
Identification of a Highly Virulent Verticillium nonalfalfae Strain Vn011 and Expression Analysis of Its Orphan Genes During Potato Inoculation

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Result
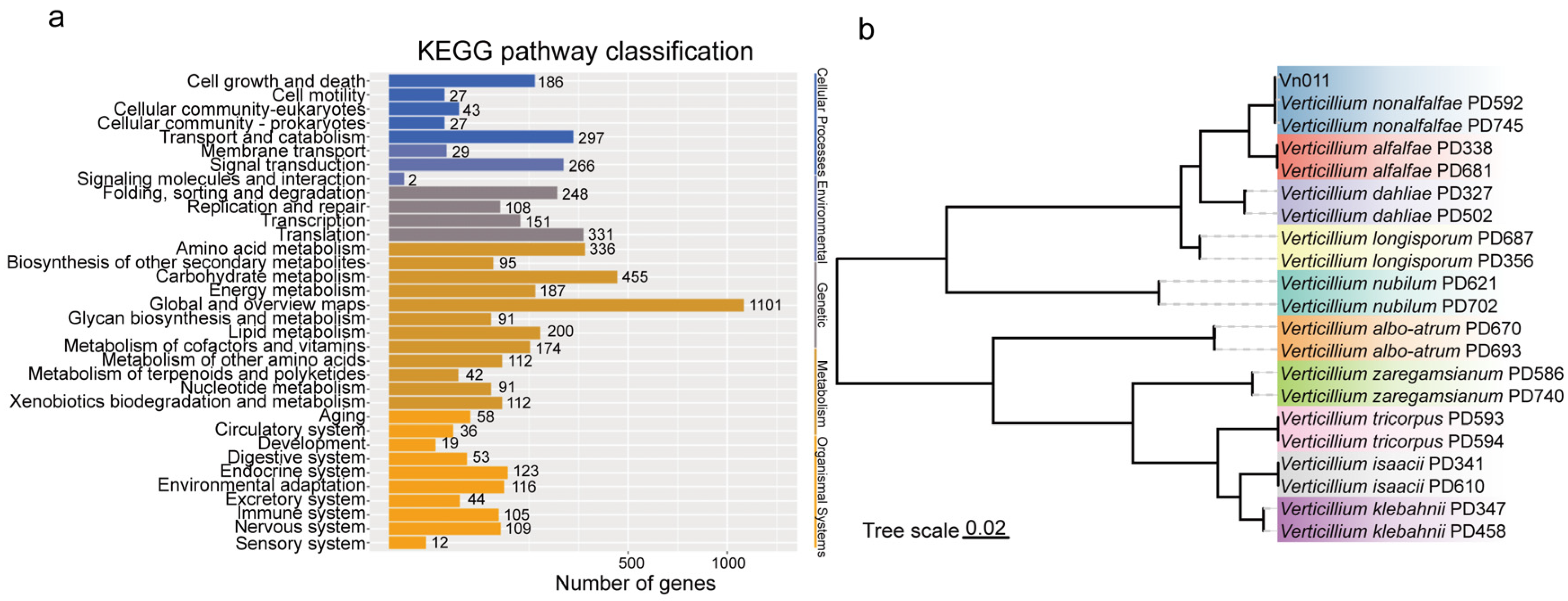
2.1. Isolation of a Highly Virulent Verticillium Wilt Pathogen from Potato Fields
2.2. Identification of V. nonalfalfae via PCR and Sequencing Analysis
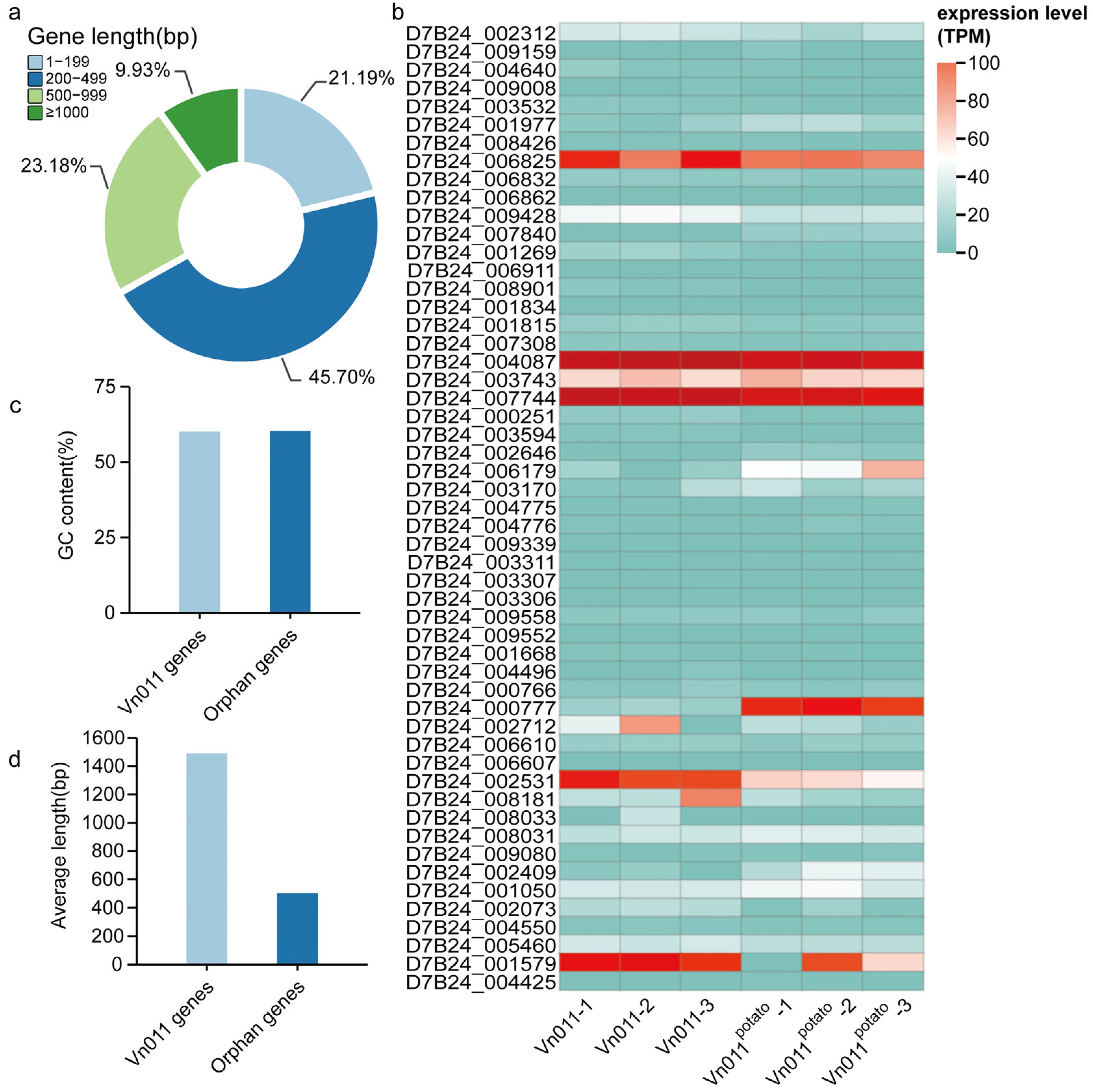
2.3. Identification of Orphan Genes
2.4. Expression Patterns of Orphan Genes After Inoculation
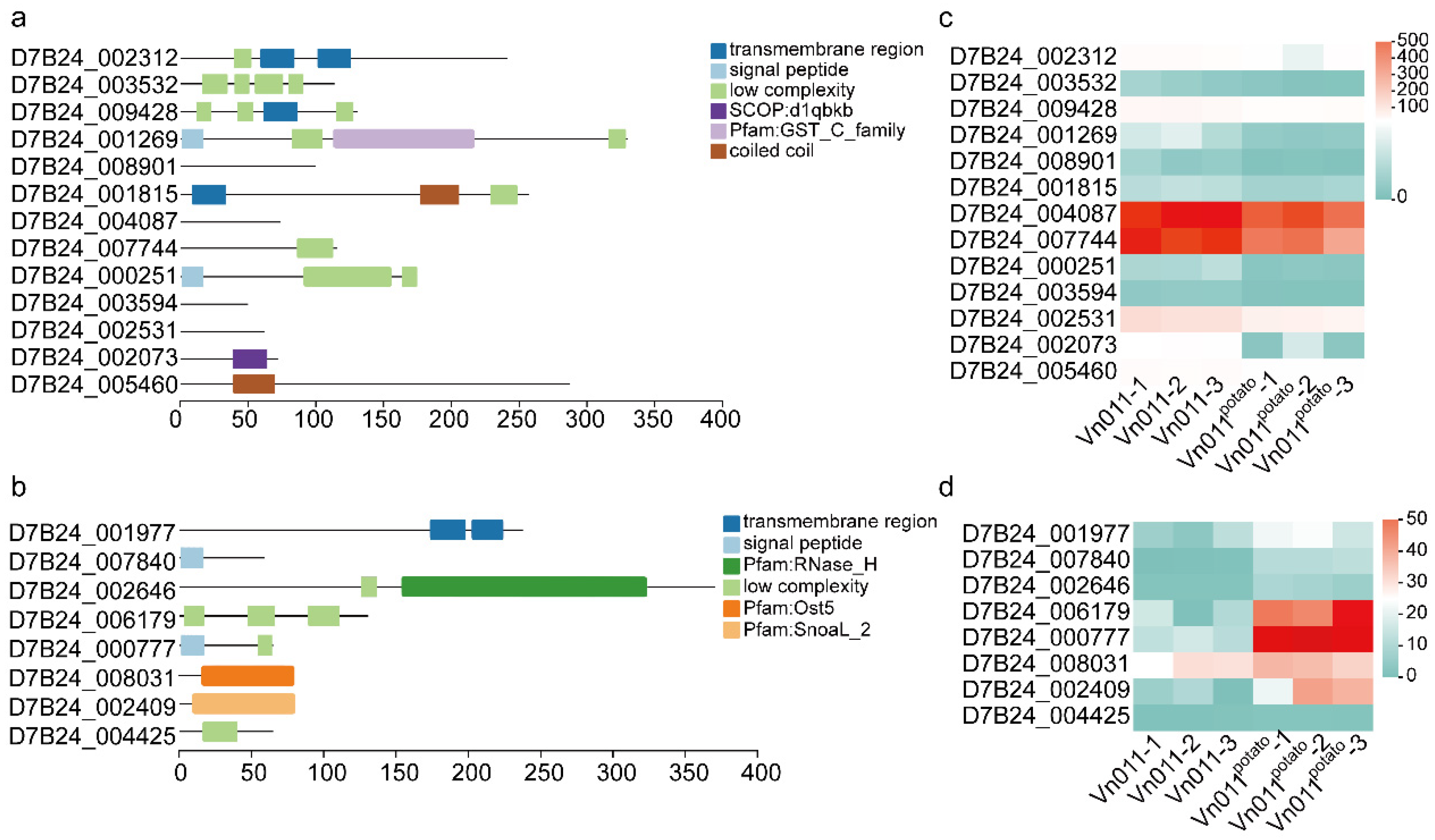
2.5. Functional Features of Orphan Genes
3. Discussion
4. Methods
4.1. Plants Cultivation
4.2. Isolation and Purification of Verticillium from Potato
4.3. Pathogen Inoculation and Identification
4.4. Genome Sequencing of V. nonalfalfae
4.5. Identification of Orphan Genes in Vn011
4.6. Differential Gene Expression Analysis
4.7. Observation of Pathogen Infection
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Birch, P.R.J.; Bryan, G.; Fenton, B.; Gilroy, E.M.; Hein, I.; Jones, J.T.; Prashar, A.; Taylor, M.A.; Torrance, L.; Toth, I.K. Crops that feed the world 8: Potato: Are the trends of increased global production sustainable? Food Secur. 2012, 4, 477–508. [Google Scholar] [CrossRef]
- Tian, J.; Chen, J.; Ye, X.; Chen, S. Health benefits of the potato affected by domestic cooking: A review. Food Chem. 2016, 202, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Yang, R.; Zhang, L.; Wei, Q.; Zhang, Y.; Wang, Y.; Shi, Y. A review of potato salt tolerance. Int. J. Mol. Sci. 2023, 24, 10726. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, J.K.; Buckseth, T.; Zinta, R.; Bhatia, N.; Dalamu, D.; Naik, S.; Poonia, A.K.; Kardile, H.B.; Challam, C.; Singh, R.K.; et al. Germplasm, breeding, and genomics in potato improvement of biotic and abiotic stresses tolerance. Front. Plant Sci. 2022, 13, 805671. [Google Scholar] [CrossRef]
- Dung, J.; Ingram, J.; Cummings, T.; Johnson, D. Impact of seed lot infection on the development of black dot and Verticillium wilt of potato in Washington. Plant Dis. 2012, 96, 1179–1184. [Google Scholar] [CrossRef]
- Smith, H.C. The morphology of Verticillium albo-atrum, V. dahliae, and V. tricorpus. N. Z. J. Agric. Res. 2012, 8, 450–478. [Google Scholar] [CrossRef]
