


Genome-Wide Analysis of the WRKY Transcription Factor Family in Roses and Their Putative Role in Defence Signalling in the Rose–Blackspot Interaction


Abstract
1. Introduction
2. Materials & Methods
2.1. Plant and Fungal Material
2.2. Inoculation Assay and Experiments
2.3. Identification of WRKY Candidates in R. chinensis and Other Rose Species
2.4. Phylogenetic Analyses
2.5. Promoter Region Analysis
2.6. Overexpression Experiments
2.7. Experimental Induction of Abiotic Stress
3. Results
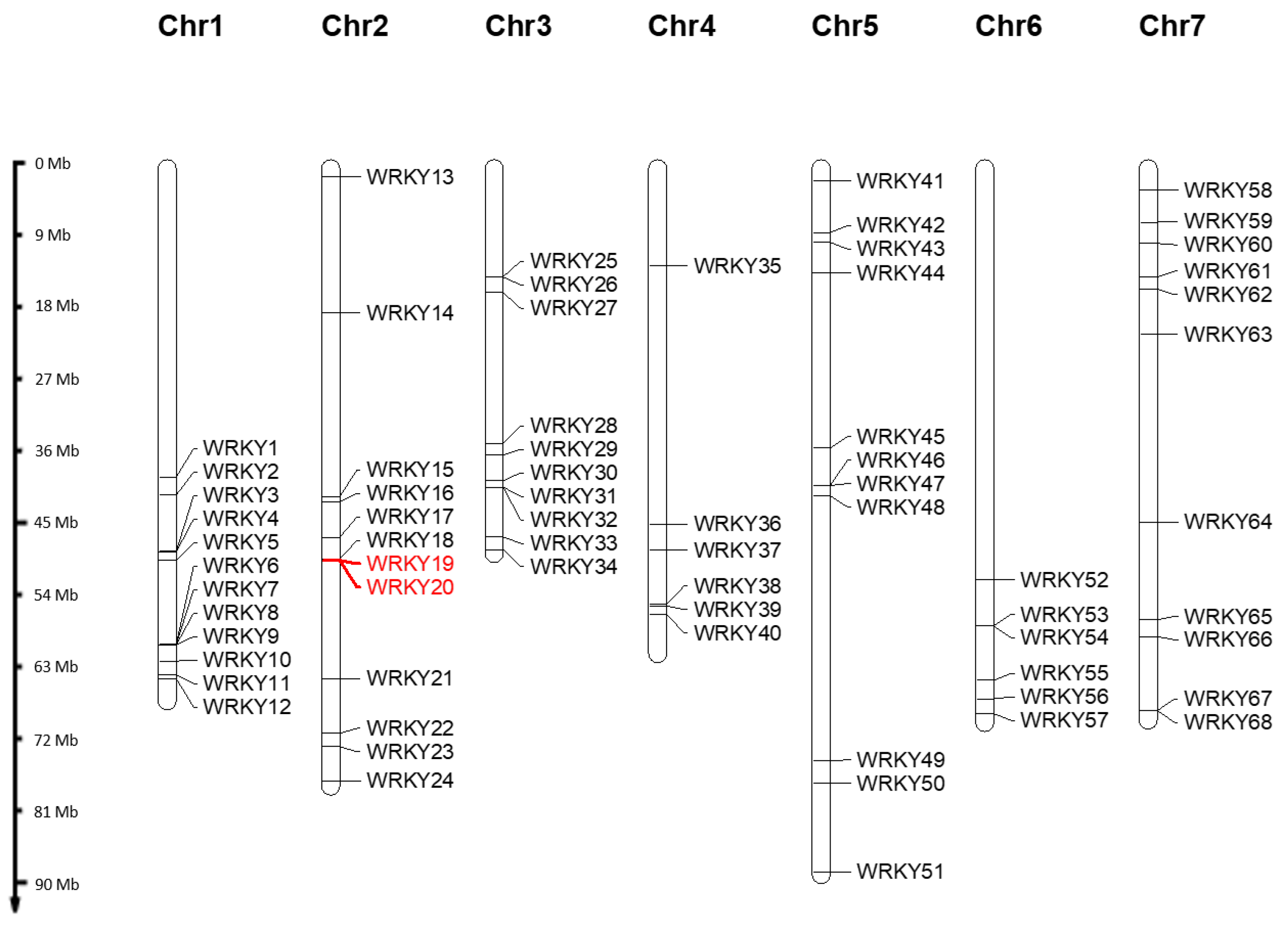
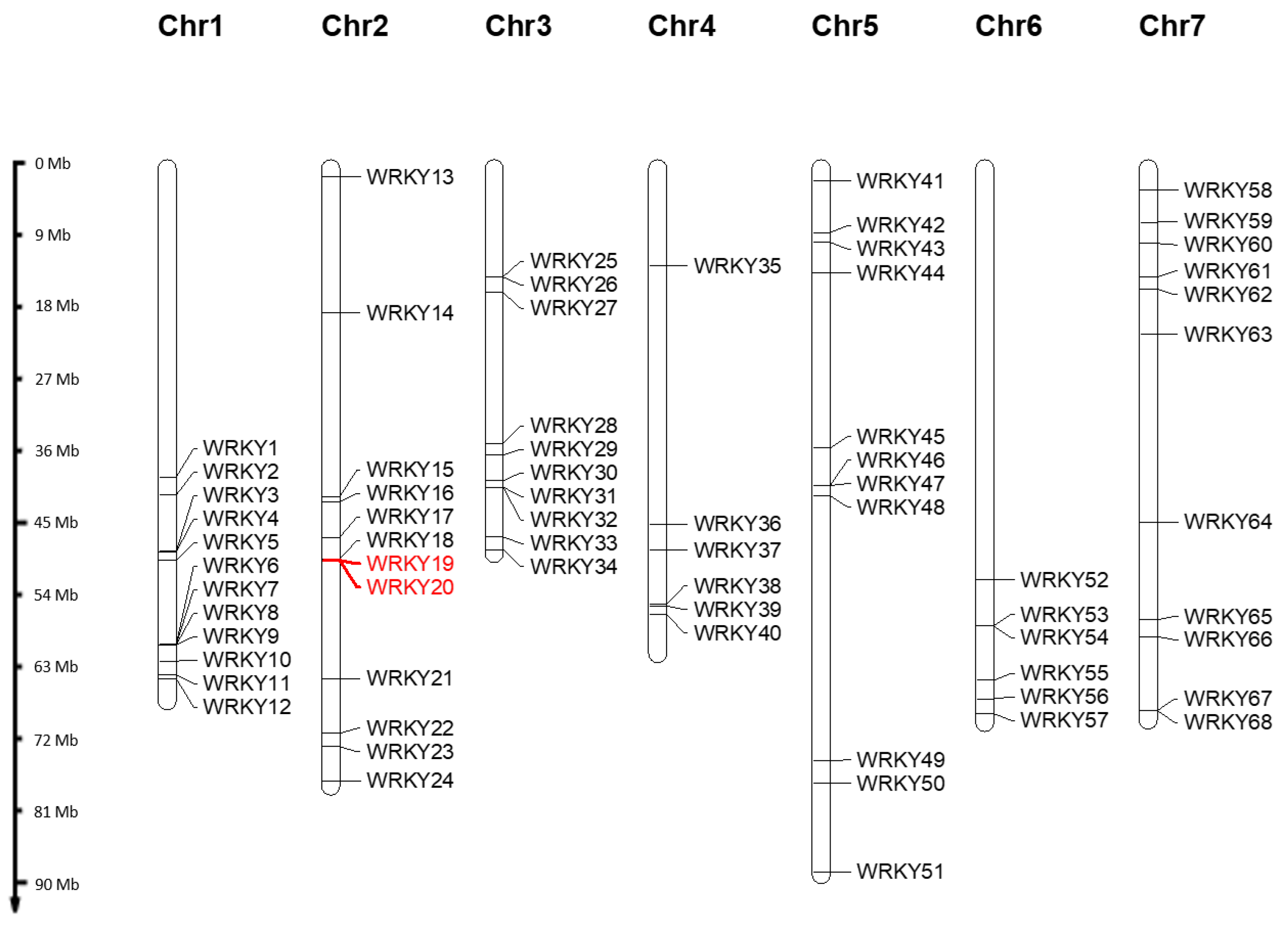
3.1. Identification of an Extended Set of WRKY Candidates by Analysing Six Rose Genomes
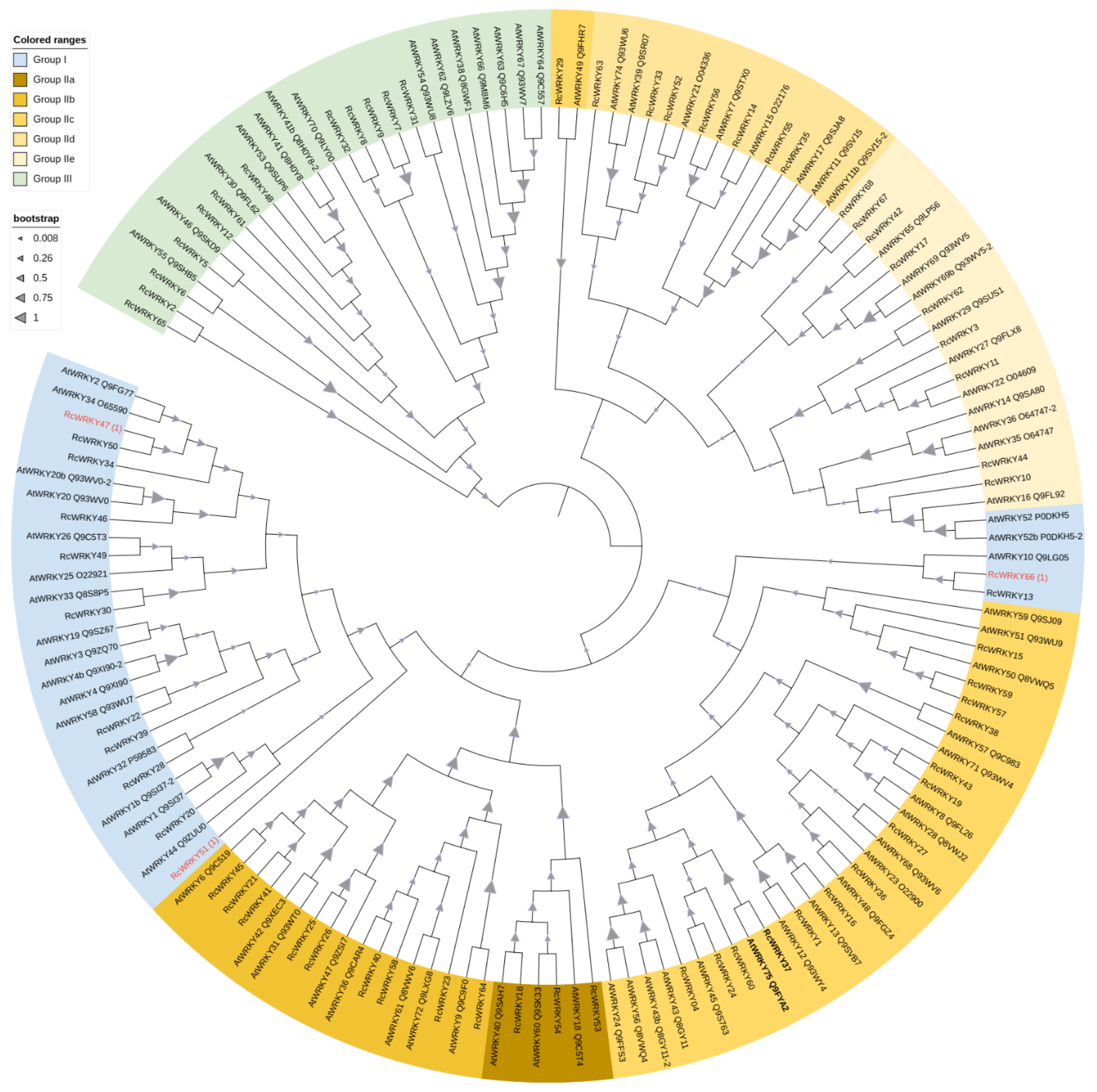
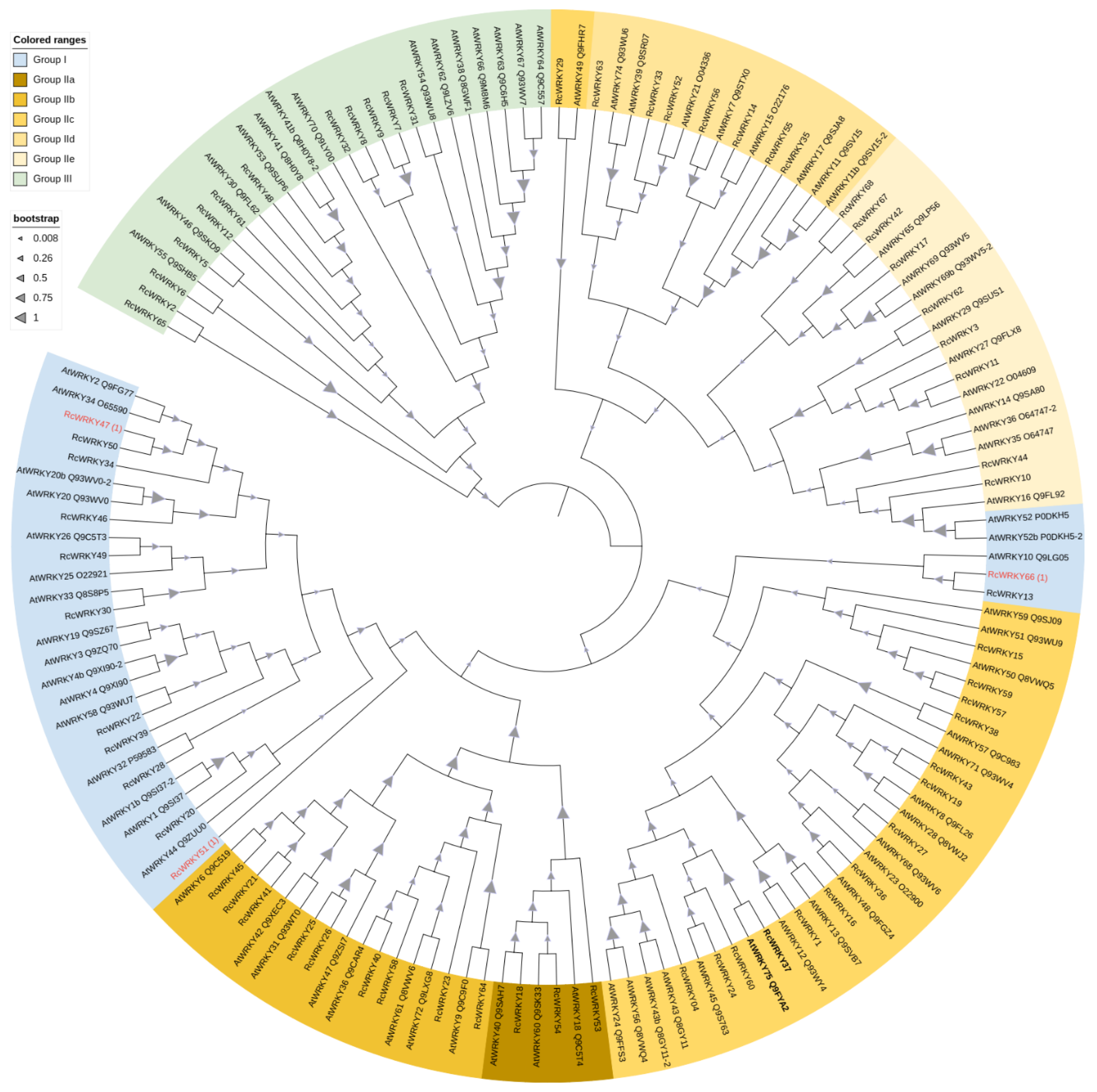
3.2. Phylogenetic Analyses
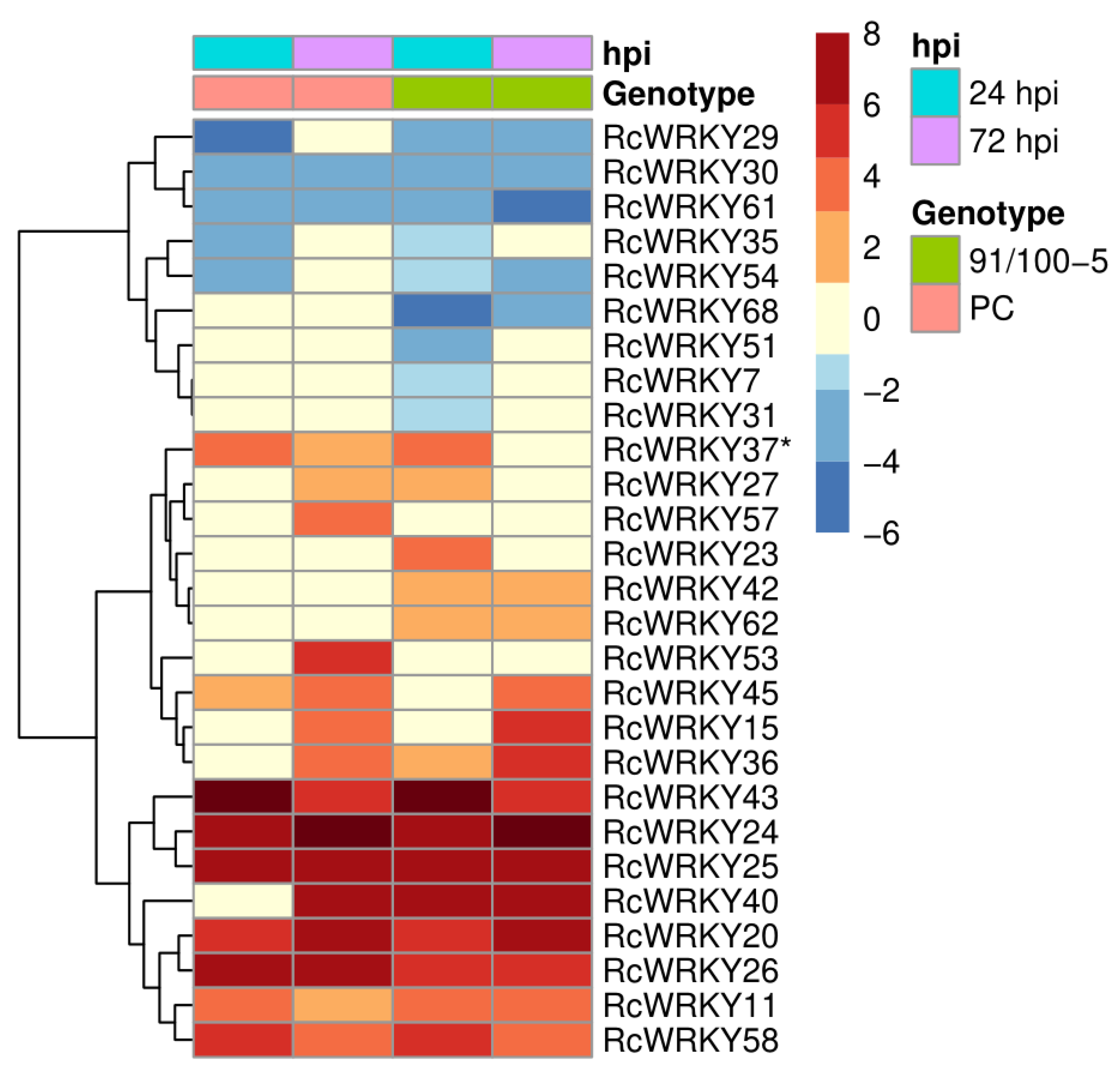
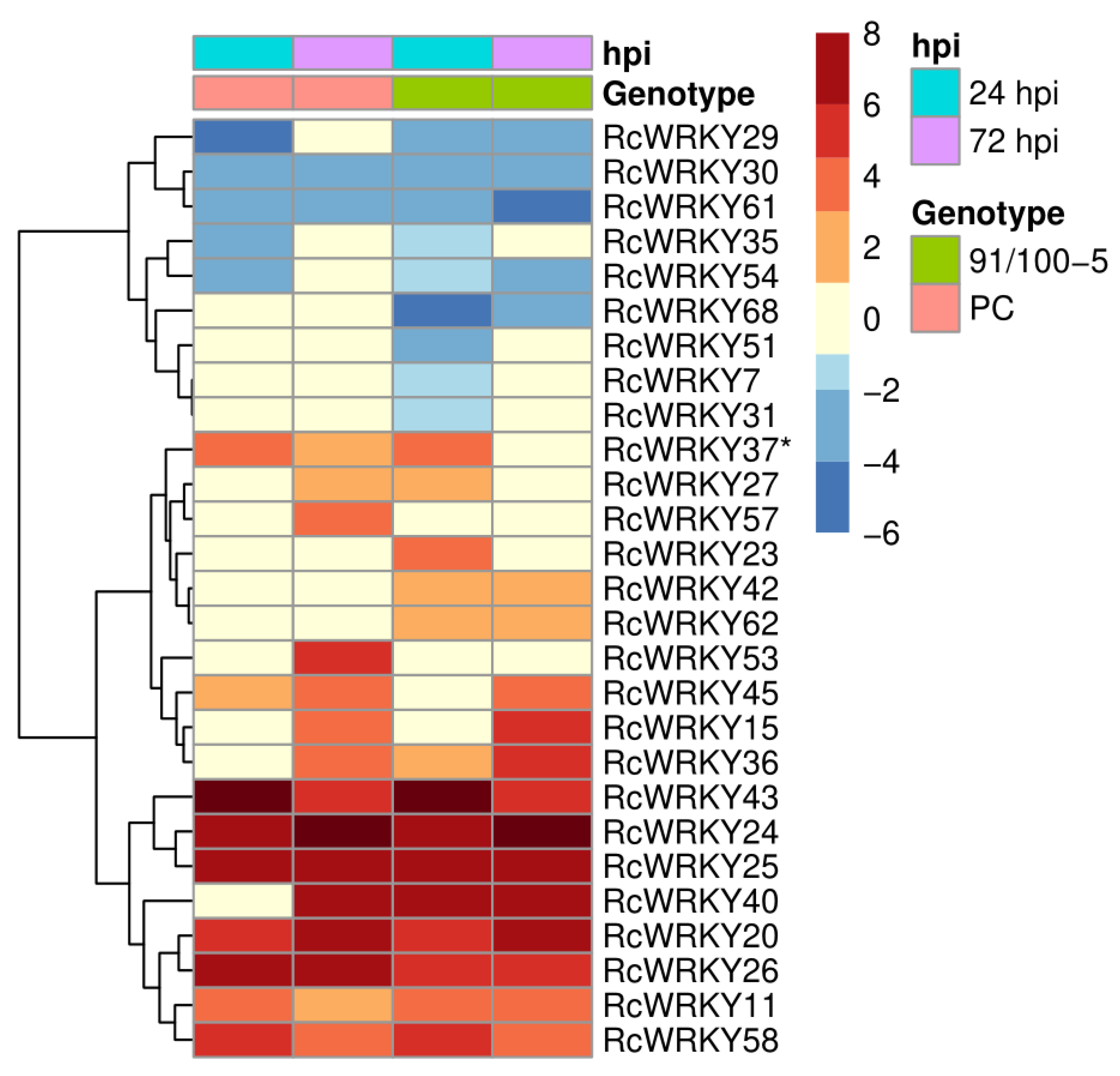
3.3. Expression of the RcWRKY Genes under Pathogen Attack
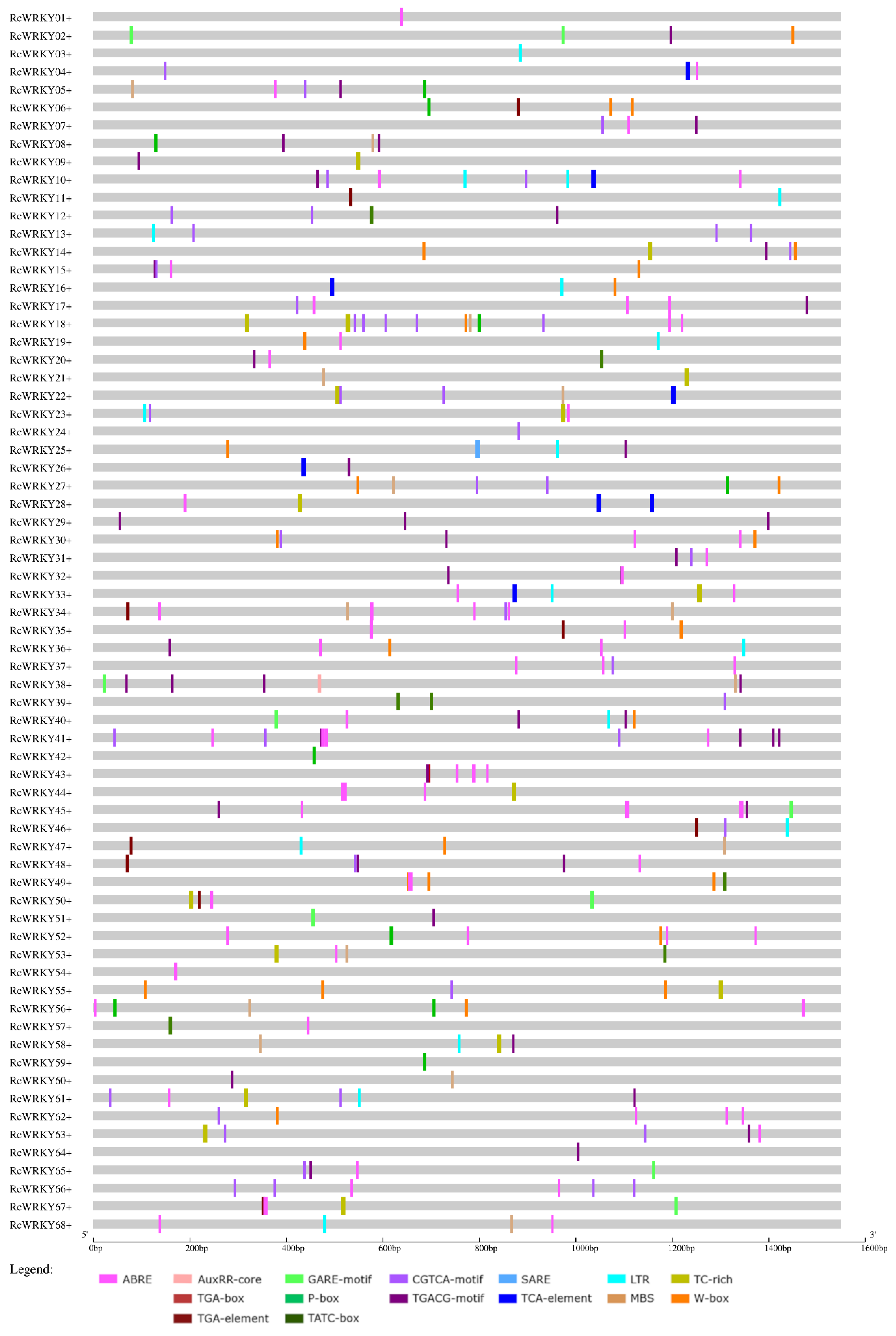
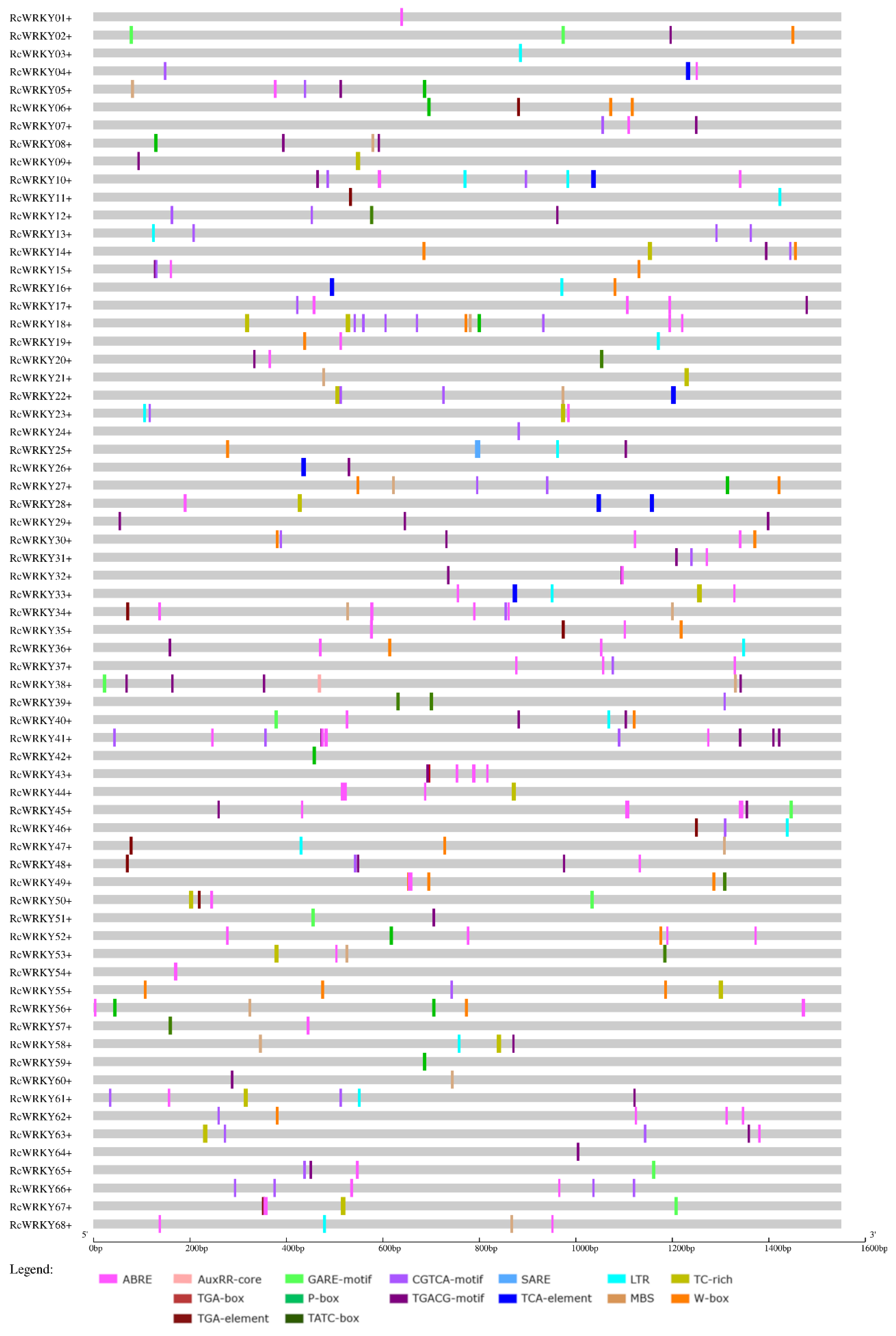
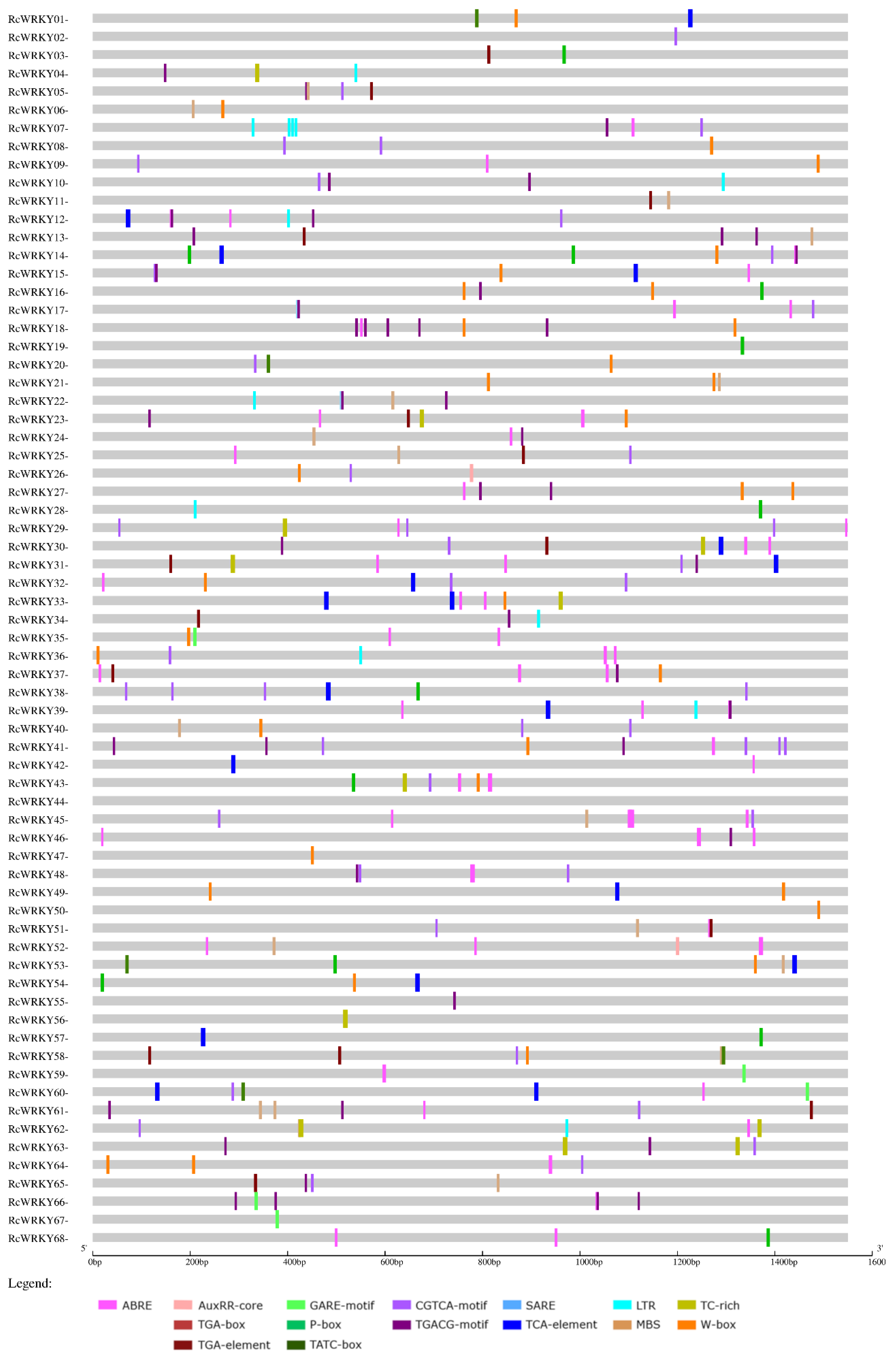
3.4. Promoter Analyses
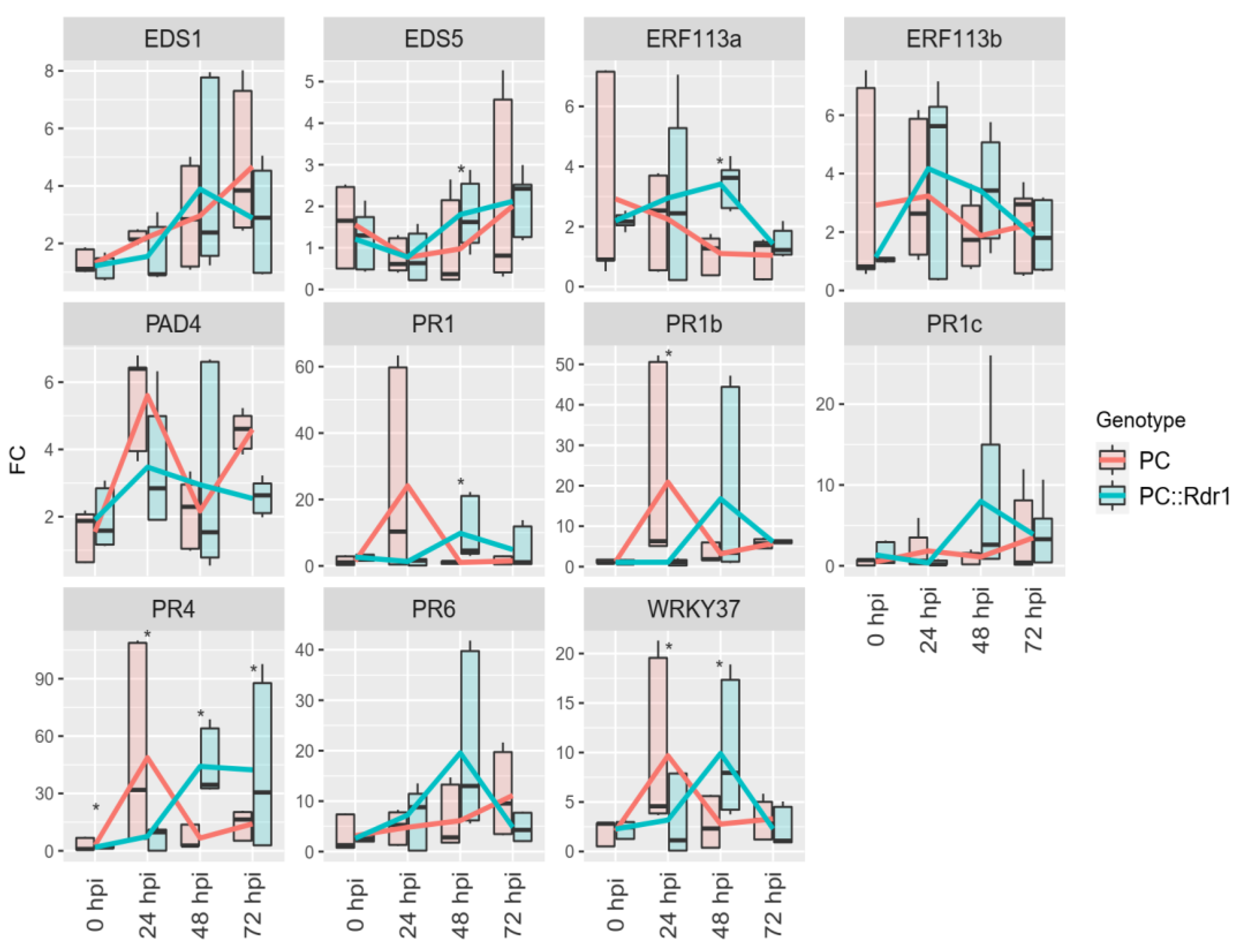
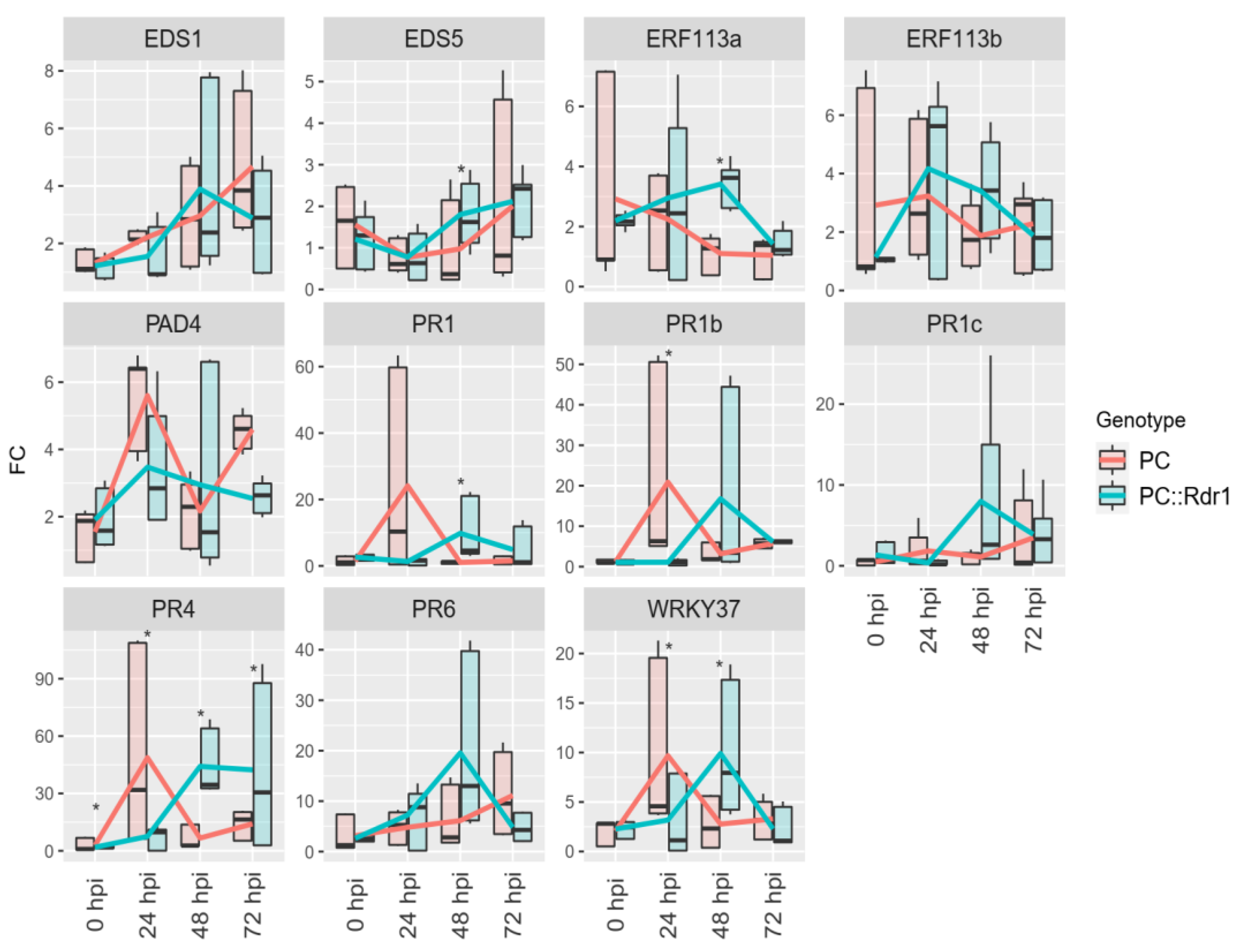
3.5. Expression of RcWRKY37 and Other Defence-Related Genes in Compatible and Incompatible Interactions with D. rosae
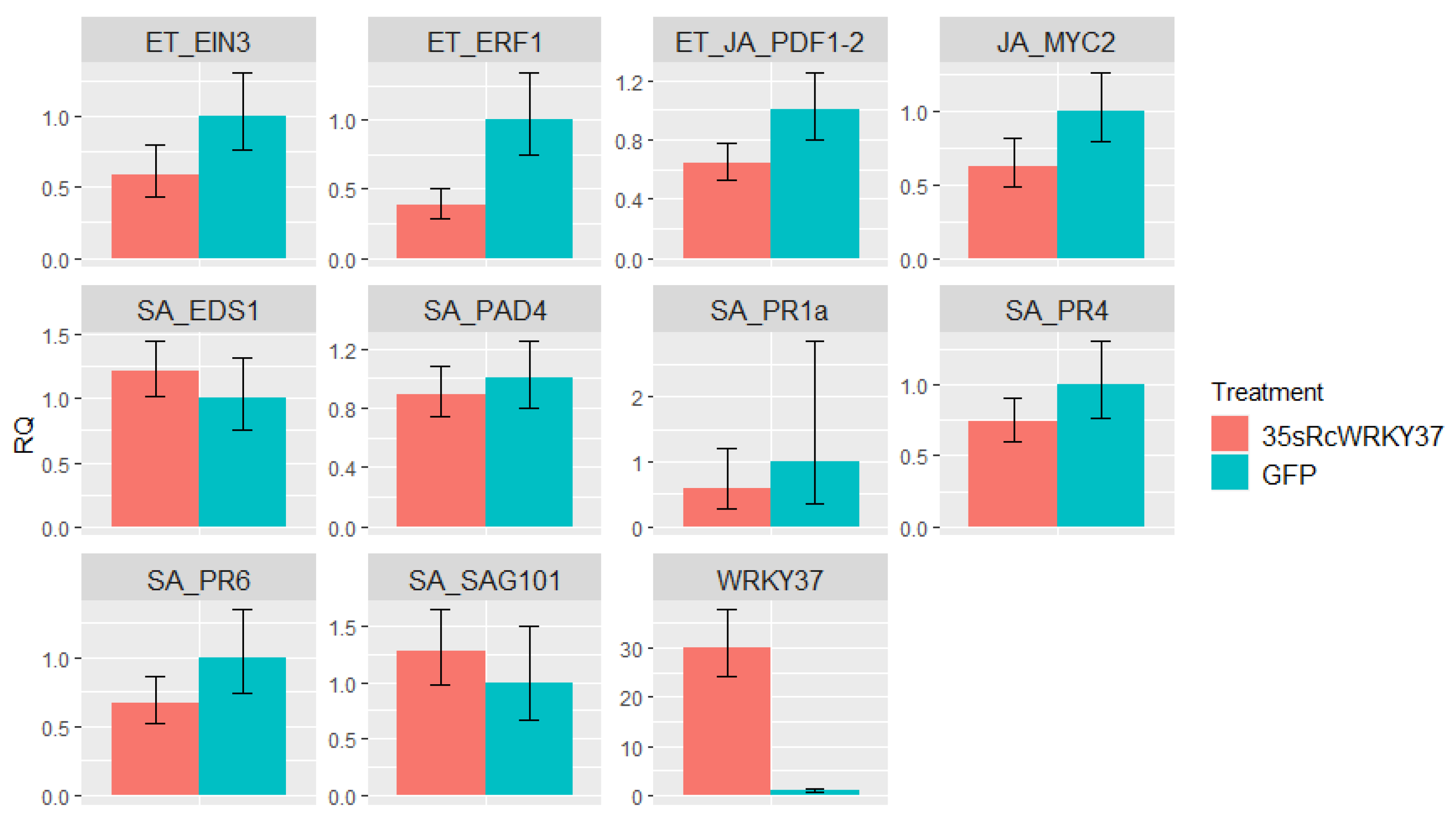
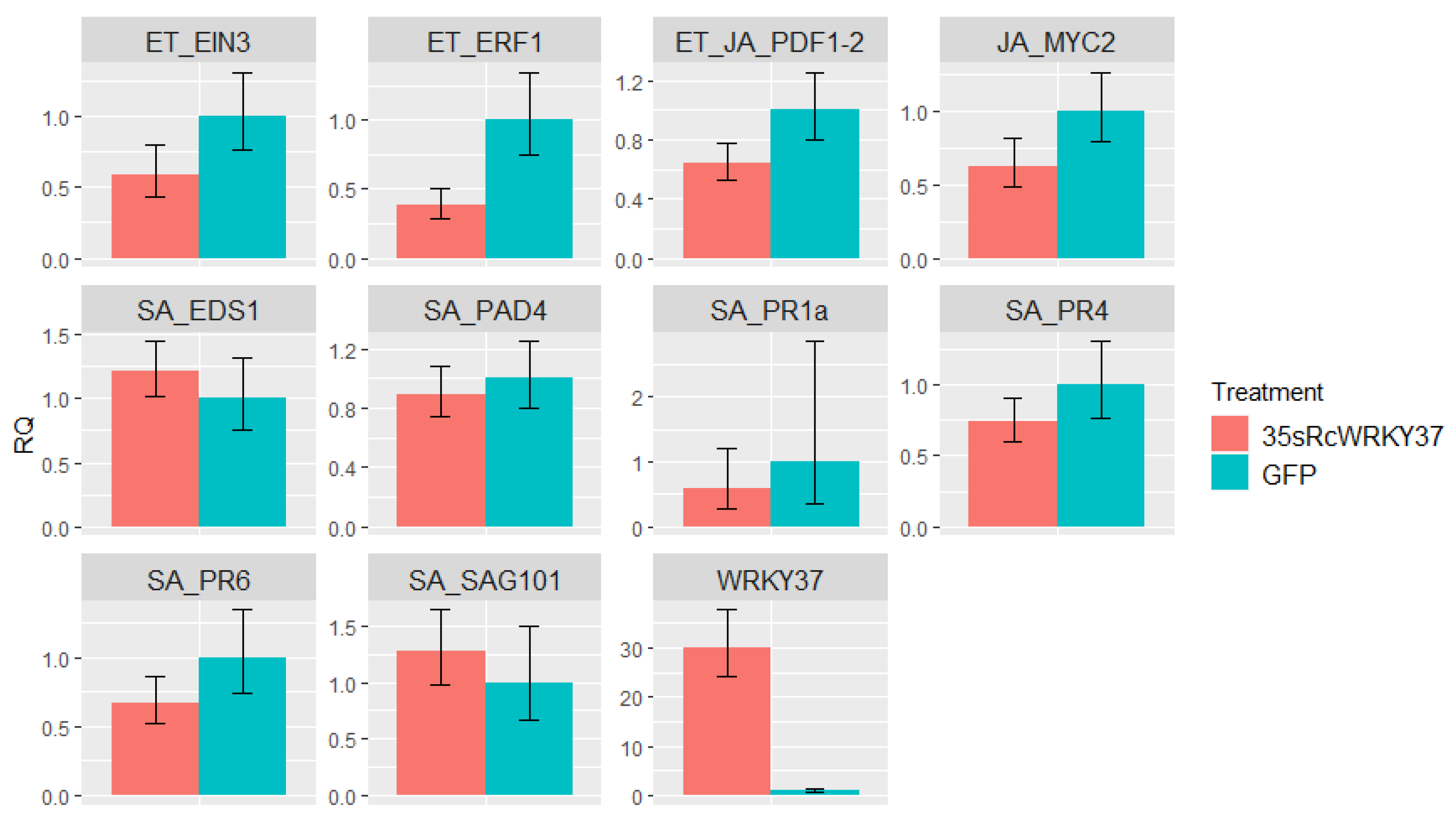
3.6. Effect of Transient Overexpression of RcWRKY37 on Defence-Related Genes
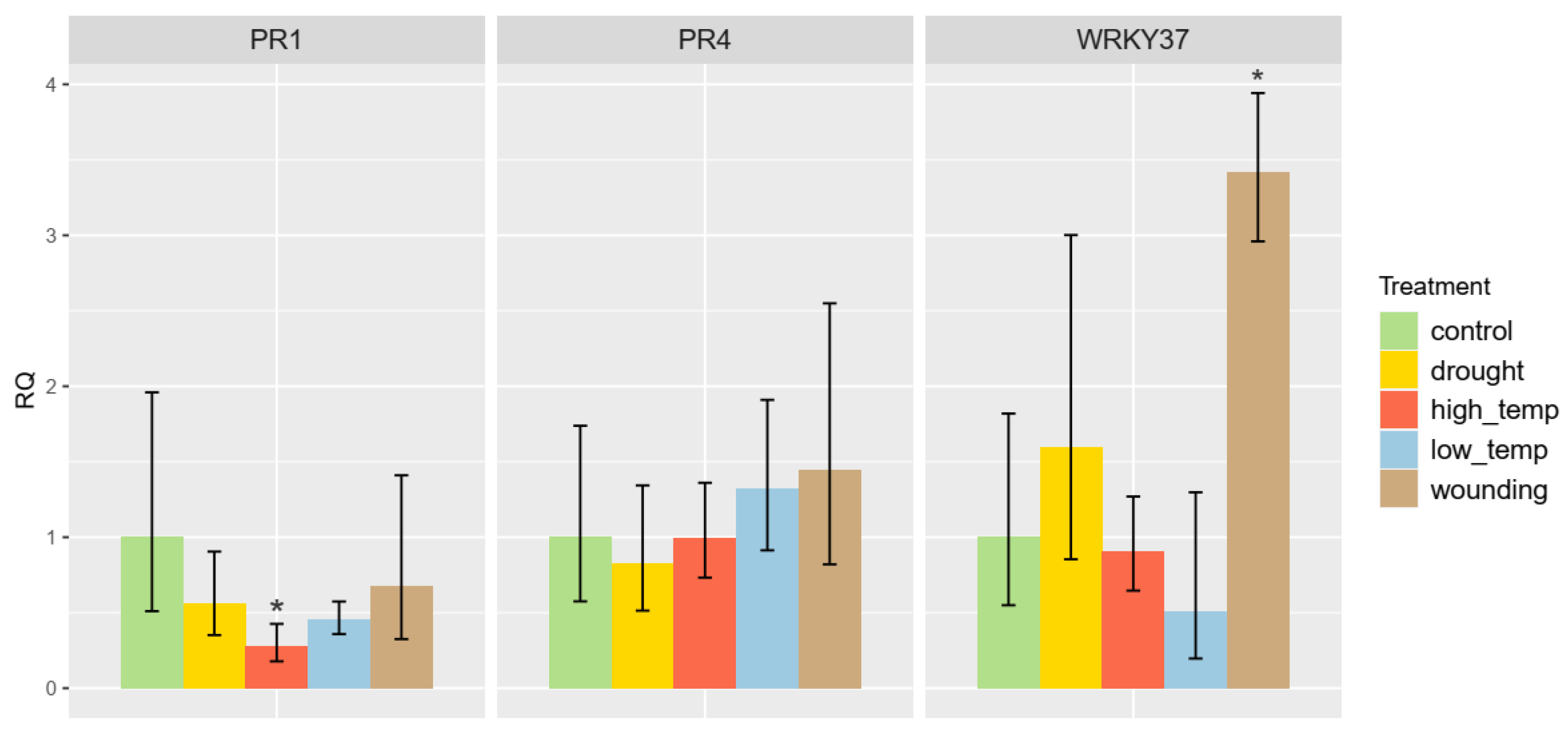
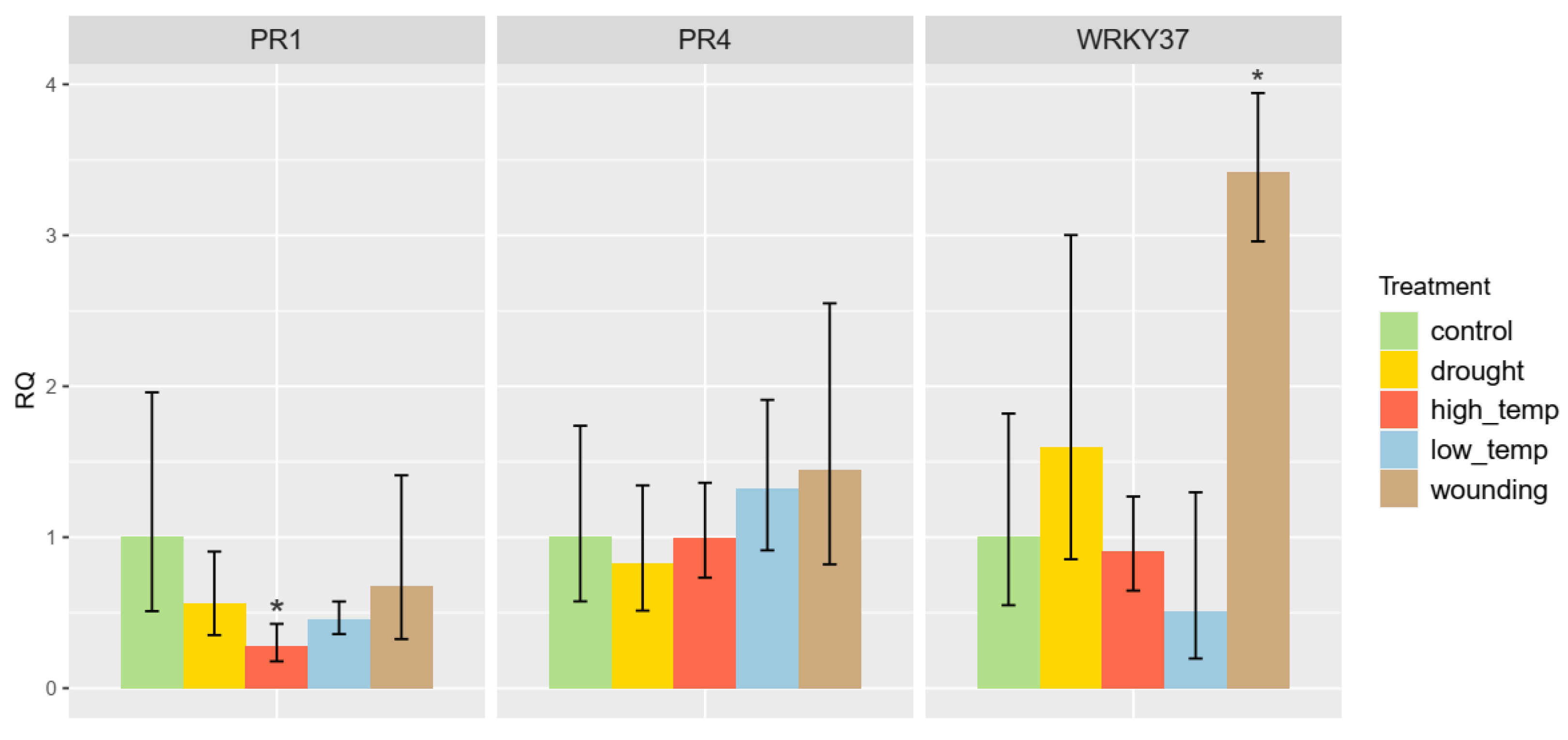
3.7. Abiotic Stress Induction of RcWRKY37
