Phylogenetic Reconstruction and Divergence Time Estimation of Blumea DC. (Asteraceae: Inuleae) in China Based on nrDNA ITS and cpDNA trnL-F Sequences

 ,
,
Abstract
1. Introduction
2. Results
2.1. Features of nrDNA ITS and cpDNA trnL-F Sequences in Blumea DC.
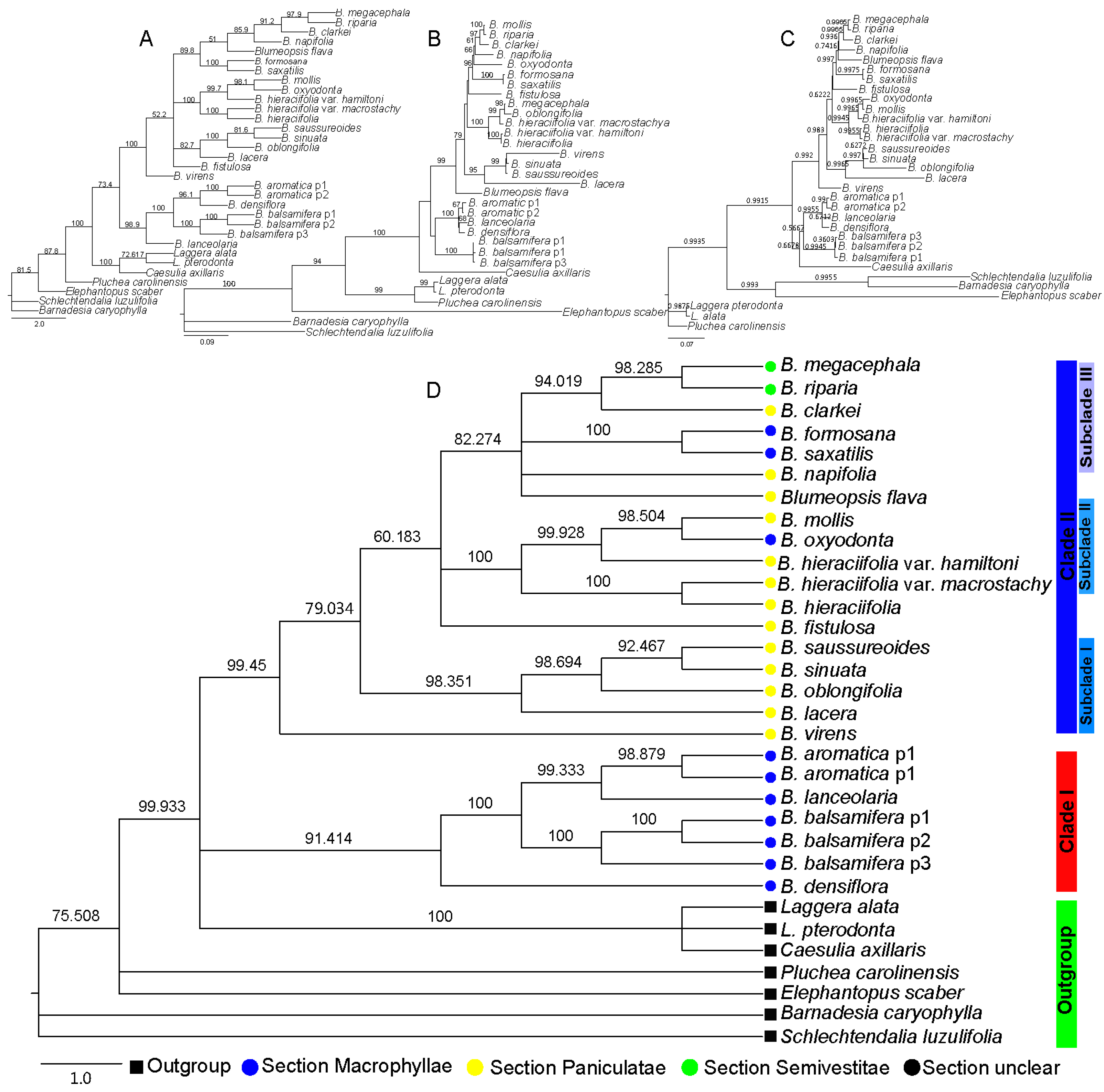
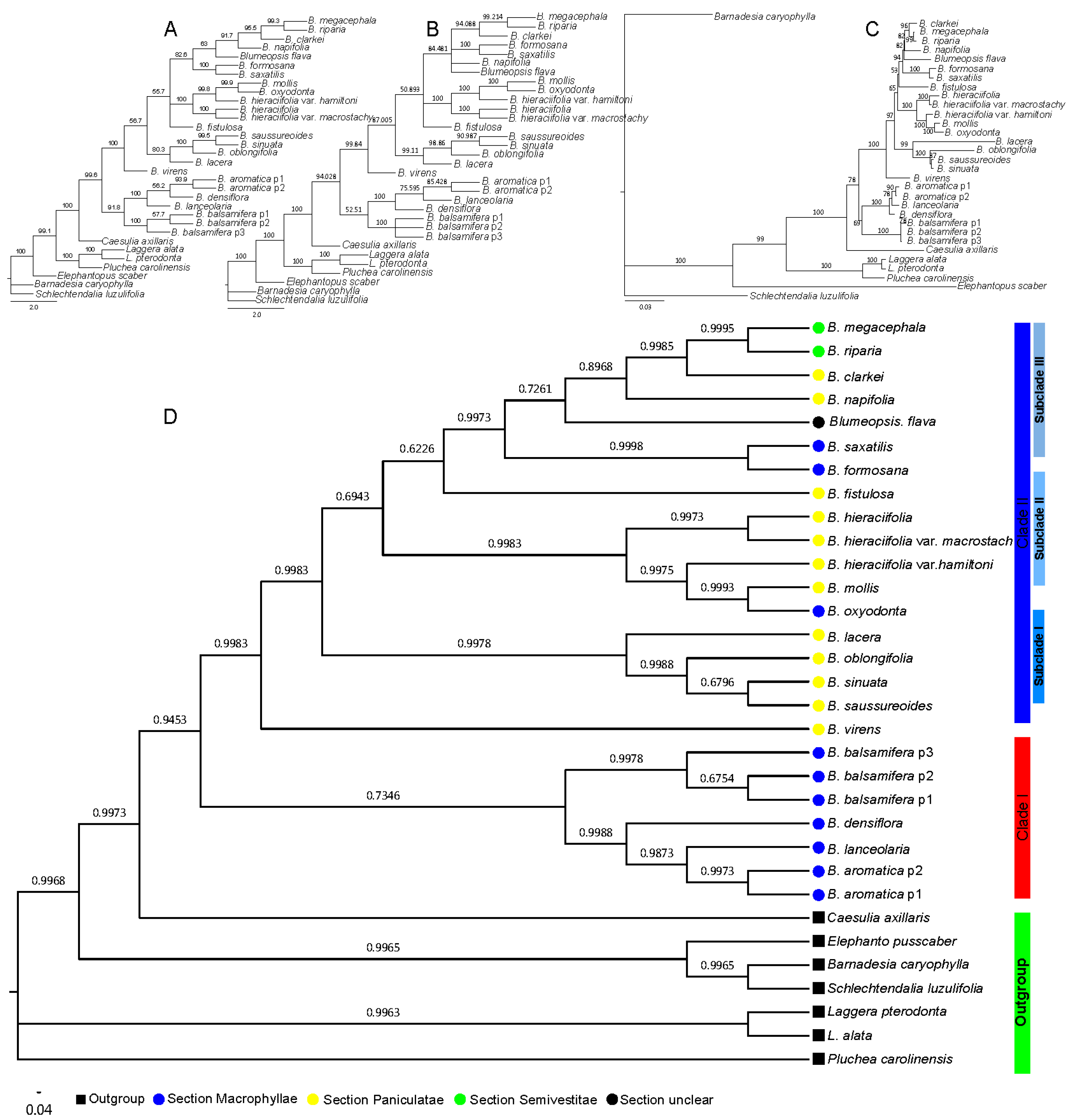
2.2. Phylogenetic Relationship of Blumea DC.
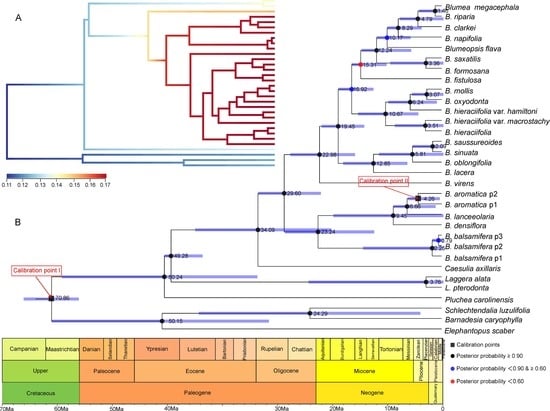
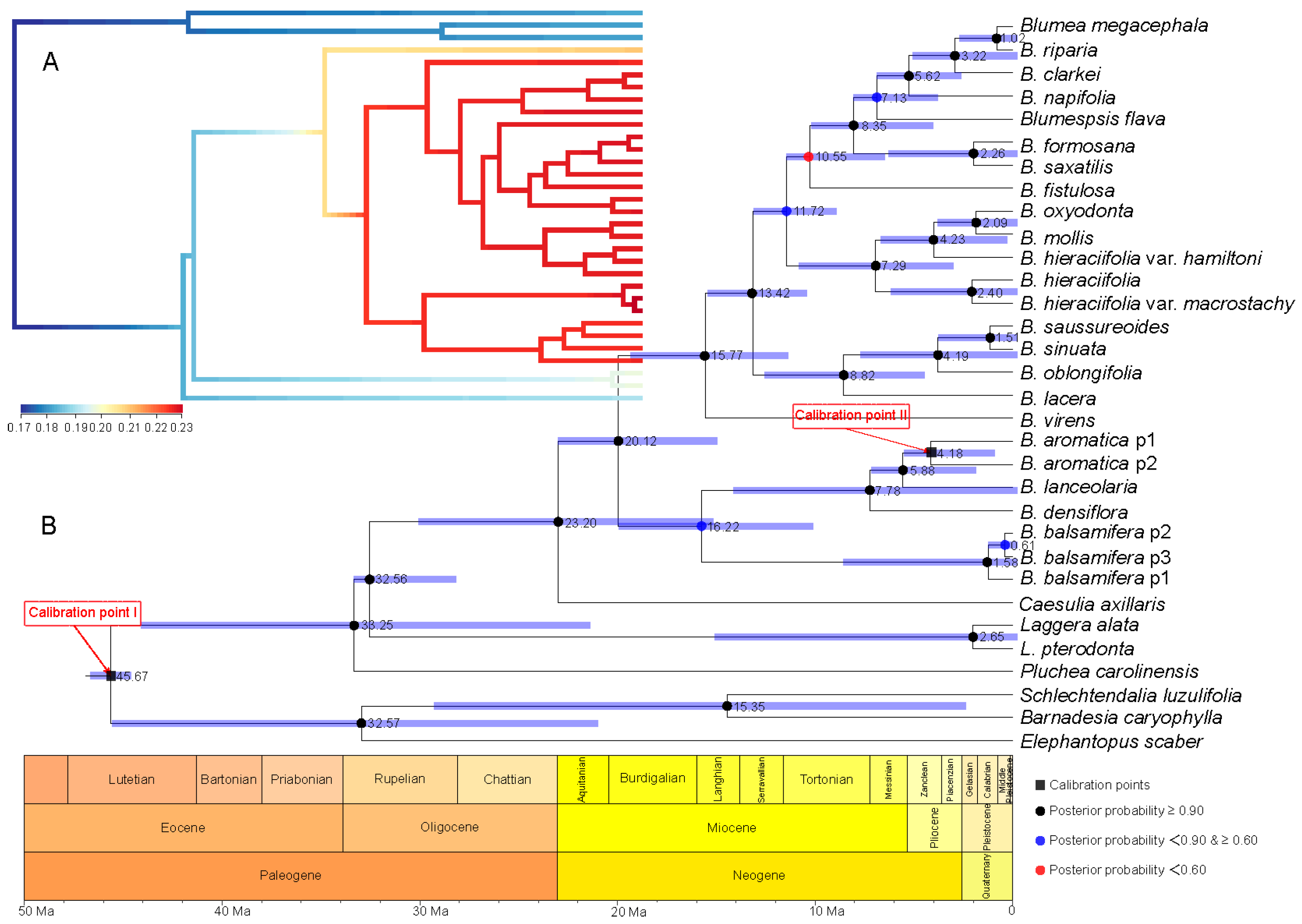
2.3. Divergence Time Estimation and Evolutionary Event Hypothesis
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. DNA Isolation
4.3. DNA Amplification and Sequencing: ITS and trnL-F Sequence Amplification and Sequencing
4.4. Sequence Processing, Conversion, and Analysis
4.4.1. Single Sequence
4.4.2. Combined Sequence
4.5. Phylogenetic Tree Construction
4.5.1. Neighbor-Joining (NJ) Method
4.5.2. Maximum Parsimony (MP) Method
4.5.3. Maximum Likelihood (ML) Method
4.5.4. Bayesian inference (BI) method
4.6. Divergence Time Estimation
4.6.1. Time Node Selection and Correction
4.6.2. Divergence Time Estimation and Evolutionary Event Hypothesis
Author Contributions
Funding
Conflicts of Interest
References
- Randeria, A.J. The compositae genus blumea: A taxonomic revision. Blumea Biodivers. Evol. Biogeogr. Plants 1960, 10, 176–317. [Google Scholar]
- Bremer, K.; Anderberg, A.A. Asteraceae: Cladistics & Classification; Timber Press: Portland, OR, USA, 1994. [Google Scholar]
- Anderberg, A.A.; Eldenäs, P. XVII. Tribe inuleae cass. In Flowering Plants. Eudicots: Asterales; Kadereit, J.W., Jeffrey, C., Eds.; Springer: Berlin, Germany, 2007; pp. 374–390. [Google Scholar]
- Chao-chien, C.; Yong-qian, T. The asteraceae: Tribe of inuleae-helenieae. In Flora Reipublicae Popularis Sinica; Yong, L., Ed.; Science: Beijing, China, 1984; Volume 75, pp. 11–44. [Google Scholar]
- Shi, Z.; Chen, Y.; Chen, Y.; Lin, Y.; Liu, S.; Ge, X.; Gao, T.; Zhu, S.; Liu, Y.; Yang, Q.; et al. Asteraceae (Compositae) [family introduction, glossary, systematic list, and key to tribes]. In Flora of China; Raven, P.H., Zhang, L., Al-Shehbaz, I.A., Turland, N.J., Guanghua, Z., Pepper, L.J., Eds.; Science Press: Beijing, China; Missouri Botanical Garden Press: St. Louis, MO, USA, 2011; Volumes 20–21, pp. 829–837. [Google Scholar]