- Borza, T.; Beaton, B.; Govindarajan, A.; Gao, X.; Liu, Y.; Ganga, Z.; Wang-Pruski, G. Incidence and abundance of Verticillium dahliae in soil from various agricultural fields in Prince Edward Island, Canada. Eur. J. Plant Pathol. 2017, 151, 825–830. [Google Scholar] [CrossRef]
- Wang, L.; Fu, G.; Ma, J.; Rziwangguli Lang, Y.; Patiguli, L.K. Isolation and Identification of the Pathogens Causing Verticillium wilt of Potato in Urumqi and Changji Area, Xinjiang. Xinjiang Agric. Sci. 2014, 51, 667–672. [Google Scholar]
- Inderbitzin, P.; Bostock, R.M.; Davis, R.M.; Usami, T.; Platt, H.W.; Subbarao, K.V. Phylogenetics and taxonomy of the fungal vascular wilt pathogen Verticillium, with the descriptions of five new species. PLoS ONE 2011, 6, e28341. [Google Scholar] [CrossRef]
- Inderbitzin, P.; Davis, R.M.; Bostock, R.M.; Subbarao, K.V. Identification and differentiation of Verticillium species and V. longisporum lineages by simplex and multiplex PCR assays. PLoS ONE 2013, 8, e65990. [Google Scholar] [CrossRef]
- Wang, D.; Wen, S.; Zhao, Z.; Long, Y.; Fan, R. Hypothetical protein VDAG_07742 is required for Verticillium dahliae pathogenicity in potato. Int. J. Mol. Sci. 2023, 24, 3630. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Addrah, M.E.; Zhang, Y.; Jia, R.; Kang, L.; Zhao, J.; Zhang, J. Identification and comparison of biological characteristics and pathogenicity of different mating types of V. dahliae isolated from potato and sunflower. Sci. Rep. 2022, 12, 12840. [Google Scholar] [CrossRef] [PubMed]
- Flajsman, M.; Mandelc, S.; Radisek, S.; Stajner, N.; Jakse, J.; Kosmelj, K.; Javornik, B. Identification of novel virulence-associated proteins secreted to xylem by Verticillium nonalfalfae during colonization of hop plants. Mol. Plant-Microbe Interact. 2016, 29, 362–373. [Google Scholar] [CrossRef]
- Lee, H.W.; Ho, W.W.H.; Alexander, B.J.R.; Baskarathevan, J. A rapid qPCR for the detection of Verticillium nonalfalfae MLST2—A highly pathogenic fungus on Kiwifruit. Plant Dis. 2022, 106, 2348–2354. [Google Scholar] [CrossRef]
