Complete Chloroplast Genomes of Ampelopsis humulifolia and Ampelopsis japonica: Molecular Structure, Comparative Analysis, and Phylogenetic Analysis


Abstract
:1. Introduction
2. Results and Discussion
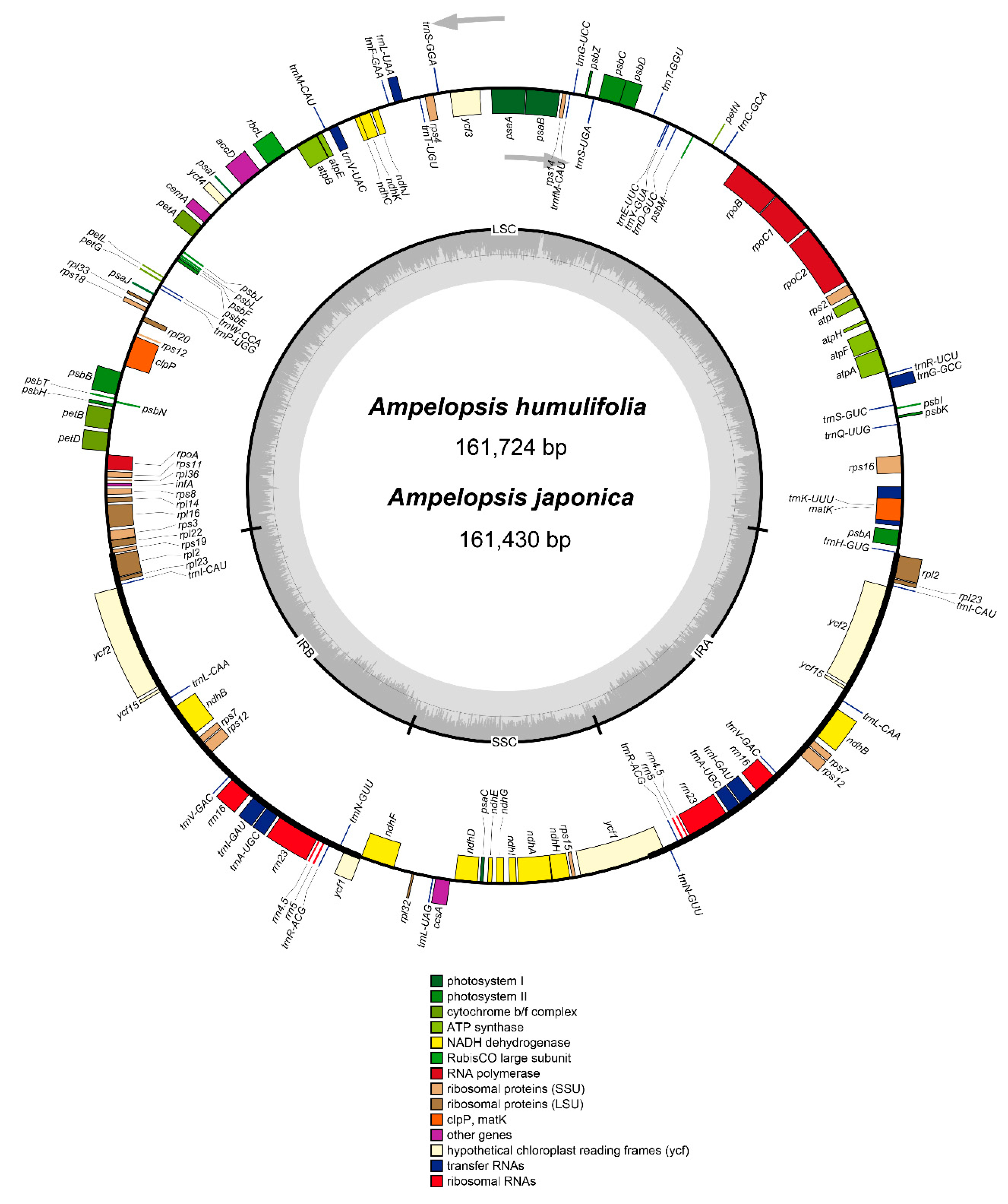
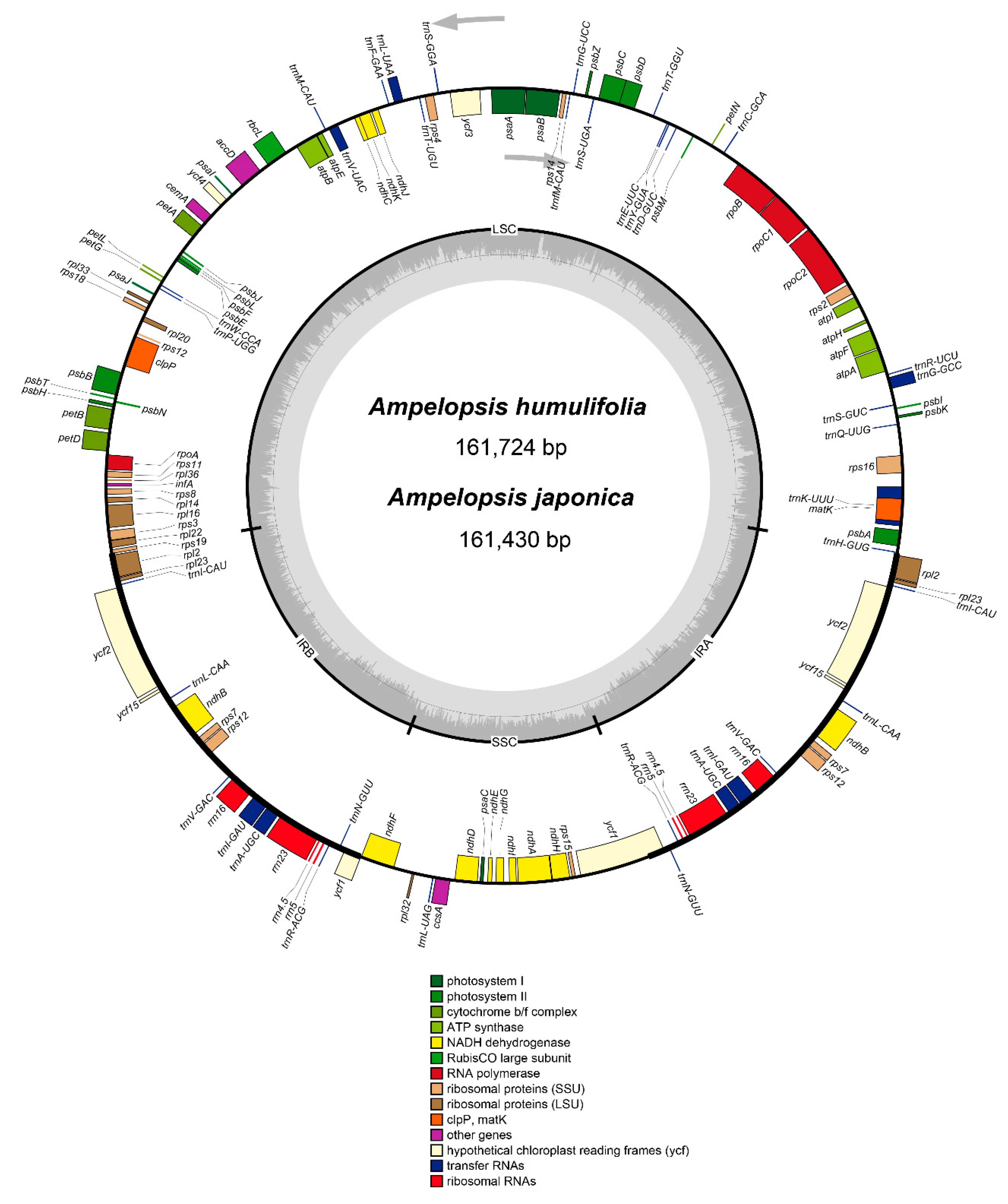
2.1. Characteristics of the Chloroplast Genomes of A. humulifolia and A. japonica
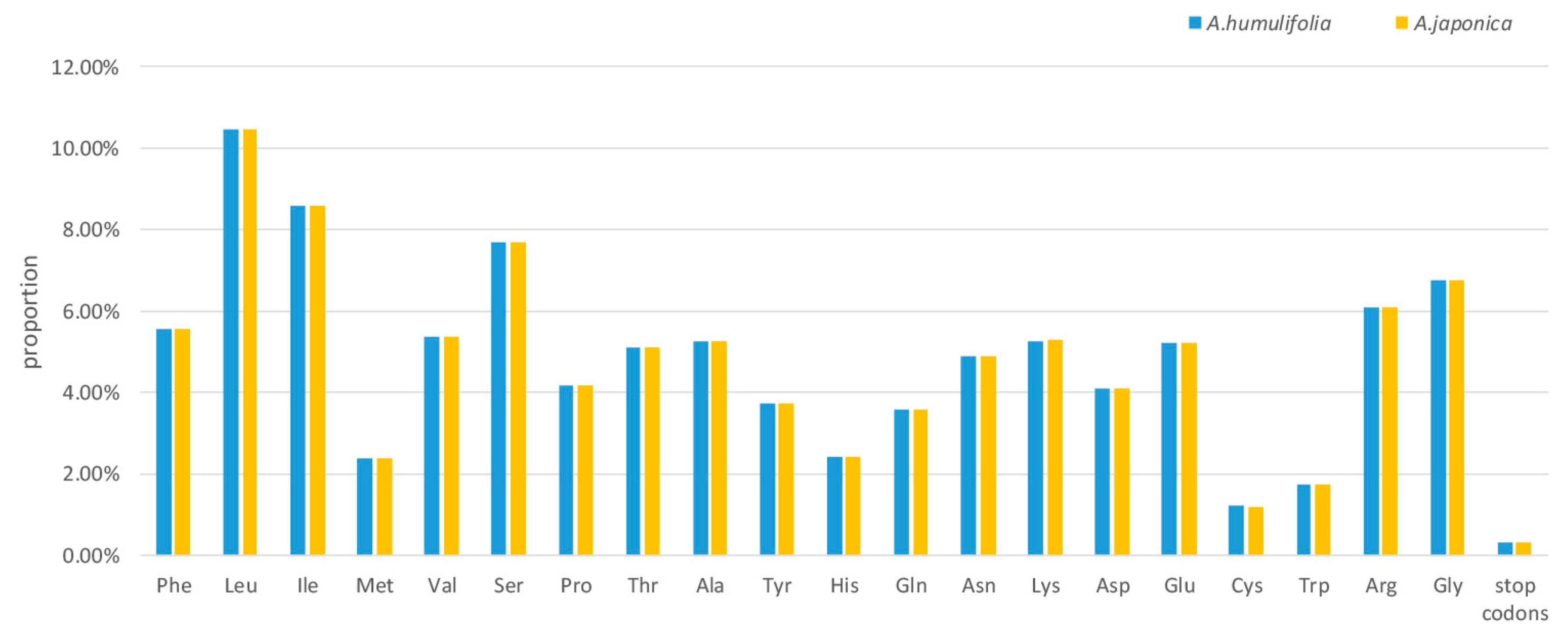
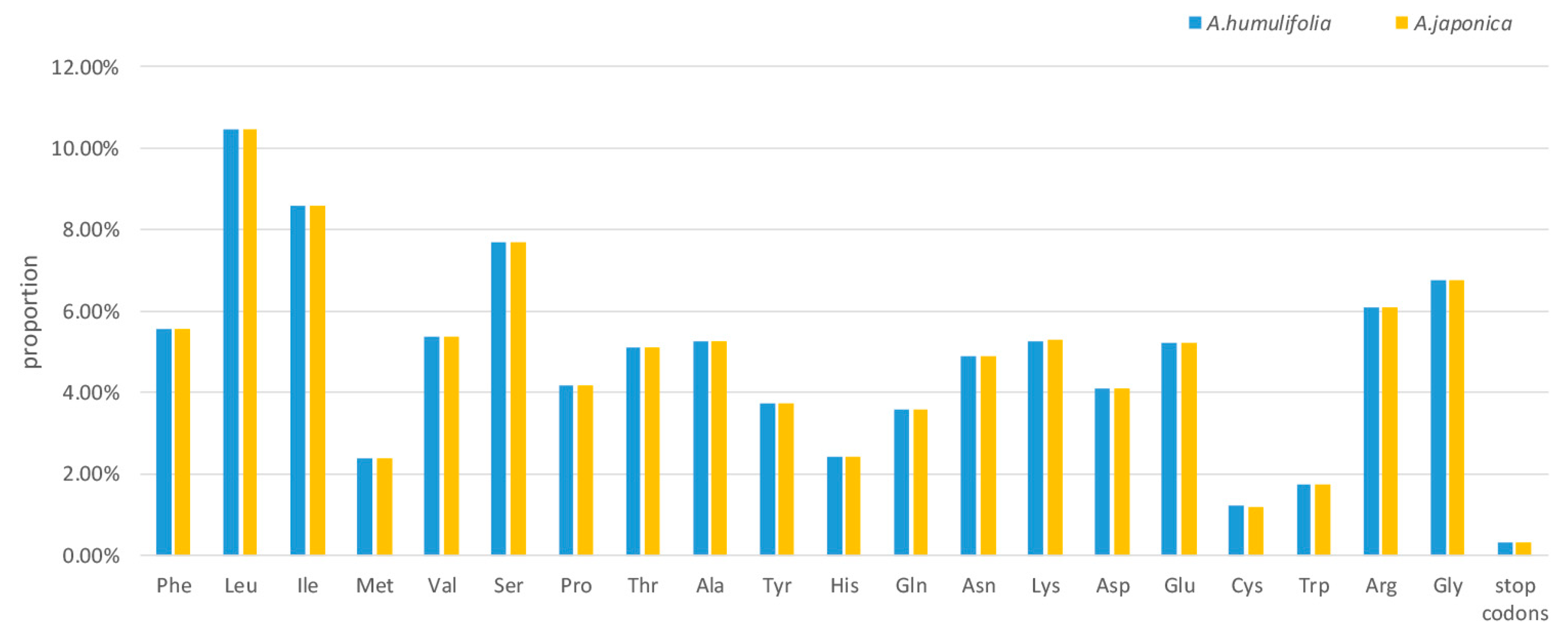
2.2. Codon Usage Analysis
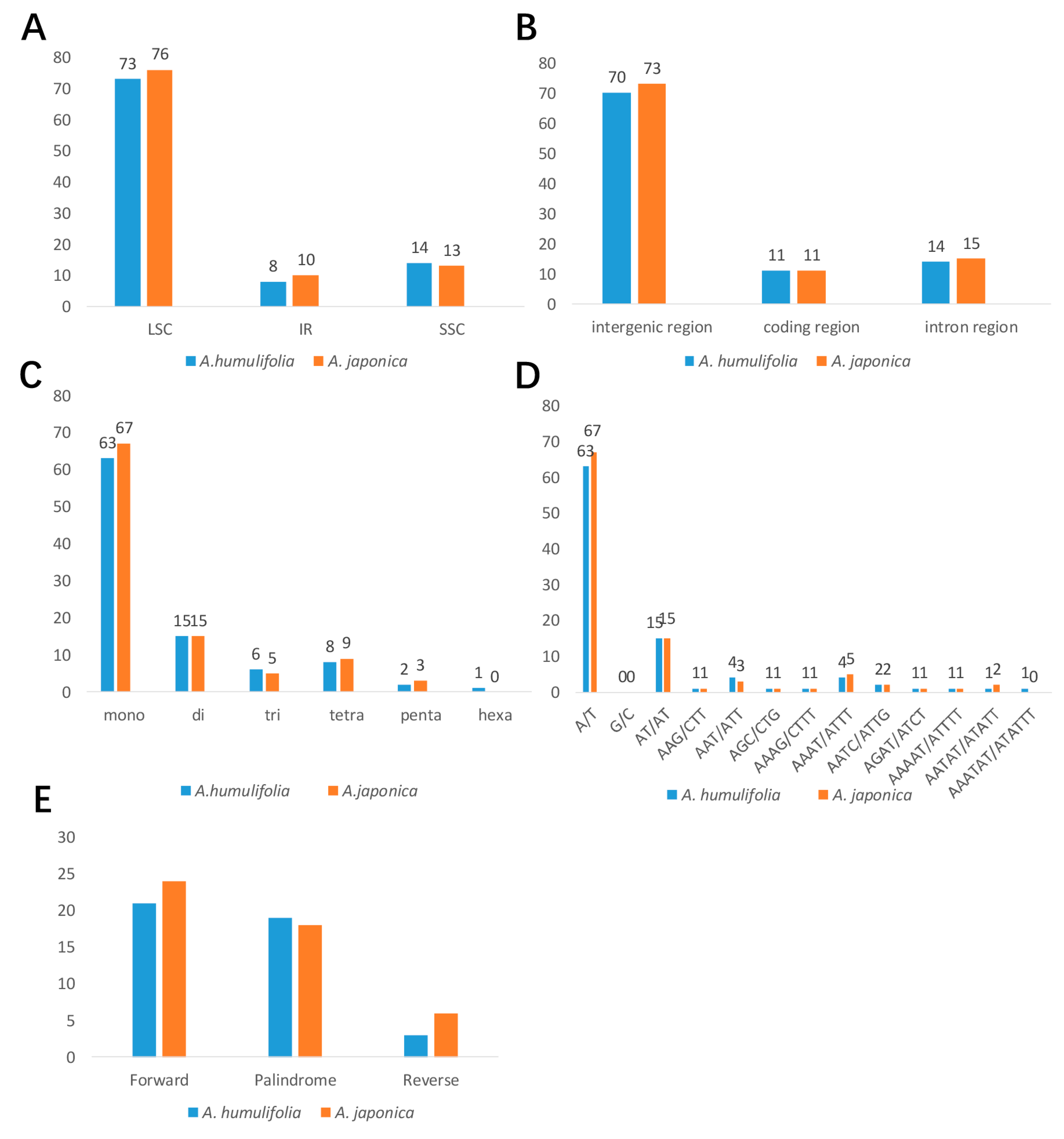
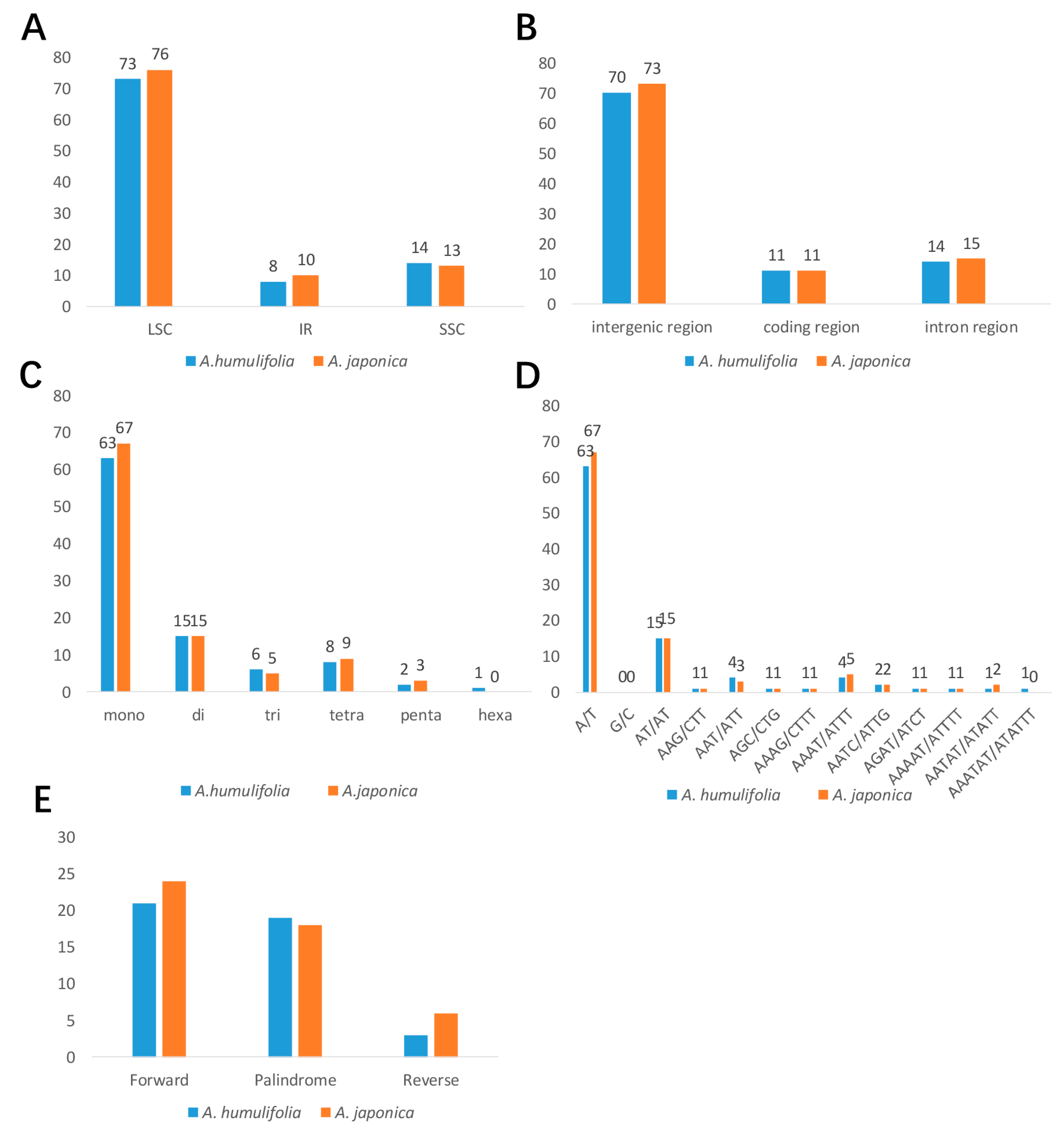
2.3. Analysis of SSRs and Long Repeats
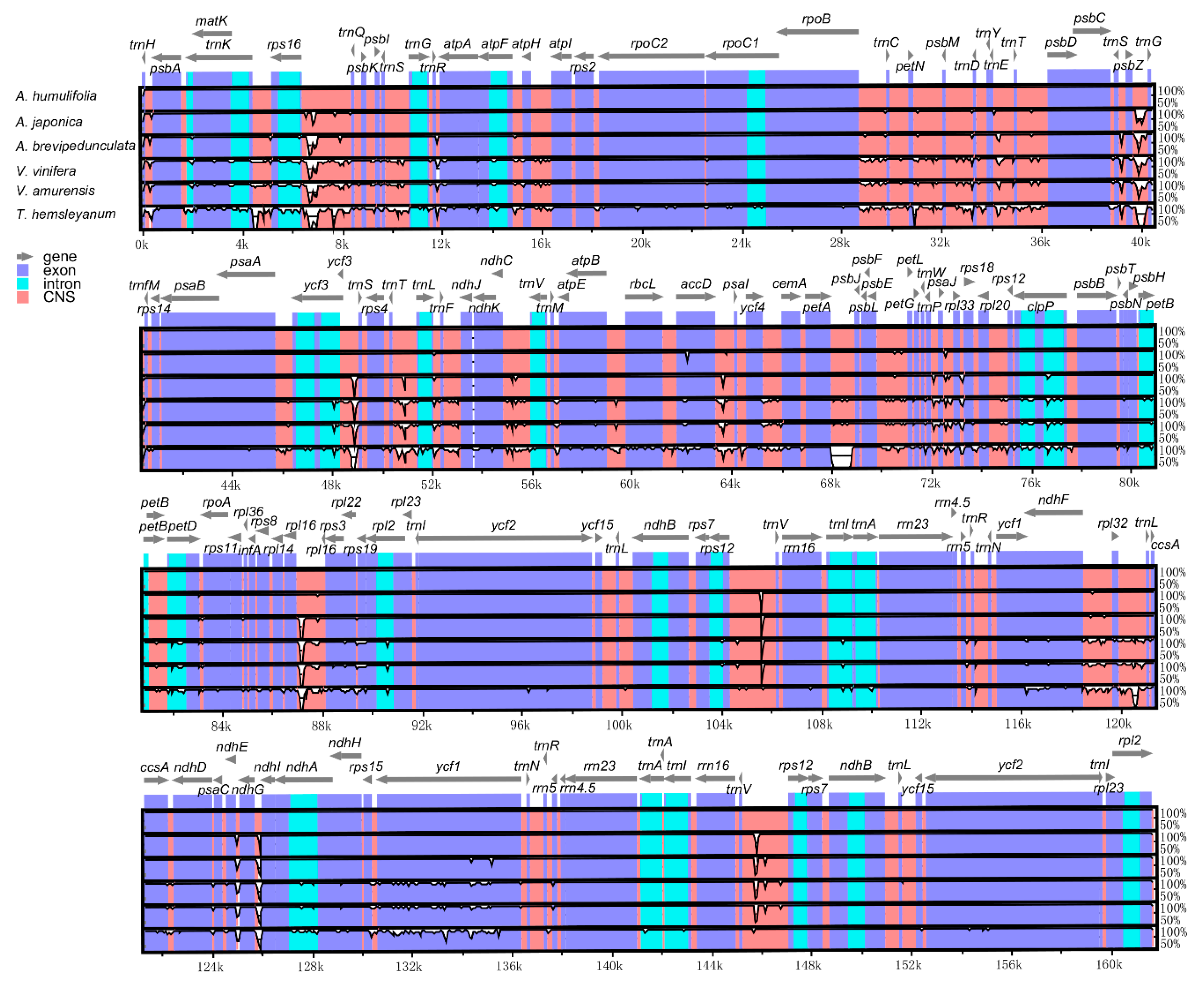
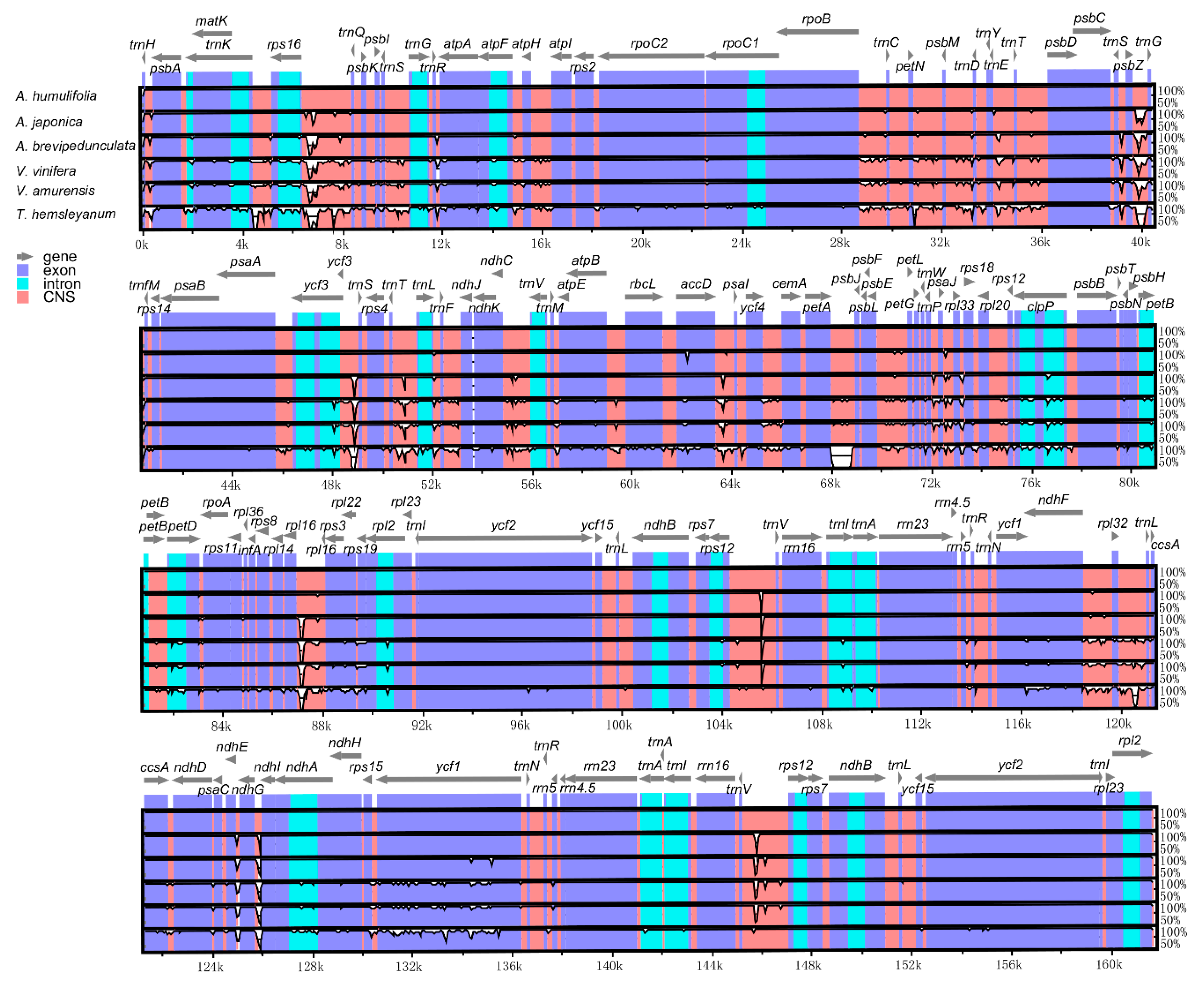
2.4. Comparative Genome Analysis
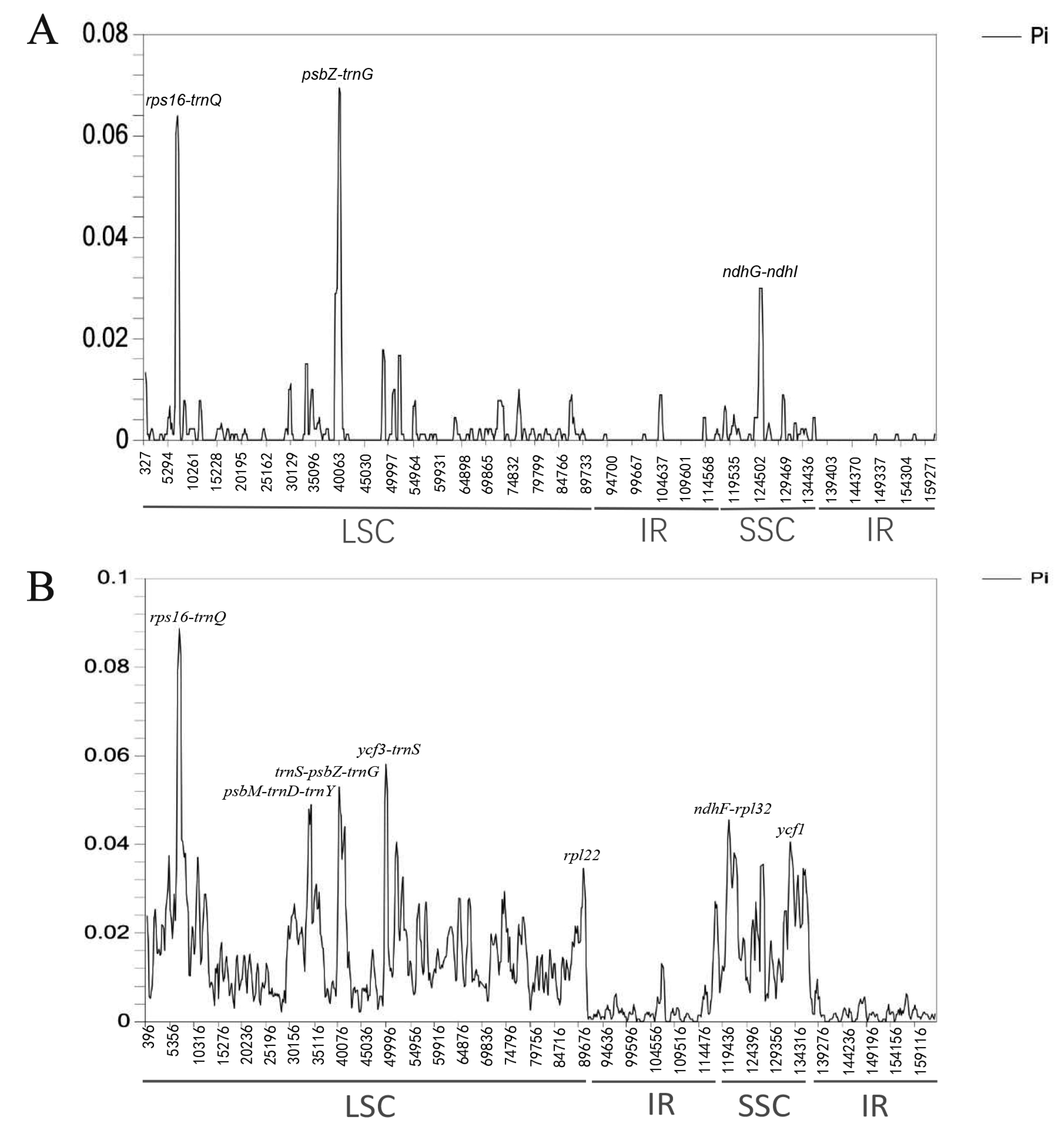
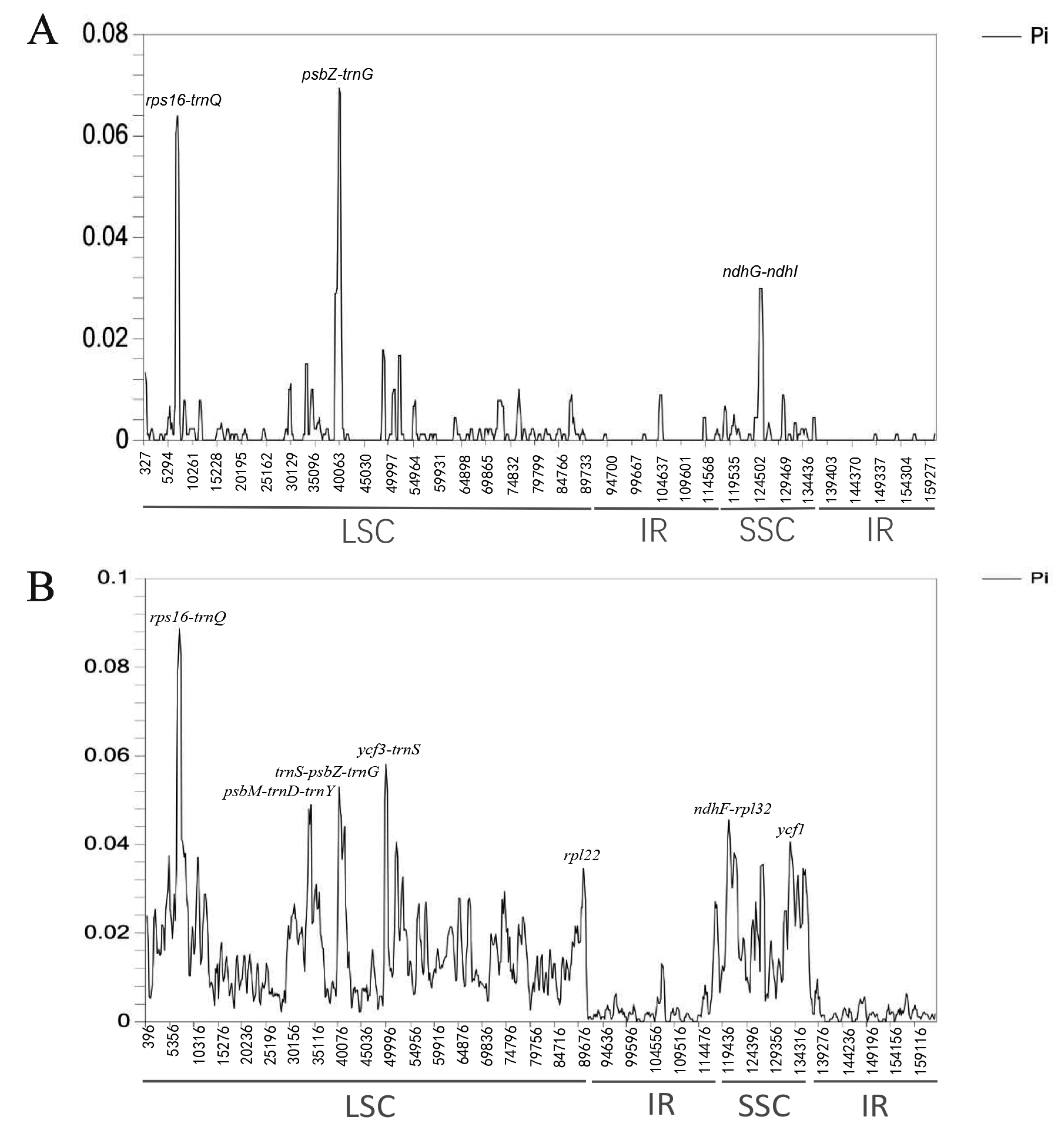
2.5. Nucleotide Diversity Analysis
2.6. Marker Comparison
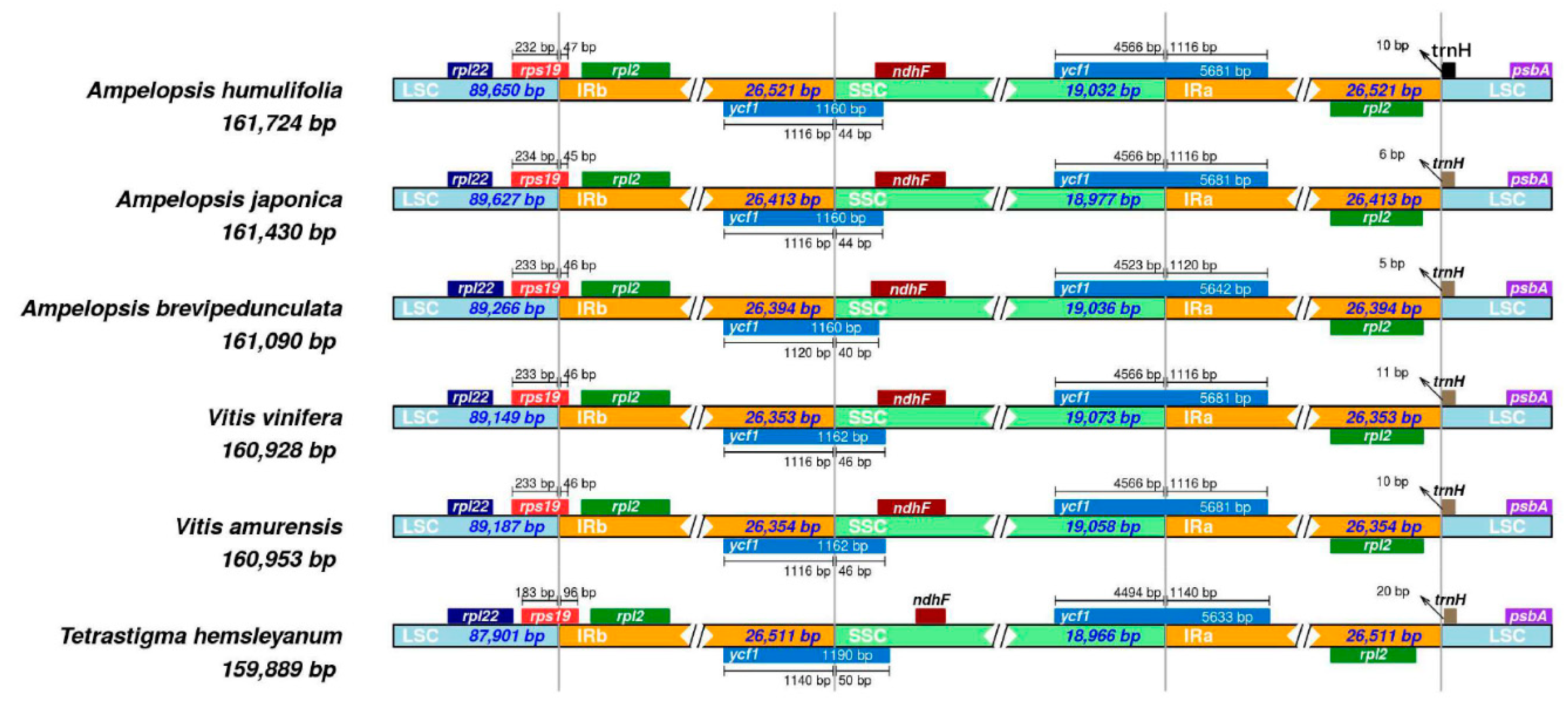
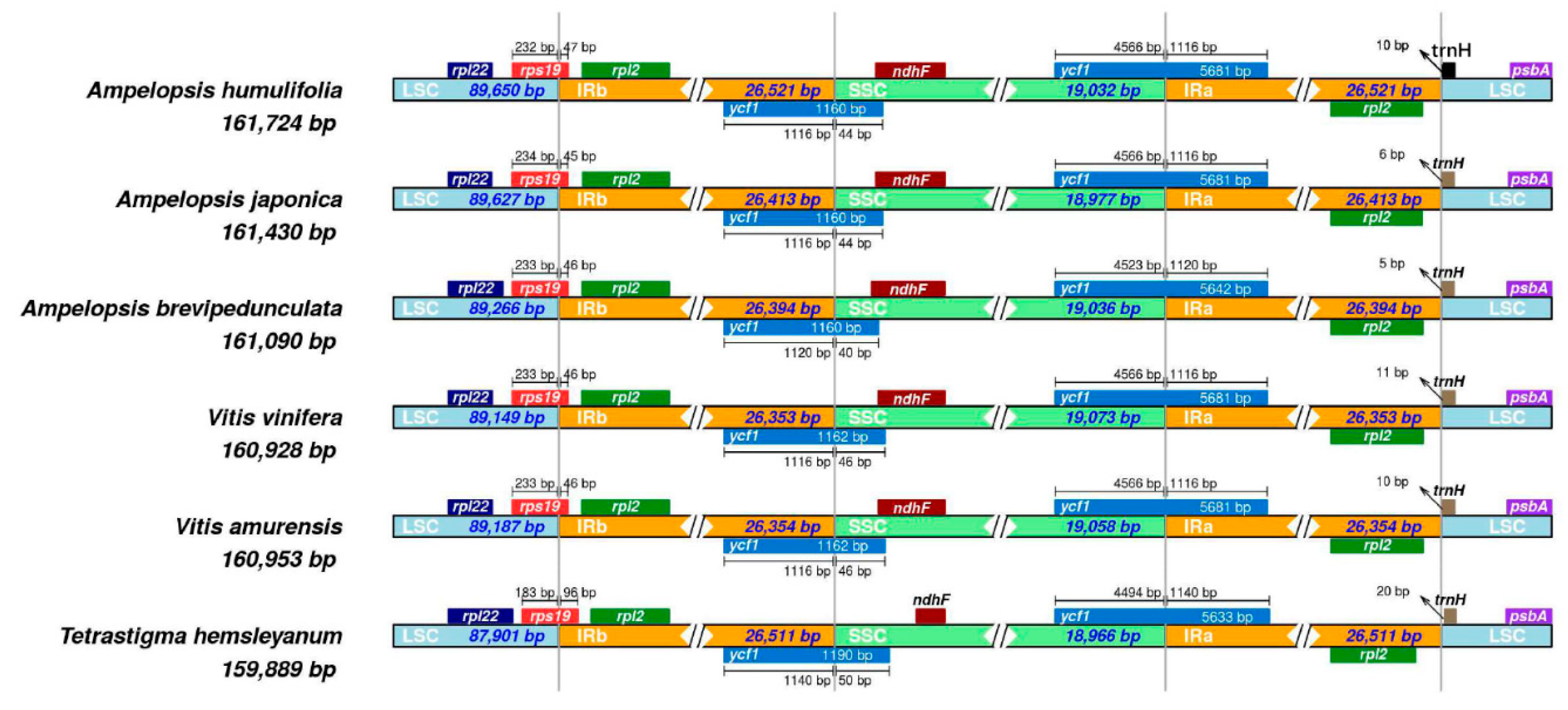
2.7. IR Expansion and Contraction
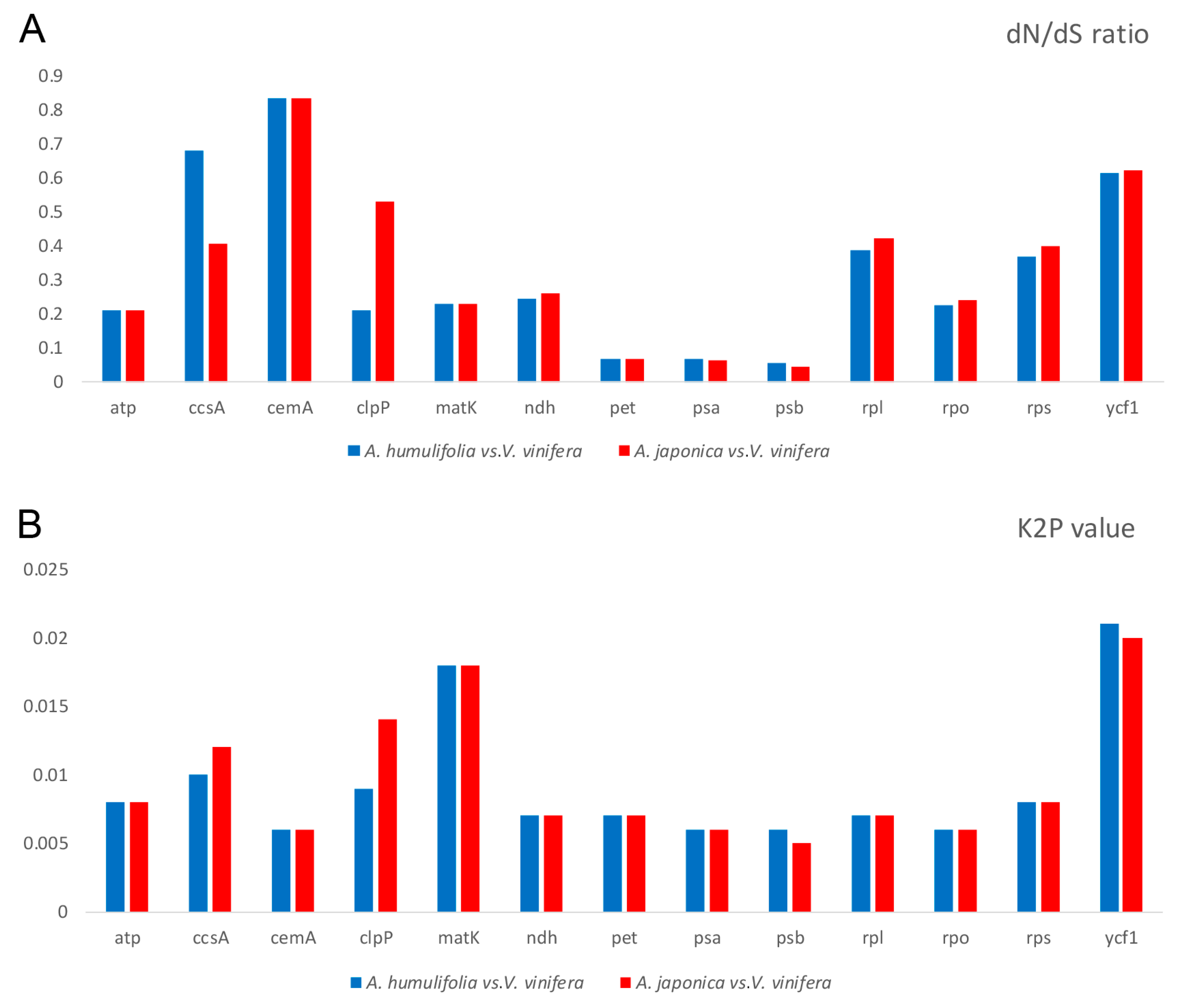
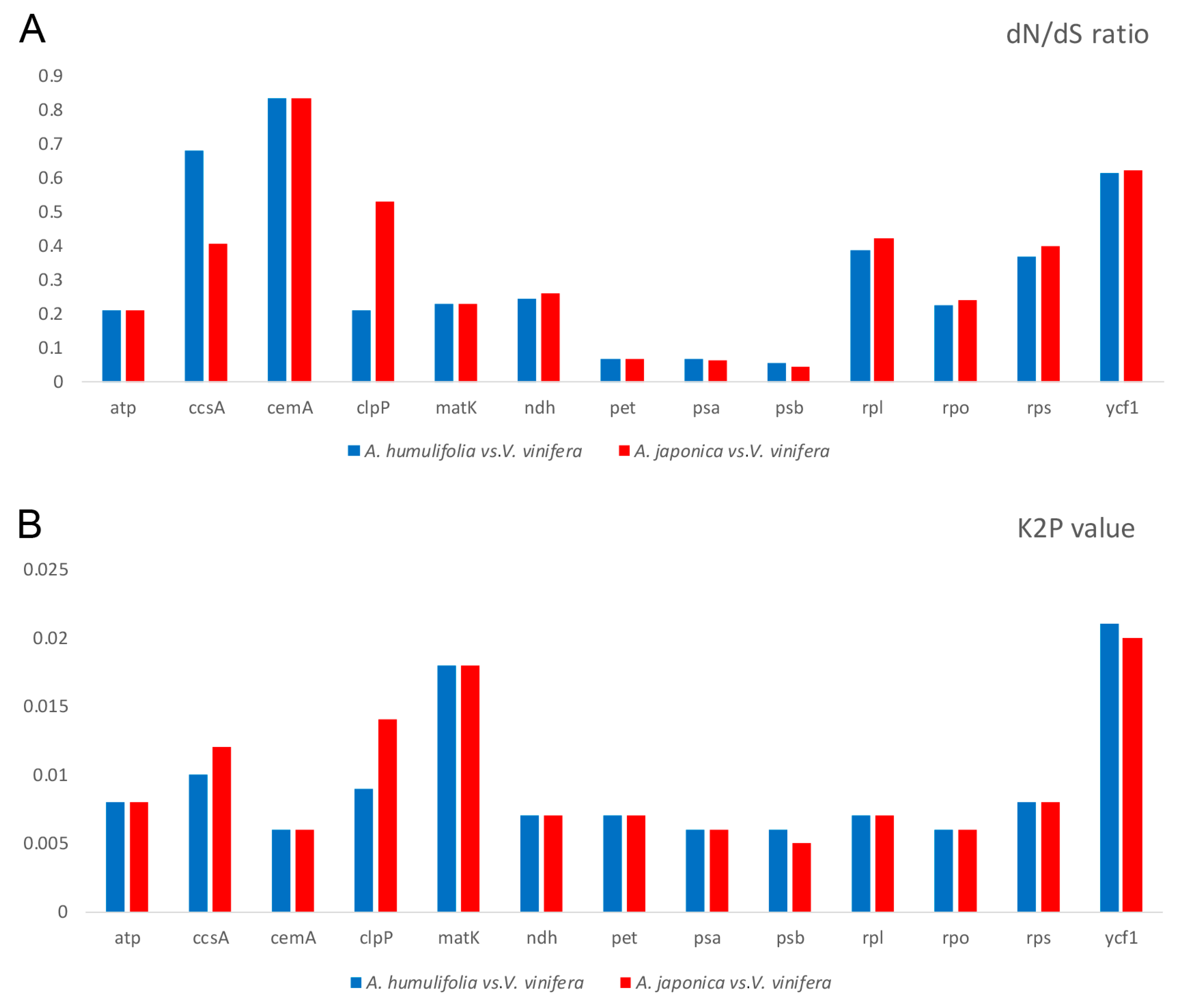
2.8. dN/dS Ratio and Kimura 2-Parameter (K2P) Genetic Distance
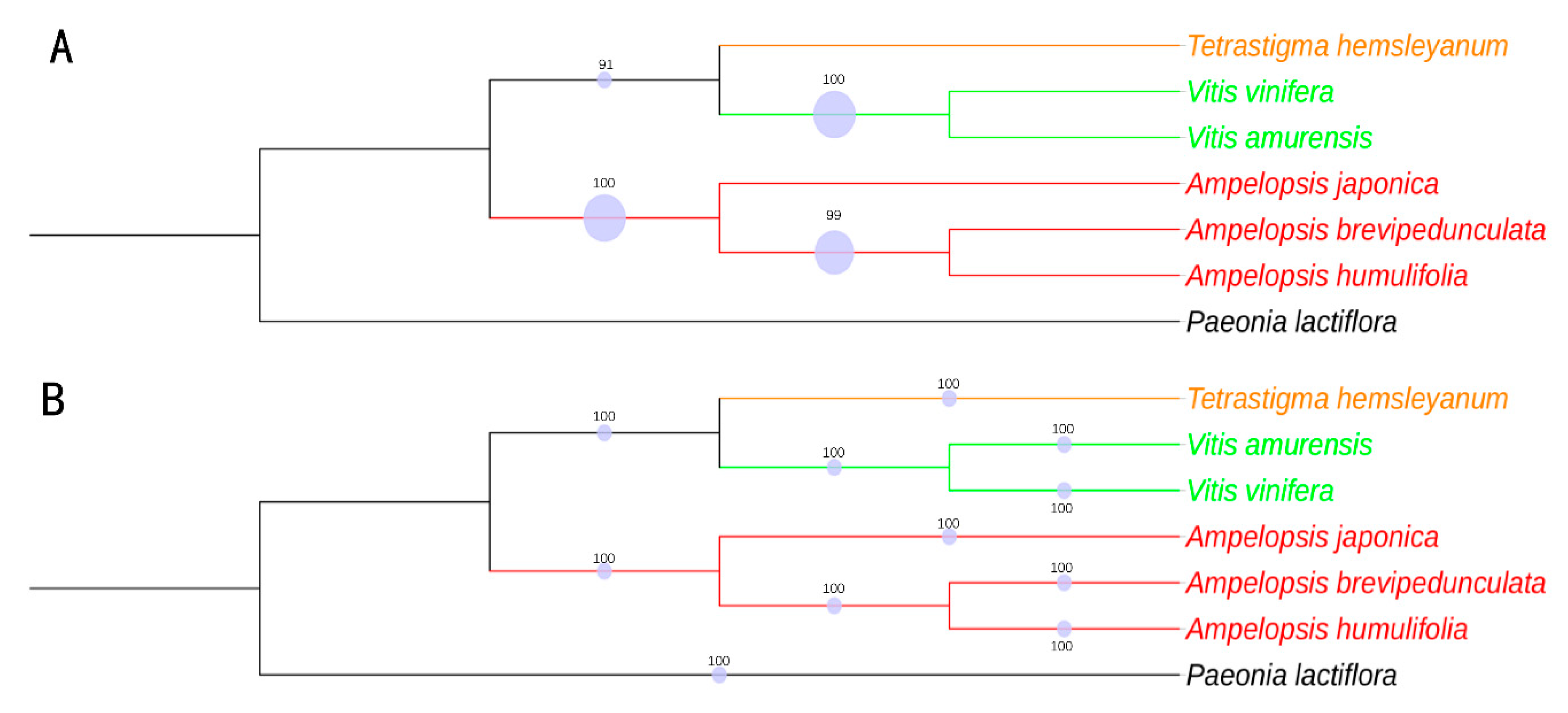
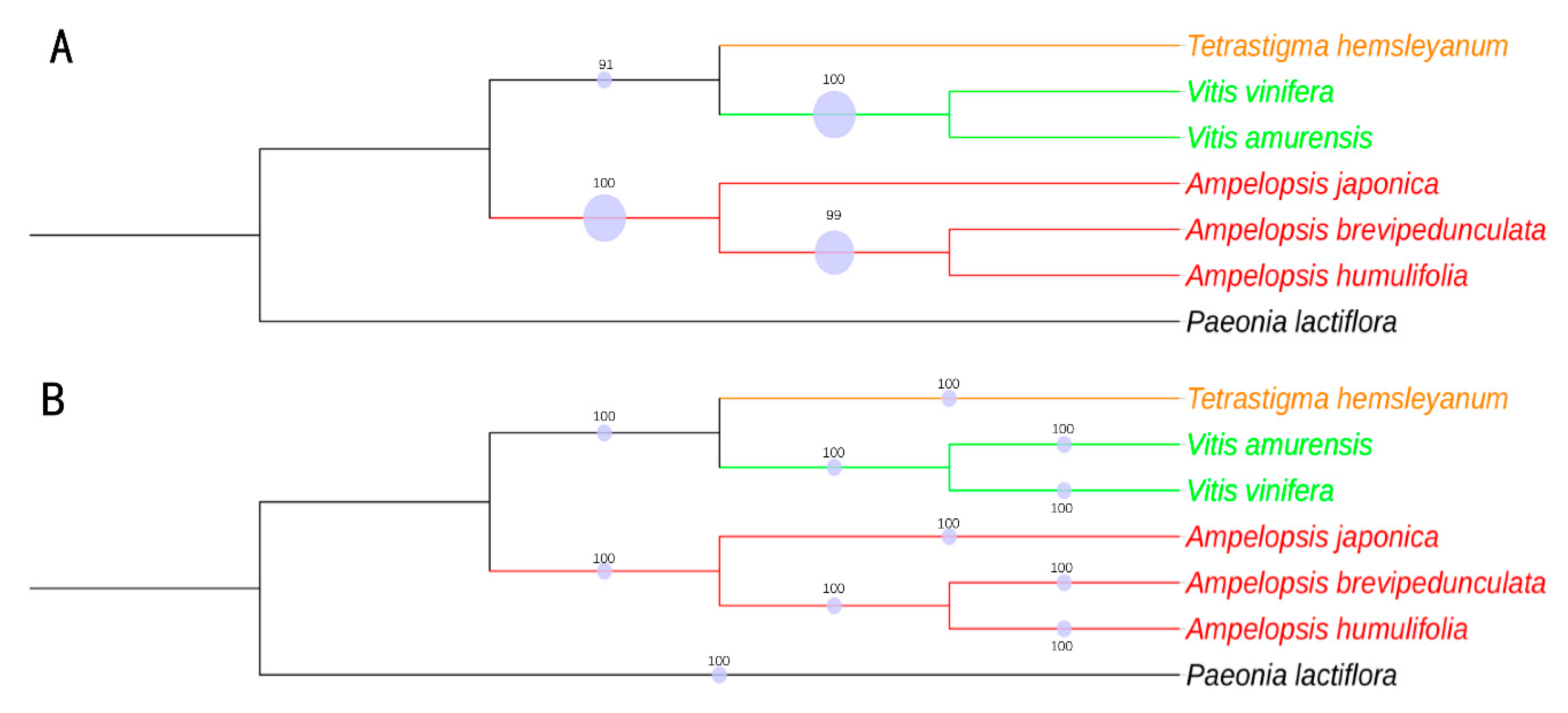
2.9. Phylogenomic Analysis
3. Materials and Methods
3.1. DNA Sequencing and Genome Assembly
3.2. Genome Annotation
3.3. Genome Structure Analysis
3.4. Genome Comparison
3.5. Analysis of dN/dS Ratio and K2P Genetic Distance
3.6. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- The Editorial Committee of Flora of China. Flora of China; Science Press: Beijing, China, 1998. [Google Scholar]