- Jia, M.; Li, X. Chinese National Pharmacology, 1st ed.; Chinese Medical Science: Beijing, China, 2005. [Google Scholar]
- Perry, L.M.; Metzger, J. Medicinal Plants of East and Southeast Asia: Attributed Properties and Uses, 1st ed.; Massachusetts Institute of Technology: Cambridge, MA, USA, 1980. [Google Scholar]
- State Pharmacopoeia Commission of the PRC. Pharmacopoeia of the People’s Republic of China; Chemical Industry Press: Beijing, China, 2000. [Google Scholar]
- Huang, T.; Di, Z.; Zhao, S. Modern Ben Cao Gang Mu, 1st ed.; Chinese Medical Science Press: Beijing, China, 2001. [Google Scholar]
- Pornpongrungrueng, P.; Borchsenius, F.; Englund, M.; Anderberg, A.A.; Gustafsson, M.H.G. Phylogenetic relationships in Blumea (Asteraceae: Inuleae) as evidenced by molecular and morphological data. Plant Syst. Evol. 2007, 269, 223–243. [Google Scholar] [CrossRef]
- Pornpongrungrueng, P.; Borchsenius, F.; Gustafsson, M.H.G. Relationships within Blumea (Inuleae, Asteraceae) and the Utility of the 5S-NTS in Species-Level Phylogeny Reconstruction. Taxon 2009, 58, 259–272. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Xia, X. DAMBE7: New and Improved Tools for Data Analysis in Molecular Biology and Evolution. Mol. Biol. Evol. 2018, 35, 1550–1552. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and high-performance computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Swofford, D.L. PAUP*; Version 4; Sinauer Associates: Sunderland, MA, USA, 2003. [Google Scholar]
- Akaike, H. Information theory and an extension of the maximum likelihood principle. In Proceedings of the 2nd International Symposium on Information Theory, Tsahkadsor, Armenia, 2–8 September 1971; Csáki, F., Petrov, B.N., Eds.; Akadémiai Kiado: Budapest, Hungary, 1973; p. 5. [Google Scholar]
- Schwarz, G.E. Estimating the Dimension of a Model. Ann. Stat. 1978, 6, 461–464. [Google Scholar] [CrossRef]