4. Discussion
4.1. Identification of WRKY Genes in R. chinensis
4.2. Transcriptional Regulation of WRKY Genes in Rose Leaves after Infection with D. rosae
4.3. Expression of RcWRKY37 and Other Defence-Related Genes in Susceptible and D. rosae-Resistant Rose Genotypes
4.4. The Role of RcWRKY37 in Different Signalling Pathways
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wang, H.; Chen, W.; Xu, Z.; Chen, M.; Yu, D. Functions of WRKYs in plant growth and development. Trends Plant Sci. 2023, 28, 630–645. [Google Scholar] [CrossRef] [PubMed]
- Rushton, P.J.; Macdonald, H.; Huttly, A.K.; Lazarus, C.M.; Hooley, R. Members of a new family of DNA-binding proteins bind to a conserved cis-element in the promoters of alpha-Amy2 genes. Plant Mol. Biol. 1995, 29, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Eulgem, T.; Rushton, P.J.; Robatzek, S.; Somssich, I.E. The WRKY superfamily of plant transcription factors. Trends Plant Sci. 2000, 5, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Rushton, P.J.; Torres, J.T.; Parniske, M.; Wernert, P.; Hahlbrock, K.; Somssich, I.E. Interaction of elicitor-induced DNA-binding proteins with elicitor response elements in the promoters of parsley PR1 genes. EMBO J. 1996, 15, 5690–5700. [Google Scholar] [CrossRef] [PubMed]
- Turck, F.; Zhou, A.; Somssich, I.E. Stimulus-Dependent, Promoter-Specific Binding of Transcription Factor WRKY1 to Its Native Promoter and the Defense-Related Gene PcPR1-1 in Parsley. Plant Cell 2004, 16, 2573–2585. [Google Scholar] [CrossRef]