- Lechner, Y.; Maschek, O.; Kirisits, T.; Halmschlager, E. Further pathogenicity testing of Verticillium nonalfalfae, a biocontrol agent against the invasive Tree of Heaven (Ailanthus altissima), on non-target tree species in Europe. Phytoparasitica 2022, 51, 113–130. [Google Scholar] [CrossRef] [PubMed]
- Fradin, E.F.; Thomma, B.P. Physiology and molecular aspects of Verticillium wilt diseases caused by V. dahliae and V. albo-atrum. Mol. Plant Pathol. 2006, 7, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Tran, V.-T.; Braus-Stromeyer, S.A.; Kusch, H.; Reusche, M.; Kaever, A.; Kühn, A.; Valerius, O.; Landesfeind, M.; Aßhauer, K.; Tech, M.; et al. Verticillium transcription activator of adhesion Vta2 suppresses microsclerotia formation and is required for systemic infection of plant roots. New Phytol. 2014, 202, 565–581. [Google Scholar] [CrossRef]
- Siew, N.; Fischer, D. Analysis of singleton ORFans in fully sequenced microbial genomes. Proteins 2003, 53, 241–251. [Google Scholar] [CrossRef]
- Vakirlis, N.; Carvunis, A.R.; McLysaght, A. Synteny-based analyses indicate that sequence divergence is not the main source of orphan genes. Elife 2020, 9, e53500. [Google Scholar] [CrossRef]
- Jiang, M.; Li, X.; Dong, X.; Zu, Y.; Zhan, Z.; Piao, Z.; Lang, H. Research advances and prospects of orphan genes in plants. Front. Plant Sci. 2022, 13, 947129. [Google Scholar] [CrossRef]
- Wissler, L.; Gadau, J.; Simola, D.F.; Helmkampf, M.; Bornberg-Bauer, E. Mechanisms and dynamics of orphan gene emergence in insect genomes. Genome Biol. Evol. 2013, 5, 439–455. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Panda, A.; Ghosh, T.C. Elucidating evolutionary features and functional implications of orphan genes in Leishmania major. Infect. Genet. Evol. 2015, 32, 330–337. [Google Scholar] [CrossRef]
- Prabh, N.; Rödelsperger, C. De Novo, Divergence, and mixed origin contribute to the emergence of orphan genes in Pristionchus Nematodes. G3 2019, 9, 2277–2286. [Google Scholar] [CrossRef]
- Wang, Z.; Jin, Q.; Li, Q.; Ou, X.; Li, S.; Liu, Z.; Huang, J. Multiplex PCR identification of Aspergillus cristatus and Aspergillus chevalieri in Liupao tea based on orphan genes. Foods 2022, 11, 2217. [Google Scholar] [CrossRef]