- Schmidt, H.A.; Petzold, E.; Vingron, M.; Von Haeseler, A. Molecular phylogenetics: Parallelized parameter estimation and quartet puzzling. J. Parallel Distrib. Comput. 2003, 63, 719–727. [Google Scholar] [CrossRef]
- Shimodaira, H.; Hasegawa, M. CONSEL: For assessing the confidence of phylogenetic tree selection. Bioinformatics 2001, 17, 1246–1247. [Google Scholar] [CrossRef]
- Ki-Joong, K.; Keung-Sun, C.; Jansen, R.K. Two chloroplast DNA inversions originated simultaneously during the early evolution of the sunflower family (Asteraceae). Mol. Biol. Evol. 2005, 22, 1783–1792. [Google Scholar]
- Torices, R. Adding time-calibrated branch lengths to the Asteraceae supertree. J. Syst. Evol. 2010, 48, 271–278. [Google Scholar] [CrossRef]
- Wagstaff, S.J.; Ilse, B.; Motomi, I. Evolution and biogeography of Pleurophyllum (Astereae, Asteraceae), a small genus of megaherbs endemic to the subantarctic islands. Am. J. Bot. 2011, 98, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Rabosky, D.L. Automatic Detection of Key Innovations, Rate Shifts, and Diversity-Dependence on Phylogenetic Trees. PLoS ONE 2014, 9, e89543. [Google Scholar] [CrossRef] [PubMed]
- Rabosky, D.L.; Grundler, M.; Anderson, C.; Title, P.; Shi, J.J.; Brown, J.W.; Huang, H.; Larson, J.G. BAMMtools: An R package for the analysis of evolutionary dynamics on phylogenetic trees. Methods Ecol. Evol. 2014, 5, 701–707. [Google Scholar] [CrossRef]
- Suchard, M.A.; Baele, G.; Lemey, P.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4. [Google Scholar] [CrossRef] [PubMed]
- Bell, M.A.; Lloyd, G.T. strap: An R package for plotting phylogenies against stratigraphy and assessing their stratigraphic congruence. Palaeontology 2015, 58, 379–389. [Google Scholar] [CrossRef]
- Barreda, V.D.; Palazzesi, L.; Tellería, M.C.; Olivero, E.B.; Raine, J.I.; Forest, F. Early evolution of the angiosperm clade Asteraceae in the Cretaceous of Antarctica. Proc. Natl. Acad. Sci. USA 2015, 112, 10989–10994. [Google Scholar] [CrossRef] [PubMed]
- Hedges, S.B.; Marin, J.; Suleski, M.; Paymer, M.; Kumar, S. Tree of Life Reveals Clock-Like Speciation and Diversification. Mol. Biol. Evol. 2015, 32, 835–845. [Google Scholar] [CrossRef]
