Physiol. 2016, 172, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Unterseer, S.; Bauer, E.; Haberer, G.; Seidel, M.; Knaak, C.; Ouzunova, M.; Meitinger, T.; Strom, T.M.; Fries, R.; Pausch, H.; et al. A powerful tool for genome analysis in maize: Development and evaluation of the high density 600 k SNP genotyping array. BMC Genom. 2014, 15, 823. [Google Scholar] [CrossRef] [PubMed]
- Messing, J.; Dooner, H.K. Organization and variability of the maize genome. Curr. Opin. Plant Biol. 2006, 9, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Frascaroli, E.; Schrag, T.A.; Melchinger, A.E. Genetic diversity analysis of elite European maize (Zea mays L.) inbred lines using AFLP, SSR, and SNP markers reveals ascertainment bias for a subset of SNPs. Theor. Appl. Genet. 2013, 126, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Glaubitz, J.C.; Casstevens, T.M.; Lu, F.; Harriman, J.; Elshire, R.J.; Sun, Q.; Buckler, E.S. TASSEL-GBS: A high capacity genotyping by sequencing analysis pipeline. PLoS ONE 2014, 9, e90346. [Google Scholar] [CrossRef]
- Le Gouis, J.; Bordes, J.; Ravel, C.; Heumez, E.; Faure, S.; Praud, S.; Galic, N.; Remoué, C.; Balfourier, F.; Allard, V.; et al. Genome-wide association analysis to identify chromosomal regions determining components of earliness in wheat. Theor. Appl. Genet. 2012, 124, 597–611. [Google Scholar] [CrossRef]
- Bauer, E.; Falque, M.; Walter, H.; Bauland, C.; Camisan, C.; Campo, L.; Meyer, N.; Ranc, N.; Rincent, R.; Schipprack, W.; et al. Intraspecific variation of recombination rate in maize. Genome Biol. 2013, 14, R103. [Google Scholar] [CrossRef]
- Springer, N.M.; Ying, K.; Fu, Y.; Ji, T.; Yeh, C.-T.; Jia, Y.; Wu, W.; Richmond, T.; Kitzman, J.; Rosenbaum, H.; et al. Maize Inbreds exhibit high levels of copy number variation (CNV) and presence/absence variation (PAV) in genome content. PLoS Genet. 2009, 5, e1000734. [Google Scholar] [CrossRef]
- Cormier, F.; Le Gouis, J.; Dubreuil, P.; Lafarge, S.; Praud, S. A genome-wide identification of chromosomal regions determining nitrogen use efficiency components in wheat (Triticum aestivum L.). Theor. Appl. Genet. 2014, 127, 2679–2693. [Google Scholar] [CrossRef]
- Ott, A.; Liu, S.; Schnable, J.C.; Yeh, C.T.E.; Wang, K.S.; Schnable, P.S. tGBS® genotyping-by-sequencing enables reliable genotyping of heterozygous loci. Nucleic Acids Res. 2017, 45, e178. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.; Choudhary, S.; Padiya, J.; Ubale, A.; Mikkilineni, V.; Char, B. Recent advances and applicability of GBS, GWAS, and GS in maize. In Genotyping by Sequencing for Crop Improvement; Sonah, H., Goyal, V., Shivaraj, S.M., Deshmukh, R.K., Eds.; Wiley Online Library: Hoboken, NJ, USA, 2022; pp. 188–217. [Google Scholar]
- Haberer, G.; Young, S.; Bharti, A.K.; Gundlach, H.; Raymond, C.; Fuks, G.; Butler, E.; Wing, R.A.; Rounsley, S.; Birren, B.; et al. Structure and architecture of the maize genome. Plant Physiol. 2005, 139, 1612–1624. [Google Scholar] [CrossRef] [PubMed]
- Moussa, A.A.; Mandozai, A.; Jin, Y.; Qu, J.; Zhang, Q.; Zhao, H.; Anwari, G.; Khalifa, M.A.S.; Lamboro, A.; Noman, M.; et al. Genome-wide association screening and verification of potential genes associated with root architectural traits in maize (Zea mays L.) at multiple seedling stages. BMC Genom. 2021, 22, 558. [Google Scholar] [CrossRef] [PubMed]
- Rashid, Z.; Sofi, M.; Harlapur, S.I.; Kachapur, R.M.; Dar, Z.A.; Singh, P.K.; Zaidi, P.H.; Vivek, B.S.; Nair, S.K. Genome-wide association studies in tropical maize germplasm reveal novel and known genomic regions for resistance to Northern corn leaf blight. Sci. Rep. 2020, 10, 21949. [Google Scholar] [CrossRef] [PubMed]