- Su, T.; Bai, J.X.; Chen, Y.J.; Wang, X.N.; Fu, X.Q.; Li, T.; Guo, H.; Zhu, P.L.; Wang, Y.; Yu, Z.L. An ethanolic extract of Ampelopsis radix exerts anti-colorectal cancer effects and potently inhibits STAT3 signaling in vitro. Front. Pharm. 2017, 8, 227. [Google Scholar] [CrossRef] [PubMed]
- Li, H.J.; Zhang, L.; Wang, S.G. Pharmacological action and clinical application of radix Ampelopsis. Food Drug. 2007, 9, 60–62. [Google Scholar]
- Nho, K.J.; Chun, J.M.; Kim, D.S.; Kim, H.K. Ampelopsis japonica ethanol extract suppresses migration and invasion in human MDAMB231 breast cancer cells. Mol. Med. Rep. 2015, 11, 3722–3728. [Google Scholar] [CrossRef] [PubMed]
- Chinese Pharmacopoeia Commission. Pharmacopoeia of the People’s Republic of China; The Medicine Science and Technology Press: Beijing, China, 2015. [Google Scholar]
- Cho, M.S.; Yang, J.Y.; Yang, T.J.; Kim, S.C. Evolutionary comparison of the chloroplast genome in the woody Sonchus Alliance (Asteraceae) on the canary islands. Genes 2019, 10, 217. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, N.; Tamburino, R.; Cantarella, C.; De Carluccio, V.; Sannino, L.; Cozzolino, S.; Cardi, T.; Scotti, N. The complete plastome sequences of eleven capsicum genotypes: Insights into DNA variation and molecular evolution. Genes 2018, 9, 503. [Google Scholar] [CrossRef]
- Raman, G.; Park, S. The complete chloroplast genome sequence of Ampelopsis: Gene organization, comparative analysis, and phylogenetic relationships to other Angiosperms. Front. Plant. Sci. 2016, 7. [Google Scholar] [CrossRef]
- Liu, J.; Qi, Z.C.; Zhao, Y.P.; Fu, C.X.; Jenny Xiang, Q.Y. Complete cpDNA genome sequence of Smilax china and phylogenetic placement of Liliales—Influences of gene partitions and taxon sampling. Mol. Phylogenet. Evol. 2012, 64, 545–562. [Google Scholar] [CrossRef]