- Lisiecki, L.E.; Raymo, M.E. A Pliocene-Pleistocene stack of 57 globally distributed benthic δ18O records. Paleoceanogr. Paleoclimatol. 2005, 20. [Google Scholar] [CrossRef]
- Miller, K.G.; Browning, J.V.; Aubry, M.P.; Wade, B.S.; Katz, M.E.; Kulpecz, A.A.; Wright, J.D. Eocene-Oligocene global climate and sea-level changes: St. Stephens Quarry, Alabama. Geol. Soc. Am. Bull. 2008, 120, 34–53. [Google Scholar] [CrossRef]
- Smedmark, J.E.E.; Anderberg, A.A. Boreotropical migration explains hybridization between geographically distant lineages in the pantropical clade Sideroxyleae (Sapotaceae). Am. J. Bot. 2007, 94, 1491–1505. [Google Scholar] [CrossRef] [PubMed]
- Lu, W. A Systematic Study of Blumea DC. (Asteraceae) of Taiwan; National Taiwan University: Taibei, Taiwan, 1996. [Google Scholar]
- Pang, Y.; Wang, D.; Fan, Z.; Chen, X.; Yu, F.; Hu, X.; Wang, K.; Yuan, L. Blumea balsamifera—A phytochemical and pharmacological review. Molecules 2014, 19, 9453–9477. [Google Scholar] [CrossRef] [PubMed]
- Lanyon, S.M.; Omland, K.E. A Molecular Phylogeny of the Blackbirds (Icteridae): Five Lineages Revealed by Cytochrome-B Sequence Data. Auk Ornithol. Adv. 1999, 116, 629–639. [Google Scholar] [CrossRef]
- Benton, M. Fossil Record 2, 1st ed.; Springer: Dordrecht, The Netherlands, 1993. [Google Scholar]
- Graham, A. A contribution to the geologic history of the Compositae. In Proceedings of the International Compositae Conference, Kew, UK, 24 July–5 August 1994; Hind, D.J.N., Beentje, H., Eds.; Royal Botanic Gardens: London, UK, 1996; Volume 1, pp. 123–140. [Google Scholar]
- De-xin, J.; Hu-qiu, Y. A research on Tertiary Palynology from the Qaidam Basin, Qinghai Province (in Chinese). Acta Bot. Sin. 1998, 1, 77–82. [Google Scholar]
- Hind, D.J.N.; Jeffrey, C.; Pope, G.V. Advances in compositae systematics. In Distributed for Royal Botanic Gardens, Kew; Royal Botanic Gardens, Kew: London, UK, 1995. [Google Scholar]
- Zhao, H.; Wang, L.; Yuan, J. Origin and time of Qiongzhou strait. Mar. Quat. Geol. 2007, 27, 33–38. [Google Scholar]