- Yang, S.; Zhou, L.; Miao, L.; Shi, J.; Sun, C.; Fan, W.; Lan, J.; Chen, H.; Liu, L.; Dou, S.; et al. The expression and binding properties of the rice WRKY68 protein in the Xa21-mediated resistance response to Xanthomonas oryzae pv. Oryzae. J. Integr. Agric. 2016, 15, 2451–2460. [Google Scholar] [CrossRef]
- Anisimova, O.K.; Shchennikova, A.V.; Kochieva, E.Z.; Filyushin, M.A. Pathogenesis-Related Genes of PR1, PR2, PR4, and PR5 Families Are Involved in the Response to Fusarium Infection in Garlic (Allium sativum L.). Int. J. Mol. Sci. 2021, 22, 6688. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Chen, C.; Chen, Z. Expression profiles of the Arabidopsis WRKY gene superfamily during plant defense response. Plant Mol. Biol. 2003, 51, 21–37. [Google Scholar] [CrossRef]
- Wang, D.; Amornsiripanitch, N.; Dong, X. A genomic approach to identify regulatory nodes in the transcriptional network of systemic acquired resistance in plants. PLoS Pathog. 2006, 2, e123. [Google Scholar] [CrossRef] [PubMed]
- Andreasson, E.; Jenkins, T.; Brodersen, P.; Thorgrimsen, S.; Petersen, N.H.T.; Zhu, S.; Qiu, J.-L.; Micheelsen, P.; Rocher, A.; Petersen, M.; et al. The MAP kinase substrate MKS1 is a regulator of plant defense responses. EMBO J. 2005, 24, 2579–2589. [Google Scholar] [CrossRef]
- Qiu, J.-L.; Fiil, B.K.; Petersen, K.; Nielsen, H.B.; Botanga, C.J.; Thorgrimsen, S.; Palma, K.; Suarez-Rodriguez, M.C.; Sandbech-Clausen, S.; Lichota, J.; et al. Arabidopsis MAP kinase 4 regulates gene expression through transcription factor release in the nucleus. EMBO J. 2008, 27, 2214–2221. [Google Scholar] [CrossRef] [PubMed]