- Provan, J.; Powell, W.; Hollingsworth, P.M. Chloroplast microsatellites: New tools for studies in plant ecology and evolution. Trends Ecol. Evol. 2001, 16, 142–147. [Google Scholar] [CrossRef]
- Ravi, V.; Khurana, J.P.; Tyagi, A.K.; Khurana, P. An update on chloroplast genomes. Plant Syst. Evol. 2008, 271, 101–122. [Google Scholar] [CrossRef]
- Abdullah; Mehmood, F.; Shahzadi, I.; Waseem, S.; Mirza, B.; Ahmed, I.; Waheed, M.T. Chloroplast genome of Hibiscus rosa-sinensis (Malvaceae): Comparative analyses and identification of mutational hotspots. Genomics 2019. [Google Scholar] [CrossRef]
- Yan, M.; Zhao, X.; Zhou, J.; Huo, Y.; Ding, Y.; Yuan, Z. The complete chloroplast genomes of Punica granatum and a comparison with other species in Lythraceae. Int. J. Mol. Sci. 2019, 20, 2886. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Xu, C.; Li, C.; Sun, J.; Zuo, Y.; Shi, S.; Cheng, T.; Guo, J.; Zhou, S. ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 2015, 5, 8348. [Google Scholar] [CrossRef] [PubMed]
- Group, C.P.W. A DNA barcode for land plants. Proc. Natl. Acad. Sci. USA 2009, 106, 12794–12797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, B.; Li, D.; Li, X. DNA barcoding of Actinidia (Actinidiaceae) using internal transcribed spacer, matK, rbcL and trnH-psbA, and its taxonomic implication. N.-Z. J. Bot. 2018, 56, 1–12. [Google Scholar] [CrossRef]
- Yang, H.; Dong, Y.; Gu, Z.-J.; Liang, N.; Yang, J.-B. A preliminary assessment of matK, rbcL and trnH—psbA as DNA Barcodes for Calamus (Arecaceae) species in China with a note on ITS. Ann. Bot. Fenn. 2012, 49, 5–6. [Google Scholar] [CrossRef]