- Yong, H.; Guo, X.; Ho, S.Y.W.; Shi, H.; Li, J.; Li, J.; Bo, C.; Wang, Y. Diversification and Demography of the Oriental Garden Lizard (Calotes versicolor) on Hainan Island and the Adjacent Mainland. PLoS ONE 2013, 8, e64754. [Google Scholar]
- Zhu, H. Biogeographical Evidences Help Revealing the Origin of Hainan Island. PLoS ONE 2016, 11, e0151941. [Google Scholar] [CrossRef] [PubMed]
- Taberlet, P.; Gielly, L.; Pautou, G.; Bouvet, J. Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Mol. Biol. 1991, 17, 1105–1109. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef]
- Talavera, G.; Castresana, J. Improvement of Phylogenies after Removing Divergent and Ambiguously Aligned Blocks from Protein Sequence Alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef]
- Panero, J.L.; Funk, V.A. The value of sampling anomalous taxa in phylogenetic studies: Major clades of the Asteraceae revealed. Mol. Phylogenetics Evol. 2008, 47, 757–782. [Google Scholar] [CrossRef] [PubMed]
- Funk, V.A.; Sancho, G.; Roque, N.; Kelloff, C.L.; Ventosa-Rodríguez, I.; Diazgranados, M.; Bonifacino, J.M.; Chan, R. A phylogeny of the Gochnatieae: Understanding a critically placed tribe in the Compositae. Taxon 2014, 63, 859–882. [Google Scholar] [CrossRef]
- Farris, J.S.; Källersjö, M.; Kluge, A.G.; Bult, C. Testing Significance of Incongruence. Cladistics Int. J. Willi Hennig Soc. 2010, 10, 315–319. [Google Scholar] [CrossRef]
- Vaidya, G.; Lohman, D.J.; Meier, R. SequenceMatrix: Concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics Int. J. Willi Hennig Soc. 2011, 27, 171–180. [Google Scholar] [CrossRef]
- Lam-Tung, N.; Schmidt, H.A.; Arndt, V.H.; Bui Quang, M. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar]
- Chernomor, O.; Haeseler, A.V.; Minh, B.Q. Terrace Aware Data Structure for Phylogenomic Inference from Supermatrices. Syst. Biol. 2016, 65, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ronquist, F. Mrbayes: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Phylogenies from molecular sequences: Inference and reliability. Annu. Rev. Genet. 1988, 22, 521–565. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DNA Region | Number of Characters | Number of Constant Characters | Number of Variable Characters | Number of Informative Characters | Percent of Informative Sites (%) | Best Substitution Model | Model Matrix | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Rates | Ncat | P-invar | Gamma Shape | |||||||