- Popescu, S.C.; Popescu, G.V.; Bachan, S.; Zhang, Z.; Gerstein, M.; Snyder, M.; Dinesh-Kumar, S.P. MAPK target networks in Arabidopsis thaliana revealed using functional protein microarrays. Genes Dev. 2009, 23, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Mao, G.; Meng, X.; Liu, Y.; Zheng, Z.; Chen, Z.; Zhang, S. Phosphorylation of a WRKY transcription factor by two pathogen-responsive MAPKs drives phytoalexin biosynthesis in Arabidopsis. Plant Cell 2011, 23, 1639–1653. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Chen, Y.; Bi, D.; Cao, Y.; Tong, J. Deciphering Evolutionary Dynamics of WRKY I Genes in Rosaceae Species. Front. Ecol. Evol. 2021, 9, 857. [Google Scholar] [CrossRef]
- Bao, F.; Ding, A.; Cheng, T.; Wang, J.; Zhang, Q. Genome-Wide Analysis of Members of the WRKY Gene Family and Their Cold Stress Response in Prunus mume. Genes 2019, 10, 911. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Gala, J.; Higuera, J.-J.; Rodríguez-Franco, A.; Muñoz-Blanco, J.; Amil-Ruiz, F.; Caballero, J.L. A Comprehensive Study of the WRKY Transcription Factor Family in Strawberry. Plants 2022, 11, 1585. [Google Scholar] [CrossRef] [PubMed]
- Raymond, O.; Gouzy, J.; Just, J.; Badouin, H.; Verdenaud, M.; Lemainque, A.; Vergne, P.; Moja, S.; Choisne, N.; Pont, C.; et al. The Rosa genome provides new insights into the domestication of modern roses. Nat. Genet. 2018, 50, 772–777. [Google Scholar] [CrossRef]