- Liu, Q.; Xue, Q. Comparative studies on codon usage pattern of chloroplasts and their host nuclear genes in four plant species. J. Genet. 2005, 84, 55–62. [Google Scholar] [CrossRef]
- Park, I.; Kim, W.J.; Yeo, S.M.; Choi, G.; Kang, Y.M.; Piao, R.; Moon, B.C. The complete chloroplast genome sequences of Fritillaria ussuriensis Maxim. and Fritillaria cirrhosa D. Don, and comparative analysis with other Fritillaria species. Molecules 2017, 22, 982. [Google Scholar] [CrossRef]
- Qi, W.; Lin, F.; Liu, Y.; Huang, B.; Cheng, J.; Zhang, W.; Zhao, H. High-throughput development of simple sequence repeat markers for genetic diversity research in Crambe abyssinica. BMC Plant. Biol. 2016, 16, 139. [Google Scholar] [CrossRef]
- Yu, J.; Dossa, K.; Wang, L.; Zhang, Y.; Wei, X.; Liao, B.; Zhang, X. PMDBase: A database for studying microsatellite DNA and marker development in plants. Nucleic Acids Res. 2017, 45, 1046–1053. [Google Scholar] [CrossRef]
- Chen, Y.L.; Fan, G.; Liu, Y.; Song, C.; Zhang, Y.; Xiang, L.; Lai, X.R. Identification of traditional chinese medicine rubiae radix et and its adulterants using DNA barcodes. Chin. Pharm. J. 2015, 50, 1266–1272. [Google Scholar]
- Wang, W.; Messing, J. High-throughput sequencing of three Lemnoideae (duckweeds) chloroplast genomes from total DNA. PLoS ONE 2011, 6, e24670. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, C.; Miao, H.; Xiong, S. Insights from the complete chloroplast genome into the evolution of Sesamum indicum L. PLoS ONE 2013, 8, e80508. [Google Scholar] [CrossRef] [PubMed]