- Entwistle, S.; Li, X.; Yin, Y. Orphan genes shared by pathogenic genomes are more associated with bacterial pathogenicity. mSystems 2019, 4, 1128. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Hei, R.; Yang, Y.; Zhang, S.; Wang, Q.; Wang, W.; Zhang, Q.; Yan, M.; Zhu, G.; Huang, P.; et al. An orphan protein of Fusarium graminearum modulates host immunity by mediating proteasomal degradation of TaSnRK1α. Nat. Commun. 2020, 11, 4382. [Google Scholar] [CrossRef]
- Merényi, Z.; Krizsán, K.; Sahu, N.; Liu, X.-B.; Bálint, B.; Stajich, J.E.; Spatafora, J.W.; Nagy, L.G. Genomes of fungi and relatives reveal delayed loss of ancestral gene families and evolution of key fungal traits. Nat. Ecol. Evol. 2023, 7, 1221–1231. [Google Scholar] [CrossRef]
- Lan, Y.; Cong, Q.; Yu, Q.; Liu, L.; Cui, X.; Li, X.; Wang, Q.; Yang, S.; Yu, H.; Kong, Y. Genome sequencing of three pathogenic fungi provides insights into the evolution and pathogenic mechanisms of the cobweb disease on cultivated mushrooms. Foods 2024, 13, 2779. [Google Scholar] [CrossRef]
- Batista, T.M.; Hilario, H.O.; de Brito, G.A.M.; Moreira, R.G.; Furtado, C.; de Menezes, G.C.A.; Rosa, C.A.; Rosa, L.H.; Franco, G.R. Whole-genome sequencing of the endemic Antarctic fungus Antarctomyces pellizariae reveals an ice-binding protein, a scarce set of secondary metabolites gene clusters and provides insights on Thelebolales phylogeny. Genomics 2020, 112, 2915–2921. [Google Scholar] [CrossRef]
- Vidha, S.; Kuntal, P.; Hsuan, P.; Maria Victoria, A.-P.; Aileen, B.; Avinash, K.; Antonio, D.P.; Amey, R. Molecular dialogue during host manipulation by the vascular wilt fungus Fusarium oxysporum. Annu. Rev. Phytopathol. 2024, 62, 97–126. [Google Scholar] [CrossRef]
- Oliveira-Garcia, E.; Yan, X.; Oses-Ruiz, M.; de Paula, S.; Talbot, N.J. Effector-triggered susceptibility by the rice blast fungus Magnaporthe oryzae. New Phytol. 2023, 241, 1007–1020. [Google Scholar] [CrossRef]
- Kanja, C.; Hammond-Kosack, K.E. Proteinaceous effector discovery and characterization in filamentous plant pathogens. Mol. Plant Pathol. 2020, 21, 1353–1376. [Google Scholar] [CrossRef]
- Sauerborn, E.; Corredor, N.C.; Reska, T.; Perlas, A.; Atum, S.V.d.F.; Goldman, N.; Wantia, N.; da Costa, C.P.; Foster-Nyarko, E.; Urban, L. Detection of hidden antibiotic resistance through real-time genomics. Nat. Commun. 2024, 15, 5494. [Google Scholar] [CrossRef] [PubMed]