- Liu, X.; Li, D.; Zhang, S.; Xu, Y.; Zhang, Z. Genome-wide characterization of the rose (Rosa chinensis) WRKY family and role of RcWRKY41 in gray mold resistance. BMC Plant Biol. 2019, 19, 522. [Google Scholar] [CrossRef]
- Encinas-Villarejo, S.; Maldonado, A.M.; Amil-Ruiz, F.; de los Santos, B.; Romero, F.; Pliego-Alfaro, F.; Muñoz-Blanco, J.; Caballero, J.L. Evidence for a positive regulatory role of strawberry (Fragaria × ananassa) Fa WRKY1 and Arabidopsis at WRKY75 proteins in resistance. J. Exp. Bot. 2009, 60, 3043–3065. [Google Scholar] [CrossRef]
- Wei, W.; Hu, Y.; Han, Y.-T.; Zhang, K.; Zhao, F.-L.; Feng, J.-Y. The WRKY transcription factors in the diploid woodland strawberry Fragaria vesca: Identification and expression analysis under biotic and abiotic stresses. Plant Physiol. Biochem. PPB 2016, 105, 129–144. [Google Scholar] [CrossRef]
- Chen, X.; Liu, J.; Lin, G.; Wang, A.; Wang, Z.; Lu, G. Overexpression of AtWRKY28 and AtWRKY75 in Arabidopsis enhances resistance to oxalic acid and Sclerotinia sclerotiorum. Plant Cell Rep. 2013, 32, 1589–1599. [Google Scholar] [CrossRef]
- Hibrand Saint-Oyant, L.; Ruttink, T.; Hamama, L.; Kirov, I.; Lakwani, D.; Zhou, N.-N.; Bourke, P.M.; Daccord, N.; Leus, L.; Schulz, D.; et al. A high-quality sequence of Rosa chinensis to elucidate genome structure and ornamental traits. Nat. Plants 2018, 4, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Su, L.; Hu, S.; Xue, J.