| nrDNA ITS | 657 | 230 | 427 | 314 | 47.79 | SYM + I + G a | Gamma | 4 | 0.2390 | 2.1770 |
| cpDNA trnL-F | 858 | 700 | 158 | 70 | 8.16 | TVM + G b | Gamma | 4 | 0.0 | 0.9240 |
| Combined sequence | 1545 | 960 | 551 | 375 | 24.27 | GTR + I + G a | Gamma | 4 | 0.0 | 0.7302 |
| DNA Region | Tree Method | Rank | obsa | aub | npc | bpd | ppe | khf | shg | wkhh | wshi |
|---|---|---|---|---|---|---|---|---|---|---|---|
| nrDNA ITS | NJa | 3 | 2.6 | 0.358 | 0.331 | 0.095 | 0.067 | 0.337 | 0.557 | 0.285 | 0.635 |
| MPb | 4 | 26.5 | 0.002 | 0.002 | 0.002 | 3.000 × 10−12 | 0.013 | 0.025 | 0.013 | 0.019 | |
| MLc | 1 | −2.6 | 0.681 | 0.669 | 0.674 | 0.866 | 0.662 | 0.859 | 0.662 | 0.885 | |
| BId | 2 | 2.6 | 0.357 | 0.331 | 0.229 | 0.067 | 0.338 | 0.558 | 0.338 | 0.648 | |
| cpDNA trnL-F | NJ | 4 | 0.4 | 0.337 | 0.274 | 0.275 | 0.176 | 0.300 | 0.309 | 0.300 | 0.309 |
| MP | 3 | 0.0 | 0.410 | 0.213 | 0.210 | 0.274 | 0.326 | 0.743 | 0.326 | 0.620 | |
| ML | 2 | 0.0 | 0.503 | 0.357 | 0.351 | 0.274 | 0.453 | 0.682 | 0.453 | 0.655 | |
| BI | 1 | 0.0 | 0.731 | 0.163 | 0.164 | 0.275 | 0.547 | 0.997 | 0.547 | 0.992 | |
| Combined sequence | NJ | 4 | 8.5 | 0.097 | 0.077 | 0.077 | 1.000 × 10−4 | 0.125 | 0.229 | 0.125 | 0.187 |
| MP | 3 | 0.0 | 0.328 | 0.293 | 0.301 | 0.009 | 0.297 | 0.435 | 0.297 | 0.413 | |
| ML | 2 | 0.0 | 0.165 | 0.055 | 0.056 | 0.495 | 0.085 | 0.753 | 0.085 | 0.691 | |
| BI | 1 | 0.0 | 0.867 | 0.578 | 0.566 | 0.496 | 0.915 | 0.982 | 0.915 | 0.986 |
| Section | Code | Latin Name | Locality | GenBank Accession Number | Reference | |
|---|---|---|---|---|---|---|
| ITS | trnL-F | |||||
| Semivestitae (2 spp.) | B. megacephala | Blumea megacephala (Randeria) Chang & Tseng | Guangxi, China | KP052666 | KP052682 | This research |
| B. riparia | B. riparia (Bl.) DC. | Yunnan, China | KP052668 | KP052685 | This research | |