- Aci, M.M.; Lupini, A.; Mauceri, A.; Morsli, A.; Khelifi, L.; Sunseri, F. Genetic variation and structure of maize populations from Saoura and Gourara oasis in Algerian Sahara. BMC Genet. 2018, 19, 51. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Shah, T.; Hao, Z.; Taba, S.; Zhang, S.; Gao, S.; Liu, J.; Cao, M.; Wang, J.; Prakash, A.B.; et al. Comparative SNP and haplotype analysis reveals a higher genetic diversity and rapider LD decay in tropical than temperate germplasm in maize. PLoS ONE 2011, 6, e24861. [Google Scholar] [CrossRef]
- Chittò, A.; Bertolini, M.; Hartings, H.; Verderio, A.; Motto, M. AFLP-based genetic relationships among maize inbred lines selected in a climatically temperate location. Maydica 2000, 45, 257–266. [Google Scholar]
- Losa, A.; Hartings, H.; Verderio, A.; Motto, M. Assesment of genetic diversity and relationships among maize inbred lines developed in Italy. Maydica 2012, 56, 1. [Google Scholar]
- Bouaziz, M.; Paccard, C.; Guedj, M.; Ambroise, C. SHIPS: Spectral hierarchical clustering for the inference of population structure in genetic studies. PLoS ONE 2012, 7, e45685. [Google Scholar] [CrossRef]
- Meirmans, P.G. AMOVA-based clustering of population genetic data. J. Hered. 2012, 103, 744–750. [Google Scholar] [CrossRef]
- Alhusain, L.; Hafez, A.M. Nonparametric approaches for population structure analysis. Hum. Genet. 2018, 12, 25. [Google Scholar] [CrossRef]
- Kobak, D.; Berens, P. The art of using t-SNE for single-cell transcriptomics. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- López-Cortés, X.A.; Matamala, F.; Maldonado, C.; Mora-Poblete, F.; Scapim, C.A. A Deep Learning Approach to Population Structure Inference in Inbred Lines of Maize. Front. Genet. 2020, 11, 543459. [Google Scholar] [CrossRef] [PubMed]
- Brandolini, A.; Brandolini, A.; Negri, A. Origine e diffusione. In Il Mais; Maggiore, T., Angelini, R., Eds.; ART Servizi Editoriali: Bologna, Italy, 2008; pp. 45–83. [Google Scholar]
- Ayesiga, S.B.; Rubaihayo, P.; Oloka, B.M.; Dramadri, I.O.; Edema, R.; Sserumaga, J.P. Genetic variation among tropical maize inbred lines from NARS and CGIAR breeding programs. Plant Mol. Biol. Rep. 2023, 41, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Naveenkumar, K.L.; Sen, D.; Vashum, S.; Sanjenbam, M. Genetic characterization and divergence studies of maize (Zea mays L.) lines developed from landraces indigenous to North Eastern Hill Region (NEHR) of India. Plant Genet. Res. Charact. Util. 2020, 18, 231–242. [Google Scholar] [CrossRef]
- Mazzucotelli, E.; Sciara, G.; Mastrangelo, A.M.; Desiderio, F.; Xu, S.S.; Faris, J.; Hayden, M.J.; Tricker, P.J.; Ozkan, H.; Echenique, V.; et al. The Global Durum Wheat Panel (GDP): An international platform to identify and exchange beneficial alleles. Front. Plant Sci. 2020, 11, 569905. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, s13742-015. [Google Scholar] [CrossRef]
- Browning, B.L.; Zhou, Y.; Browning, S.R. A one-penny imputed genome from next-generation reference panels. Am. J. Human Genet. 2018, 103, 338–348. [Google Scholar] [CrossRef]
- Rexroad, C.E.; Vallejo, R.L. Estimates of linkage disequilibrium and effective population size in rainbow trout. BMC Genet. 2009, 10, 83. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- MacQueen, J.B. Some methods for classification and analysis of multivariate observations. In Proceedings of the 5-th Berkeley Symposium on Mathematical Statistics and Probability; University of California Press: Berkeley, CA, USA, 1967; Volume 1, pp. 281–297. [Google Scholar]