-Y.; Liu, H.; Liu, G.; Jiang, Y.; Du, J.; Qiao, Y.; Fan, Y.; et al. A chromosome-level genome assembly of rugged rose (Rosa rugosa) provides insights into its evolution, ecology, and floral characteristics. Hortic. Res. 2021, 8, 141. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, N.; Hirakawa, H.; Sato, S.; Otagaki, S.; Matsumoto, S.; Tabata, S.; Tanaka, Y. Genome structure of Rosa multiflora, a wild ancestor of cultivated roses. DNA Res. 2018, 25, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Zong, D.; Liu, H.; Gan, P.; Ma, S.; Liang, H.; Yu, J.; Li, P.; Jiang, T.; Sahu, S.K.; Yang, Q.; et al. Chromosomal-scale genomes of two Rosa species provide insights into genome evolution and ascorbate accumulation. Plant J. 2024, 117, 1264–1280. [Google Scholar] [CrossRef] [PubMed]
- Drewes-Alvarez, R. DISEASE|Black Spot. In Encyclopedia of Rose Science, Three-Volume Set; Debener, T., Gudin, S., Roberts, A., Eds.; Academic Press: Boston, MA, USA, 2003; pp. 148–153. [Google Scholar]
- von Malek, B.; Debener, T. Genetic analysis of resistance to blackspot (Diplocarpon rosae) in tetraploid roses. Theor. Appl. Genet. 1998, 96, 228–231. [Google Scholar] [CrossRef]
- Blechert, O.; Debener, T. Morphological characterization of the interaction between Diplocarpon rosae and various rose species. Plant Pathol. 2005, 54, 82–90. [Google Scholar] [CrossRef]
- Gachomo, E.W.; Dehne, H.-W.; Steiner, U. Microscopic evidence for the hemibiotrophic nature of Diplocarpon rosae, cause of black spot disease of rose. Physiol. Mol. Plant Pathol. 2006, 69, 86–92. [Google Scholar] [CrossRef]