- Drouin, G.; Daoud, H.; Xia, J. Relative rates of synonymous substitutions in the mitochondrial, chloroplast and nuclear genomes of seed plants. Mol. Phylogenet. Evol. 2008, 49, 827–831. [Google Scholar] [CrossRef] [PubMed]
- Nakai, M. YCF1: A Green TIC? Plant. Cell 2015, 27, 1827–1833. [Google Scholar] [CrossRef] [Green Version]
- Goremykin, V.V.; Nikiforova, S.V.; Cavalieri, D.; Pindo, M.; Lockhart, P. The root of flowering plants and total Evidence. Syst. Biol. 2015, 64, 879–891. [Google Scholar] [CrossRef]
- Sun, L.; Fang, L.; Zhang, Z.; Chang, X.; Penny, D.; Zhong, B. Chloroplast phylogenomic inference of green algae relationships. Sci. Rep. 2016, 6, 20528. [Google Scholar] [CrossRef]
- Sanderson, M.J.; Shaffer., H.B. Troubleshooting molecular phylogenetic analyses. Annu. Rev. Ecol. Syst. 2002, 33, 49–72. [Google Scholar] [CrossRef]
- Dong, W.; Xu, C.; Wu, P.; Cheng, T.; Yu, J.; Zhou, S.; Hong, D.Y. Resolving the systematic positions of enigmatic taxa: Manipulating the chloroplast genome data of Saxifragales. Mol. Phylogenet. Evol. 2018, 126, 321–330. [Google Scholar] [CrossRef]
- Nie, Z.L.; Sun, H.; Manchester, S.R.; Meng, Y.; Luke, Q.; Wen, J. Evolution of the intercontinental disjunctions in six continents in the Ampelopsis clade of the grape family (Vitaceae). BMC Evol. Biol. 2012, 12, 17. [Google Scholar] [CrossRef]
- Trias Blasi, A.; Parnell, J.; Hodkinson, T. Multi-gene region phylogenetic analysis of the grape family (Vitaceae). Syst. Bot. 2012, 37, 941–950. [Google Scholar] [CrossRef]
- Zhang, N.; Wen, J.; Zimmer, E.A. Congruent deep relationships in the grape family (Vitaceae) based on sequences of chloroplast genomes and mitochondrial genes via genome skimming. PLoS ONE 2015, 10, e0144701. [Google Scholar] [CrossRef] [PubMed]
