Multi-Target β-Protease Inhibitors from Andrographis paniculata: In Silico and In Vitro Studies


Abstract
:1. Introduction
2. Results and Discussion
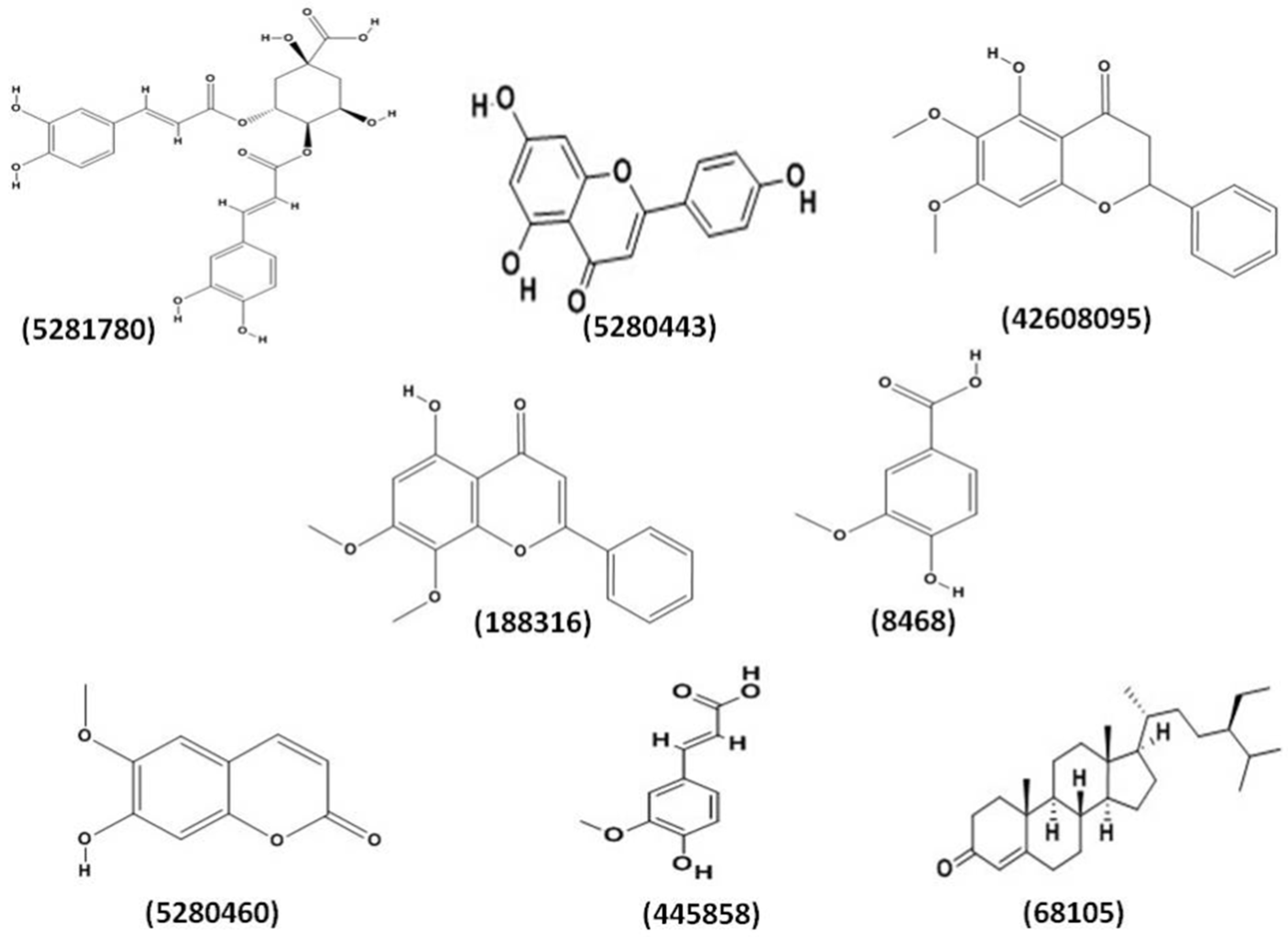
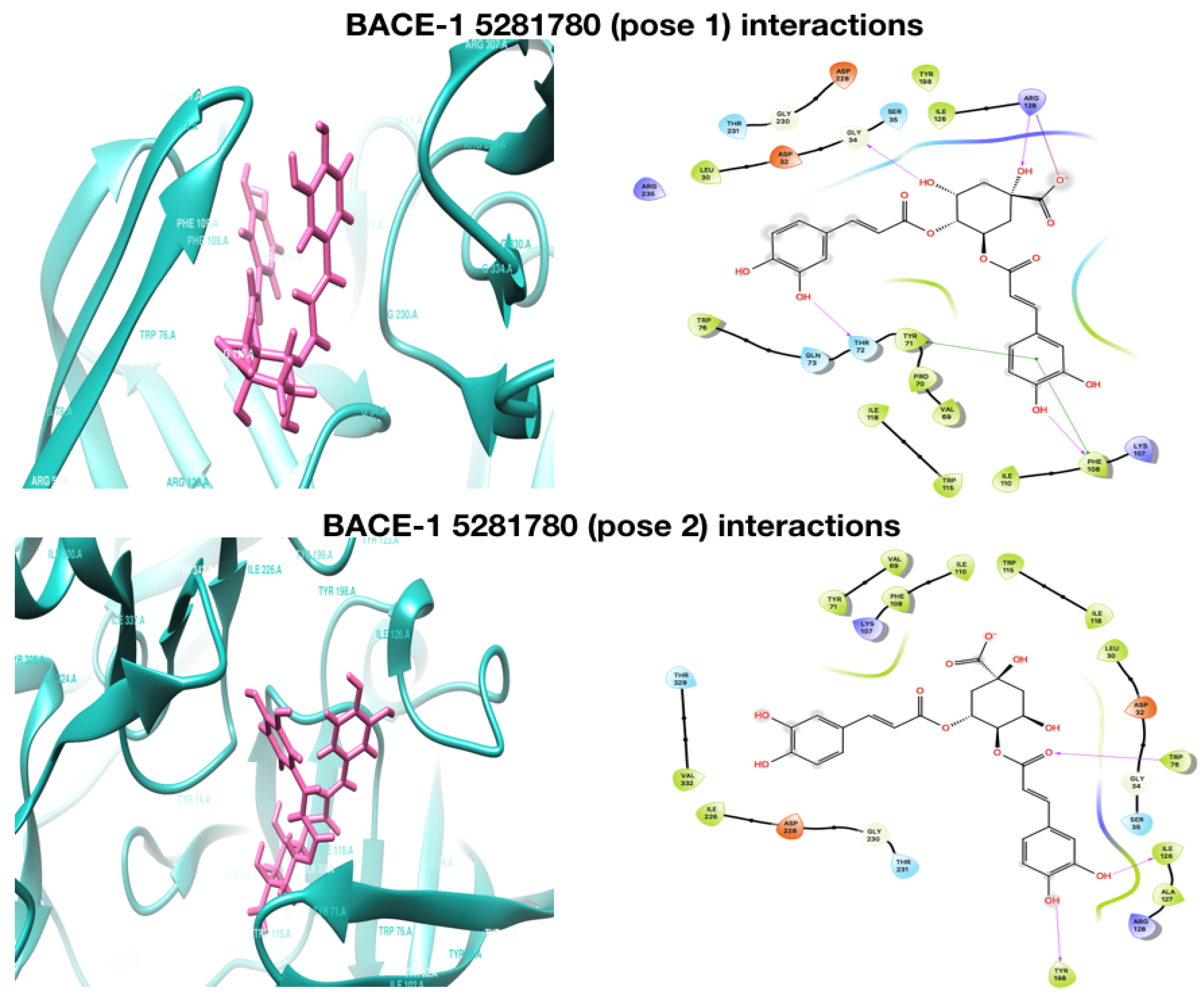