- Kaufman, L.; Rousseeuw, P.J. Finding Groups in data: An Introduction to Cluster Analysis, 1st ed.; John Wiley & Sons: New York, NY, USA, 1990; ISBN 0-471-87876-6. [Google Scholar]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Cohen, J. A coefficient of agreement for nominal scales. Educ. Psychol. Meas. 1960, 20, 37–46. [Google Scholar] [CrossRef]
- Stone, M. Cross-validatory choice and assessment of statistical predictions. J. R. Stat. Soc. Ser. B 1974, 36, 111–147. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Moore, R.M.; Harrison, A.O.; McAllister, S.M.; Polson, S.W.; Wommack, K.E. Iroki: Automatic customization and visualization of phylogenetic trees. Peer J. 2020, 8, e8584. [Google Scholar] [CrossRef] [PubMed]
- Wright, S. The interpretation of population structure by F-statistics with special regard to systems of mating. Evolution 1965, 19, 395–420. [Google Scholar] [CrossRef]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 1980, 32, 314–331. [Google Scholar]
- Nei, M. Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. USA 1973, 70, 3321–3323. [Google Scholar] [CrossRef]
- Nei, M. Genetic distance between populations. Am. Nat. 1972, 106, 283–292. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosome | SNPs | SNPs/Mb | Chromosome Coverage (%) |
|---|---|---|---|
| 1 | 2315 | 7.57 | 99.61 |
| 2 | 1864 | 7.66 | 99.57 |
| 3 | 1719 | 7.32 | 99.70 |
| 4 | 1631 | 6.62 | 99.68 |
| 5 | 1742 | 7.79 | 99.85 |
| 6 | 1189 | 6.85 | 99.67 |
| 7 | 1391 | 7.64 | 99.85 |
| 8 | 1529 | 8.46 | 99.78 |
| 9 | 1233 | 7.73 | 99.90 |
| 10 | 1259 | 8.37 | 99.60 |
| Total | 15,872 | - | - |
| Mean | 1587.2 | 7.60 | 99.72 |
| A. | ||||
| Source of variation | d.f. | Sum of squares | Variance components | Percentage of variation |
| Among populations | 3 | 18,361.74 | 32.18 | 3.25 |
| Within populations | 716 | 685,463.41 | 957.35 | 96.75 |
| Total | 719 | 703,825.14 | 989.53 | |
| Fst | 0.032 | |||
| B. | ||||
| Maize groups | No accessions | No polymorphic loci over 15,872 | Nei’s gene diversity | Mean number of pairwise differences |
| K4-1 | 119 | 15,868 | 0.1312 | 2081.88 |
| K4-2 | 63 | 15,243 | 0.1340 | 2042.19 |
| K4-3 | 22 | 12,864 | 0.1685 | 2167.74 |
| K4-4 | 156 | 15,857 | 0.1073 | 1701.07 |
| Mean value | 0.1352 | 1998.22 | ||
| LSD (p = 0.05) | 0.0010 | 14.97 | ||
| LSD (p = 0.001) | 0.0016 | 23.58 | ||
| C. | ||||
| Group | 1 | 2 | 3 | 4 |
| 1 | 2081.882 | 2090.928 | 2212.779 | 1936.086 |
| 2 | 0.014 | 2042.191 | 2222.115 | 1955.579 |
| 3 | 0.041 | 0.054 | 2167.744 | 2078.851 |
| 4 | 0.023 | 0.045 | 0.078 | 1701.074 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mastrangelo, A.M.; Hartings, H.; Lanzanova, C.; Balconi, C.; Locatelli, S.; Cassol, H.; Valoti, P.; Petruzzino, G.; Pecchioni, N. Genetic Diversity within a Collection of Italian Maize Inbred Lines: A Resource for Maize Genomics and Breeding. Plants 2024, 13, 336. https://doi.org/10.3390/plants13030336
Mastrangelo AM, Hartings H, Lanzanova C, Balconi C, Locatelli S, Cassol H, Valoti P, Petruzzino G, Pecchioni N. Genetic Diversity within a Collection of Italian Maize Inbred Lines: A Resource for Maize Genomics and Breeding. Plants. 2024; 13(3):336. https://doi.org/10.3390/plants13030336
Chicago/Turabian StyleMastrangelo, Anna Maria, Hans Hartings, Chiara Lanzanova, Carlotta Balconi, Sabrina Locatelli, Helga Cassol, Paolo Valoti, Giuseppe Petruzzino, and Nicola Pecchioni. 2024. "Genetic Diversity within a Collection of Italian Maize Inbred Lines: A Resource for Maize Genomics and Breeding" Plants 13, no. 3: 336. https://doi.org/10.3390/plants13030336