- Ingrouille, M.J.; Chase, M.W.; Michael, F.; Diane, B.; Michelle, B.; Bruijn, A.D.E. Systematics of Vitaceae from the viewpoint of plastid rbcL DNA sequence data. Bot. J. Linn. Soc. 2002, 138, 421–432. [Google Scholar] [CrossRef]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Shi, L.; Zhu, Y.; Chen, H.; Zhang, J.; Lin, X.; Guan, X. CpGAVAS, an integrated web server for the annotation, visualization, analysis, and GenBank submission of completely sequenced chloroplast genome sequences. BMC Genom. 2012, 13, 715. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Munch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Amiryousefi, A.; Hyvonen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef]
- Chang, C.C.; Lin, H.C.; Lin, I.P.; Chow, T.Y.; Chen, H.H.; Chen, W.H.; Cheng, C.H.; Lin, C.Y.; Liu, S.M. The chloroplast genome of Phalaenopsis aphrodite (Orchidaceae): Comparative analysis of evolutionary rate with that of grasses and its phylogenetic implications. Mol. Biol. Evol. 2006, 23, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Xu, C.; Cheng, T.; Zhou, S. Complete chloroplast genome of Sedum sarmentosum and chloroplast genome evolution in Saxifragales. PLoS ONE 2013, 8, e77965. [Google Scholar] [CrossRef]
- Li, B.; Lin, F.; Huang, P.; Guo, W.; Zheng, Y. Complete chloroplast genome sequence of Decaisnea insignis: Genome organization, genomic resources and comparative analysis. Sci. Rep. 2017, 7, 10073. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Li, W.X.; Jakovlić, I.; Zou, H.; Zhang, J.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. bioRxiv 2018. [Google Scholar] [CrossRef]