- Amselem, J.; Cuomo, C.A.; van Kan, J.A.; Viaud, M.; Benito, E.P.; Couloux, A.; Coutinho, P.M.; de Vries, R.P.; Dyer, P.S.; Fillinger, S.; et al. Genomic analysis of the necrotrophic fungal pathogens Sclerotinia sclerotiorum and Botrytis cinerea. PLoS Genet. 2011, 7, e1002230. [Google Scholar] [CrossRef] [PubMed]
- Bédard, C.; Gagnon-Arsenault, I.; Boisvert, J.; Plante, S.; Dubé, A.K.; Pageau, A.; Fijarczyk, A.; Sharma, J.; Maroc, L.; Shapiro, R.S.; et al. Most azole resistance mutations in the Candida albicans drug target confer cross-resistance without intrinsic fitness cost. Nat. Microbiol. 2024, 9, 3025–3040. [Google Scholar] [CrossRef]
- Alouane, T.; Rimbert, H.; Bormann, J.; González-Montiel, G.A.; Loesgen, S.; Schäfer, W.; Freitag, M.; Langin, T.; Bonhomme, L. Comparative genomics of eight Fusarium graminearum strains with contrasting aggressiveness reveals an expanded open pangenome and extended effector content signatures. Int. J. Mol. Sci. 2021, 22, 6257. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; Klosterman, S.J.; Hu, X.P.; Dai, X.F.; Subbarao, K.V. Key insights and research prospects at the dawn of the population genomics era for Verticillium dahliae. Annu. Rev. Phytopathol. 2021, 59, 31–51. [Google Scholar] [CrossRef]
- van de Vossenberg, B.T.L.H.; Warris, S.; Nguyen, H.D.T.; van Gent-Pelzer, M.P.E.; Joly, D.L.; van de Geest, H.C.; Bonants, P.J.M.; Smith, D.S.; Lévesque, C.A.; van der Lee, T.A.J. Comparative genomics of chytrid fungi reveal insights into the obligate biotrophic and pathogenic lifestyle of Synchytrium endobioticum. Sci. Rep. 2019, 9, 8672. [Google Scholar] [CrossRef]
- Yoshida, K.; Schuenemann, V.J.; Cano, L.M.; Pais, M.; Mishra, B.; Sharma, R.; Lanz, C.; Martin, F.N.; Kamoun, S.; Krause, J.; et al. The rise and fall of the Phytophthora infestans lineage that triggered the Irish potato famine. Elife 2013, 2, e00731. [Google Scholar] [CrossRef]
- Gorrie, C.; Higgs, C.; Carter, G.; Stinear, T.P.; Howden, B. Genomics of vancomycin-resistant Enterococcus faecium. Microb. Genom. 2019, 5, e000283. [Google Scholar] [CrossRef]
- Ma, L.-J.; Geiser, D.M.; Proctor, R.H.; Rooney, A.P.; O’Donnell, K.; Trail, F.; Gardiner, D.M.; Manners, J.M.; Kazan, K. Fusarium pathogenomics. Annu. Rev. Microbiol. 2013, 67, 399–416. [Google Scholar] [CrossRef]