- Gachomo, E.W.; Kotchoni, S.O. Detailed description of developmental growth stages of Diplocarpon rosae in Rosa: A core building block for efficient disease management. Ann. Appl. Biol. 2007, 151, 233–243. [Google Scholar] [CrossRef]
- Menz, I.; Straube, J.; Linde, M.; Debener, T. The TNL gene Rdr1 confers broad-spectrum resistance to Diplocarpon rosae. Mol. Plant Pathol. 2018, 19, 1104–1113. [Google Scholar] [CrossRef]
- Debener, T.; Malek, B.V.; Schreiber, M.; Drewes-Alvarez, R. Marker assisted background selection for the introgression of black spot resistance into cultivated roses. Eur. J. Hortic. Sci. 2003, 68, 245–252. [Google Scholar]
- Debener, T.; Drewes-Alvarez, R.; Rockstroh, K. Identification of five physiological races of blackspot, Diplocarpon rosae, Wolf on roses. Plant Breed. 1998, 117, 267–270. [Google Scholar] [CrossRef]
- Neu, E.; Domes, H.S.; Menz, I.; Kaufmann, H.; Linde, M.; Debener, T. Interaction of roses with a biotrophic and a hemibiotrophic leaf pathogen leads to differences in defense transcriptome activation. Plant Mol. Biol. 2019, 99, 299–316. [Google Scholar] [CrossRef]
- Kahl, G.; Molina, C.; Rotter, B.; Jüngling, R.; Frank, A.; Krezdorn, N.; Hoffmeier, K.; Winter, P. Reduced representation sequencing of plant stress transcriptomes. J. Plant Biochem. Biotechnol. 2012, 21, 119–127. [Google Scholar] [CrossRef]
- Kolde, R. Pheatmap: Pretty Heatmaps. R-Package Version 1.0.10. 2019. Available online: https://cran.r-project.org/web/packages/pheatmap/pheatmap.pdf (accessed on 29 January 2024).