- Alexandros, S. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Santorum, J.M.; Darriba, D.; Taboada, G.L.; Posada, D. jmodeltest.org: Selection of nucleotide substitution models on the cloud. Bioinformatics 2014, 30, 1310–1311. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | A. humulifolia | A. japonica |
|---|---|---|
| Genome size (bp) | 161,724 | 161,430 |
| LSC length (bp) | 89,650 | 89,626 |
| IR length (bp) | 26,521 | 26,413 |
| SSC length (bp) | 19,032 | 18,977 |
| Number of genes | 114 | 114 |
| Protein-coding genes | 80 | 80 |
| tRNA genes | 30 | 30 |
| rRNA genes | 4 | 4 |
| Total GC content (%) | 37.3 | 37.3 |
| LSC | 35.2 | 35.2 |
| IR | 42.9 | 42.9 |
| SSC | 31.8 | 31.9 |
| Category | Gene Name |
|---|---|
| Photosystem I | psaA, psaB, psaC, psaI, psaJ |
| Photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ |
| Cytochrome b/f complex | petA, petBb, petDb, petG, petL, petN |
| ATP synthase | atpA, atpB, atpE, atpFb, atpH, atpI |
| NADH dehydrogenase | ndhAb, ndhBa,b, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK |
| RNA polymerase | rpoA, rpoB, rpoC1b, rpoC2 |
| Large subunit ribosomal proteins | rpl2a, rpl14, rpl16b, rpl20, rpl22, rpl23 a, rpl32, rpl33, rpl36 |
| Small subunit ribosomal proteins | rps2, rps3, rps4, rps7a, rps8, rps11, rps12a,c, rps14, rps15, rps16b, rps18, rps19 |
| Ribosomal RNA genes | rrn4.5a, rrn5a, rrn16a, rrn23a |