| Macrophyllae (6 spp.) | B. balsamifera p1 | B. balsamifera (L.) DC. | Guizhou, China | KP052658 | KP052674 | This research |
| B. balsamifera p2 | B. balsamifera (L.) DC. | Yunnan, China | KP052659 | KP052675 | This research | |
| B. balsamifera p3 | B. balsamifera (L.) DC. | Hainan, China | KP052660 | KP052676 | This research | |
| B. aromatic p1 | B. aromatica DC. | Guizhou, China | KP052656 | KP052672 | This research | |
| B. aromatic p2 | B. aromatica DC. | Hainan, China | KP052657 | KP052673 | This research | |
| B. oxydonta | B. oxyodonta DC. | Thailand | EU195665 | EU195630 | [11] | |
| B. densiflora | B. densiflora DC. | Thailand | EF210934 | EF211029 | [10] | |
| B. saxatilis | B. saxatilis Zoll. & Mor. | Australia | EF210945 | EF211040 | [10] | |
| B. formosana | B. formosana Kitam | Zhejiang, China | KP052665 | KP052678 | This research | |
| B. clarkei | B. clarkei Hook. f. | Thailand | EF210974 | EF211069 | [10] | |
| Paniculatae (9 spp. and 2 varieties) | B. fistulosa | B. fistulosa (Roxb.) Kurz. | Yunnan, China | KP052661 | KP052677 | This research |
| B. hieraciifolia | B. hieraciifolia (D. Don) DC. | Yunnan, China | KP052662 | KP052679 | This research | |
| B. hieraciifolia var. hamiltonii | B. hieraciifolia var. hamiltonii | Burma | EF210972 | EF211067 | [10] | |
| B. hieraciifolia var. macrostachya | B. hieraciifolia var. macrostachya | Thailand | EF210937 | EF211032 | [10] | |
| B. lanceolaria | B. lanceolaria (Roxb.) Druce. | Guangxi, China | KP052664 | KP052681 | This research | |
| B. lacera | B. lacera (Burm. f.) DC. | Zhejiang, China | KP052663 | KP052680 | This research | |
| B. mollis | B. mollis (D. Don) Merr. | Hainan, China | KP052670 | KP052683 | This research | |
| B. napifolia | B. napifolia DC. | Thailand | EF210959 | EF211054 | [10] | |
| B. oblongifolia | B. oblongifolia Kitam | Hainan, China | KP052667 | KP052684 | This research | |
| B. saussureoides | B. saussureoides Chang & Tseng | Yunan, China | KP052669 | KP052686 | This research | |
| B. sinuata | B. sinuata (Lour.) Merr. | Thailand | EF210948 | EF211043 | [10] | |