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis. Nucl. Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Chao, J.-T.; Kong, Y.-Z.; Wang, Q.; Sun, Y.-H.; Gong, D.-P.; Lv, J.; Liu, G.-S. MapGene2Chrom, a tool to draw gene physical map based on Perl and SVG languages. Yi Chuan = Hereditas 2015, 37, 91–97. [Google Scholar] [PubMed]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. CABIOS 1992, 8, 275–282. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Jin, J.; Guo, A.-Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef] [PubMed]
- Yasmin, A. Identification and Molecular Characterization of the Rdr1 Resistance Gene from Roses; Gottfried Wilhelm Leibniz Universität Hannover: Hannover, Germany, 2011. [Google Scholar]
- Untergasser, A.; Nijveen, H.; Rao, X.; Bisseling, T.; Geurts, R.; Leunissen, J.A.M. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007, 35, W71–W74. [Google Scholar] [CrossRef] [PubMed]
- Menz, I.; Lakhwani, D.; Clotault, J.; Linde, M.; Foucher, F.; Debener, T. Analysis of the Rdr1 gene family in different Rosaceae genomes reveals an origin of an R-gene cluster after the split of Rubeae within the Rosoideae subfamily. PLoS ONE 2020, 15, e0227428. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Cao, X.; Shi, S.; Zhao, N.; Li, D.; Fang, P.; Chen, X.; Qi, W.; Zhang, Z. Comparative RNA-Seq analysis reveals a critical role for brassinosteroids in rose (Rosa hybrida) petal defense against Botrytis cinerea infection. BMC Genet. 2018, 19, 62. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lai, Z.; Shi, J.; Xiao, Y.; Chen, Z.; Xu, X. Roles of arabidopsis WRKY18, WRKY40 and WRKY60 transcription factors in plant responses to abscisic acid and abiotic stress. BMC Plant Biol. 2010, 10, 281. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Chen, Z.; Liu, Y.; Zhang, H.; Zhang, M.; Liu, Q.; Hong, X.; Zhu, J.-K.; Gong, Z. ABO3, a WRKY transcription factor, mediates plant responses to abscisic acid and drought tolerance in Arabidopsis. Plant J. 2010, 63, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Rushton, D.L.; Tripathi, P.; Rabara, R.C.; Lin, J.; Ringler, P.; Boken, A.K.; Langum, T.J.; Smidt, L.; Boomsma, D.D.; Emme, N.J.; et al. WRKY transcription factors: Key components in abscisic acid signalling. Plant Biotechnol. J. 2012, 10, 2–11. [Google Scholar] [CrossRef]
- Adie, B.A.T.; Pérez-Pérez, J.; Pérez-Pérez, M.M.; Godoy, M.; Sánchez-Serrano, J.-J.; Schmelz, E.A.; Solano, R. ABA is an essential signal for plant resistance to pathogens affecting JA biosynthesis and the activation of defenses in Arabidopsis. Plant Cell 2007, 19, 1665–1681. [Google Scholar] [CrossRef]
- Hobo, T.; Asada, M.; Kowyama, Y.; Hattori, T. ACGT-containing abscisic acid response element (ABRE) and coupling element 3 (CE3) are functionally equivalent. Plant J. Cell Mol. Biol. 1999, 19, 679–689. [Google Scholar] [CrossRef]
- Hattori, T.; Totsuka, M.; Hobo, T.; Kagaya, Y.; Yamamoto-Toyoda, A. Experimentally determined sequence requirement of ACGT-containing abscisic acid response element. Plant Cell Physiol. 2002, 43, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, K.; Sakuma, Y.; Kasuga, M.; Ito, Y.; Seki, M.; Goda, H.; Shimada, Y.; Yoshida, S.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Identification of cold-inducible downstream genes of the Arabidopsis DREB1A/CBF3 transcriptional factor using two microarray systems. Plant J. Cell Mol. Biol. 2004, 38, 982–993. [Google Scholar] [CrossRef]
- Ciolkowski, I.; Wanke, D.; Birkenbihl, R.P.; Somssich, I.E. Studies on DNA-binding selectivity of WRKY transcription factors lend structural clues into WRKY-domain function. Plant Mol. Biol. 2008, 68, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Raventós, D.; Jensen, A.B.; Rask, M.B.; Casacuberta, J.M.; Mundy, J.; San Segundo, B. A 20 bp cis-acting element is both necessary and sufficient to mediate elicitor response of a maize PRms gene. Plant J. Cell Mol. Biol. 1995, 7, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Liu, Z.-Q.; Xu, Y.-H.; Lu, K.; Wang, X.-F.; Zhang, D.-P. Auto- and Cross-repression of Three Arabidopsis WRKY Transcription Factors WRKY18, WRKY40, and WRKY60 Negatively Involved in ABA Signaling. J. Plant Growth Regul. 2013, 32, 399–416. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, L.; Xiang, S.; Chen, Y.; Zhang, H.; Yu, D. The transcription factor WRKY75 positively regulates jasmonate-mediated plant defense to necrotrophic fungal pathogens. J. Exp. Bot. 2021, 72, 1473–1489. [Google Scholar] [CrossRef]
- Higuera, J.J.; Garrido-Gala, J.; Lekhbou, A.; Arjona-Girona, I.; Amil-Ruiz, F.; Mercado, J.A.; Pliego-Alfaro, F.; Muñoz-Blanco, J.; López-Herrera, C.J.; Caballero, J.L. The Strawberry FaWRKY1 Transcription Factor Negatively Regulates Resistance to Colletotrichum acutatum in Fruit Upon Infection. Front. Plant Sci. 2019, 10, 480. [Google Scholar] [CrossRef]
- Guo, P.; Li, Z.; Huang, P.; Li, B.; Fang, S.; Chu, J.; Guo, H. A Tripartite Amplification Loop Involving the Transcription Factor WRKY75, Salicylic Acid, and Reactive Oxygen Species Accelerates Leaf Senescence. Plant Cell 2017, 29, 2854–2870. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Promoters of: | Upregulated WRKYs (Total Number: 18) | Downregulated WRKYs (Total Number: 9) | Nonregulated WRKYs (Total Number: 41) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Motif in Connection with | Motif | No. of Motifs | No. of Sequences | Proportion of Sequences with These Motifs in the Promoter Region of Upregulated WRKYs (%). | No. of Motifs | No. of Sequences | Proportion of Sequences with These Motifs in the Promoter Region of Downregulated WRKYs (%). | No. of Sequences | Proportion of Sequences with These Motifs in the Promoter Region of Nonregulated WRKYs (%). |
| ABA | ABRE | 43 | 15 | 83.3 | 23 | 9 | 100 | 30 | 73.2 |
| Auxin (total) | 7 | 38.9 | 5 | 55.6 | 15 | 36.6 | |||
| AuxRR-core | 1 | 1 | 5.6 | 0 | 0 | 0 | 2 | 4.9 | |
| TGA-box | 1 | 1 | 5.6 | 0 | 0 | 0 | 2 | 4.9 | |
| TGA-element | 7 | 5 | 27.8 | 5 | 5 | 55.6 | 11 | 26.8 | |
| Gibberellin (total) | 9 | 50.0 | 4 | 44.4 | 24 | 58.5 | |||
| GARE-motif | 2 | 2 | 11.1 | 2 | 2 | 22.2 | 8 | 19.5 | |
| P-box | 6 | 6 | 33.3 | 2 | 2 | 22.2 | 12 | 29.3 | |
| TATC-box | 4 | 3 | 16.7 | 0 | 0 | 0 | 6 | 14.6 | |
| JA (total) | 13 | 72.2 | 6 | 67 | 27 | 65.9 | |||
| CGTCA-motif | 18 | 13 | 72.2 | 14 | 6 | 66.7 | 27 | 65.9 | |
| TGACG-motif | 16 | 12 | 66.7 | 13 | 6 | 66.7 | 27 | 65.9 | |
| SA (total) | 6 | 33.3 | 3 | 33.3 | 15 | 36.6 | |||
| SARE | 1 | 1 | 5.6 | 0 | 0 | 0 | 0 | 0 | |
| TCA-element | 5 | 5 | 27.8 | 3 | 3 | 33.3 | 14 | 34.1 | |
| Low temperature | LTR | 9 | 8 | 44.4 | 7 | 3 | 33.3 | 13 | 31.7 |
| Drought | MBS | 11 | 9 | 50.0 | 4 | 3 | 33.3 | 14 | 34.1 |
| Biotic stress (total) | 13 | 72.2 | 6 | 66.7 | 27 | 65.9 | |||
| TC-rich | 7 | 5 | 27.8 | 4 | 4 | 44.4 | 14 | 34.1 | |
| W-box | 19 | 13 | 72.2 | 5 | 3 | 33.3 | 20 | 48.8 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domes, H.S.; Debener, T. Genome-Wide Analysis of the WRKY Transcription Factor Family in Roses and Their Putative Role in Defence Signalling in the Rose–Blackspot Interaction. Plants 2024, 13, 1066. https://doi.org/10.3390/plants13081066
Domes HS, Debener T. Genome-Wide Analysis of the WRKY Transcription Factor Family in Roses and Their Putative Role in Defence Signalling in the Rose–Blackspot Interaction. Plants. 2024; 13(8):1066. https://doi.org/10.3390/plants13081066
Chicago/Turabian StyleDomes, Helena Sophia, and Thomas Debener. 2024. "Genome-Wide Analysis of the WRKY Transcription Factor Family in Roses and Their Putative Role in Defence Signalling in the Rose–Blackspot Interaction" Plants 13, no. 8: 1066. https://doi.org/10.3390/plants13081066
APA StyleDomes, H. S., & Debener, T. (2024). Genome-Wide Analysis of the WRKY Transcription Factor Family in Roses and Their Putative Role in Defence Signalling in the Rose–Blackspot Interaction. Plants, 13(8), 1066. https://doi.org/10.3390/plants13081066