| Transfer RNA genes | trnA-UGCa,b, trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnfM-CAU, trnG-GCC, trnG-UCCb, trnH-GUG, trnI-CAUa, trnI-GAUa,b, trnK-UUUb, trnL-CAAa, trnL-UAAb, trnL-UAG, trnM-CAU, trnN-GUUa, trnP-UGG, trnQ-UUG, trnR-ACGa, trnR-UCU, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GACa, trnV-GAU, trnV-UACb, trnW-CCA, trnY-GUA |
| Fatty acid synthesis | accD |
| Carbon metabolism | cemA |
| Cytochrome c synthesis | ccsA |
| Proteolysis | clpPc |
| Translation initiation factor | infA |
| RNA processing | matK |
| Rubisco | rbcL |
| Proteins of unknown function | ycf1a, ycf2a, ycf3c, ycf4, ycf15 a |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, X.; Tan, W.; Zhang, H.; Gao, H.; Wang, W.; Tian, X. Complete Chloroplast Genomes of Ampelopsis humulifolia and Ampelopsis japonica: Molecular Structure, Comparative Analysis, and Phylogenetic Analysis. Plants 2019, 8, 410. https://doi.org/10.3390/plants8100410
Yu X, Tan W, Zhang H, Gao H, Wang W, Tian X. Complete Chloroplast Genomes of Ampelopsis humulifolia and Ampelopsis japonica: Molecular Structure, Comparative Analysis, and Phylogenetic Analysis. Plants. 2019; 8(10):410. https://doi.org/10.3390/plants8100410
Chicago/Turabian StyleYu, Xiaolei, Wei Tan, Huanyu Zhang, Han Gao, Wenxiu Wang, and Xiaoxuan Tian. 2019. "Complete Chloroplast Genomes of Ampelopsis humulifolia and Ampelopsis japonica: Molecular Structure, Comparative Analysis, and Phylogenetic Analysis" Plants 8, no. 10: 410. https://doi.org/10.3390/plants8100410
APA StyleYu, X., Tan, W., Zhang, H., Gao, H., Wang, W., & Tian, X. (2019). Complete Chloroplast Genomes of Ampelopsis humulifolia and Ampelopsis japonica: Molecular Structure, Comparative Analysis, and Phylogenetic Analysis. Plants, 8(10), 410. https://doi.org/10.3390/plants8100410