- Li, J.; Cornelissen, B.; Rep, M. Host-specificity factors in plant pathogenic fungi. Fungal Genet. Biol. 2020, 144, 103447. [Google Scholar] [CrossRef]
- Lorrain, C.; dos Santos, K.C.G.; Germain, H.; Hecker, A.; Duplessis, S. Advances in understanding obligate biotrophy in rust fungi. New Phytol. 2018, 222, 1190–1206. [Google Scholar] [CrossRef]
- Hu, Y.; Gong, C.; Yang, Z.; Han, H.; Tian, T.; Yang, X.; Xie, W.; Wang, S.; Wu, Q.; Zhou, X.; et al. Functional divergence of plant-derived Thaumatin-like protein genes in two closely related whitefly species. Adv. Sci. 2025. [Google Scholar] [CrossRef] [PubMed]
- E Cook, D.; Kramer, H.M.; E Torres, D.; Seidl, M.F.; Thomma, B.P.H.J. A unique chromatin profile defines adaptive genomic regions in a fungal plant pathogen. Elife 2020, 9, e62208. [Google Scholar] [CrossRef]
- Chen, X.; Fang, D.; Xu, Y.; Duan, K.; Yoshida, S.; Yang, S.; Sahu, S.K.; Fu, H.; Guang, X.; Liu, M.; et al. Balanophora genomes display massively convergent evolution with other extreme holoparasites and provide novel insights into parasite-host interactions. Nat. Plants 2023, 9, 1627–1642. [Google Scholar] [CrossRef]
- Rong, Z.; Ren, T.; Yue, J.; Zhou, W.; Liang, D.; Zhang, C. Characterization, genome sequencing, and development of a rapid PCR identification primer for Fusarium oxysporum f. sp. crocus, a new forma specialis causing saffron corm rot. Plants 2024, 13, 3166. [Google Scholar] [CrossRef]
- Fouché, S.; Plissonneau, C.; Croll, D. The birth and death of effectors in rapidly evolving filamentous pathogen genomes. Curr. Opin. Microbiol. 2018, 46, 34–42. [Google Scholar] [CrossRef]
- Cheng, L.; Han, Q.; Hao, Y.; Qiao, Z.; Li, M.; Liu, D.; Yin, H.; Li, T.; Long, W.; Luo, S.; et al. Genome assembly of Stewartia sinensis reveals origin and evolution of orphan genes in Theaceae. Commun. Biol. 2025, 8, 354. [Google Scholar] [CrossRef]
- Dai, J.; Patzke, C.; Liakath-Ali, K.; Seigneur, E.; Südhof, T.C. GluD1 is a signal transduction device disguised as an ionotropic receptor. Nature 2021, 595, 261–265. [Google Scholar] [CrossRef]
- Xia, S.; Chen, J.; Arsala, D.; Emerson, J.J.; Long, M. Functional innovation through new genes as a general evolutionary process. Nat. Genet. 2025, 57, 295–309. [Google Scholar] [CrossRef] [PubMed]