| B. virens | B. virens DC. | Thailand | EF210957 | EF211052 | [10] | |
| Uncertainty | Blumeposis flava | Blumeposis flava Gagnep. | Thailand | EF210960 | EF211055 | [10] |
| Outgroup | Caesulia axillaris | Caesulia axillaris Roxb. | India | EF210949 | EF211044 | [10] |
| Pluchea carolinensis | Pluchea carolinensis (Jacq.) G. Don. | Taiwan, China | AF437850 | EU385104 | [45] | |
| Laggera alata | Laggera alata (D. Don) Sch. Bip. ex Oliv. | Thailand | EF210930 | EF211025 | [10] | |
| L. pterodonta | L. pterodonta (DC.) Sch. Bip. ex Oliv. | Thailand | EF210929 | EF211024 | [10] | |
| Schlechtendalia luzulifolia | Schlechtendalia luzulifolia | Australia | KF989506 | KF989612 | [46] | |
| Barnadesia caryophylla | Barnadesia caryophylla | Australia | AY504686 | AY504768 | [46] | |
| Elephantopus scaber | Elephantopus scaber L. | Hainan, China | KP052671 | KP052687 | This research | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.-b.; Yuan, Y.; Pang, Y.-x.; Yu, F.-l.; Yuan, C.; Wang, D.; Hu, X. Phylogenetic Reconstruction and Divergence Time Estimation of Blumea DC. (Asteraceae: Inuleae) in China Based on nrDNA ITS and cpDNA trnL-F Sequences. Plants 2019, 8, 210. https://doi.org/10.3390/plants8070210
Zhang Y-b, Yuan Y, Pang Y-x, Yu F-l, Yuan C, Wang D, Hu X. Phylogenetic Reconstruction and Divergence Time Estimation of Blumea DC. (Asteraceae: Inuleae) in China Based on nrDNA ITS and cpDNA trnL-F Sequences. Plants. 2019; 8(7):210. https://doi.org/10.3390/plants8070210
Chicago/Turabian StyleZhang, Ying-bo, Yuan Yuan, Yu-xin Pang, Fu-lai Yu, Chao Yuan, Dan Wang, and Xuan Hu. 2019. "Phylogenetic Reconstruction and Divergence Time Estimation of Blumea DC. (Asteraceae: Inuleae) in China Based on nrDNA ITS and cpDNA trnL-F Sequences" Plants 8, no. 7: 210. https://doi.org/10.3390/plants8070210
APA StyleZhang, Y.-b., Yuan, Y., Pang, Y.-x., Yu, F.-l., Yuan, C., Wang, D., & Hu, X. (2019). Phylogenetic Reconstruction and Divergence Time Estimation of Blumea DC. (Asteraceae: Inuleae) in China Based on nrDNA ITS and cpDNA trnL-F Sequences. Plants, 8(7), 210. https://doi.org/10.3390/plants8070210