- O’Conner, S.; Li, L. Mitochondrial fostering: The mitochondrial genome may play a role in plant orphan gene evolution. Front. Plant Sci. 2020, 11, 600117. [Google Scholar] [CrossRef]
- Athanasouli, M.; Akduman, N.; Röseler, W.; Theam, P.; Rödelsperger, C. Thousands of Pristionchus pacificus orphan genes were integrated into developmental networks that respond to diverse environmental microbiota. PLoS Genet. 2023, 19, e1010832. [Google Scholar] [CrossRef] [PubMed]
- Shang, S.; Liu, G.; Zhang, S.; Liang, X.; Zhang, R.; Sun, G. A fungal CFEM-containing effector targets NPR1 regulator NIMIN2 to suppress plant immunity. Plant Biotechnol. J. 2024, 22, 82–97. [Google Scholar] [CrossRef]
- Zhu, J.; Qiao, Q.; Sun, Y.; Xu, Y.; Shu, H.; Zhang, Z.; Liu, F.; Wang, H.; Ye, W.; Dong, S.; et al. Divergent sequences of tetraspanins enable plants to specifically recognize microbe-derived extracellular vesicles. Nat. Commun. 2023, 14, 4877. [Google Scholar] [CrossRef]
- Alberti, S.; Hyman, A.A. Biomolecular condensates at the nexus of cellular stress, protein aggregation disease and ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 196–213. [Google Scholar] [CrossRef]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef]
- Zhou, B.; Jia, P.; Gao, F.; Guo, H. Molecular characterization and functional analysis of a necrosis- and ethylene-Inducing, protein-encoding gene family from Verticillium dahliae. Mol. Plant-Microbe Interact. 2012, 25, 964–975. [Google Scholar] [CrossRef]
- Zhang, T.; Zhao, Y.L.; Zhao, J.H.; Wang, S.; Jin, Y.; Chen, Z.Q.; Fang, Y.Y.; Hua, C.L.; Ding, S.W.; Guo, H.S. Cotton plants export microRNAs to inhibit virulence gene expression in a fungal pathogen. Nat. Plants 2016, 2, 16153. [Google Scholar] [CrossRef]
- Li, R.; Zhu, H.; Ruan, J.; Qian, W.; Fang, X.; Shi, Z.; Li, Y.; Li, S.; Shan, G.; Kristiansen, K.; et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010, 20, 265–272. [Google Scholar] [CrossRef]
- Ter-Hovhannisyan, V.; Lomsadze, A.; Chernoff, Y.O.; Borodovsky, M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008, 18, 1979–1990. [Google Scholar] [CrossRef]
- Stanke, M.; Diekhans, M.; Baertsch, R.; Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 2008, 24, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Partners, C.-N.M.a. Database resources of the national genomics data center, China National Center for Bioinformation in 2022. Nucleic Acids Res. 2022, 50, D27–D38. [Google Scholar] [CrossRef] [PubMed]
- Donoghue, M.T.; Keshavaiah, C.; Swamidatta, S.H.; Spillane, C. Evolutionary origins of Brassicaceae specific genes in Arabidopsis thaliana. BMC Evol. Biol. 2011, 11, 47. [Google Scholar] [CrossRef]
- Zhang, M.; Zhu, M.; Lang, H.; Wang, W.; Li, X.; Jiang, M. Genome-Wide identification, characterization, and expression analysis of orphan genes within coriander. Plants 2025, 14, 778. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Zu, Y.; Jiang, M.; Zhan, Z.; Li, X.; Piao, Z. Orphan gene BR2 positively regulates bolting resistance through the vernalization pathway in Chinese cabbage. Hortic. Res. 2024, 11, uhae216. [Google Scholar] [CrossRef]
- Ji, X.; Tang, J.; Zhang, J. Effects of salt stress on the morphology, growth and physiological parameters of Juglansmicrocarpa L. seedlings. Plants 2022, 11, 2381. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wan, M.; Chen, X.; Yi, X.; Fu, Y.; Jin, Y.; Lyu, D. Identification of a Highly Virulent Verticillium nonalfalfae Strain Vn011 and Expression Analysis of Its Orphan Genes During Potato Inoculation. Plants 2025, 14, 1281. https://doi.org/10.3390/plants14091281
Wan M, Chen X, Yi X, Fu Y, Jin Y, Lyu D. Identification of a Highly Virulent Verticillium nonalfalfae Strain Vn011 and Expression Analysis of Its Orphan Genes During Potato Inoculation. Plants. 2025; 14(9):1281. https://doi.org/10.3390/plants14091281
Chicago/Turabian StyleWan, Mengyuan, Xinlong Chen, Xiaoxi Yi, Yi Fu, Yuanliang Jin, and Dianqiu Lyu. 2025. "Identification of a Highly Virulent Verticillium nonalfalfae Strain Vn011 and Expression Analysis of Its Orphan Genes During Potato Inoculation" Plants 14, no. 9: 1281. https://doi.org/10.3390/plants14091281
APA StyleWan, M., Chen, X., Yi, X., Fu, Y., Jin, Y., & Lyu, D. (2025). Identification of a Highly Virulent Verticillium nonalfalfae Strain Vn011 and Expression Analysis of Its Orphan Genes During Potato Inoculation. Plants, 14(9), 1281. https://doi.org/10.3390/plants14091281