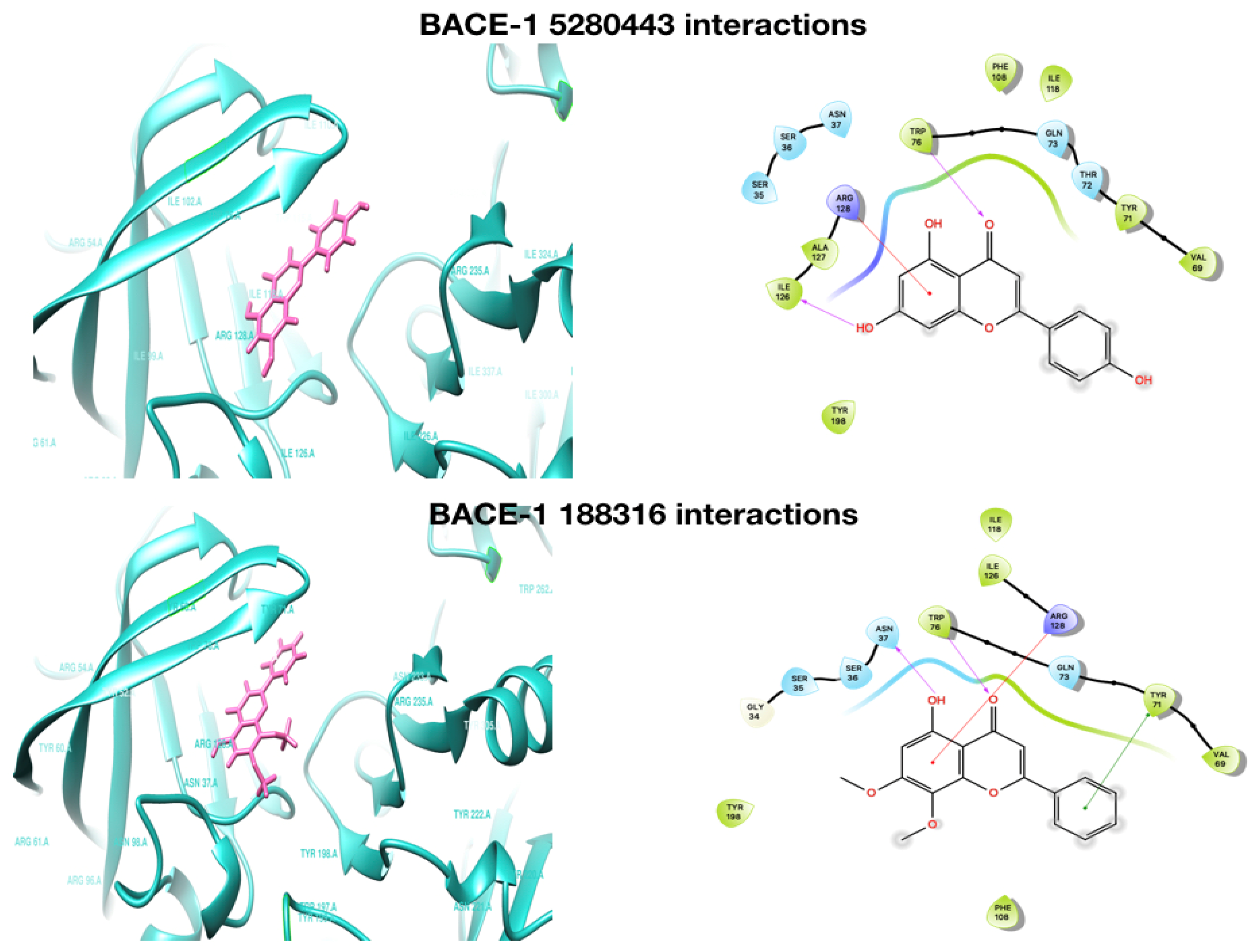
2.1. Molecular Docking Analysis
2.2. ADME Test
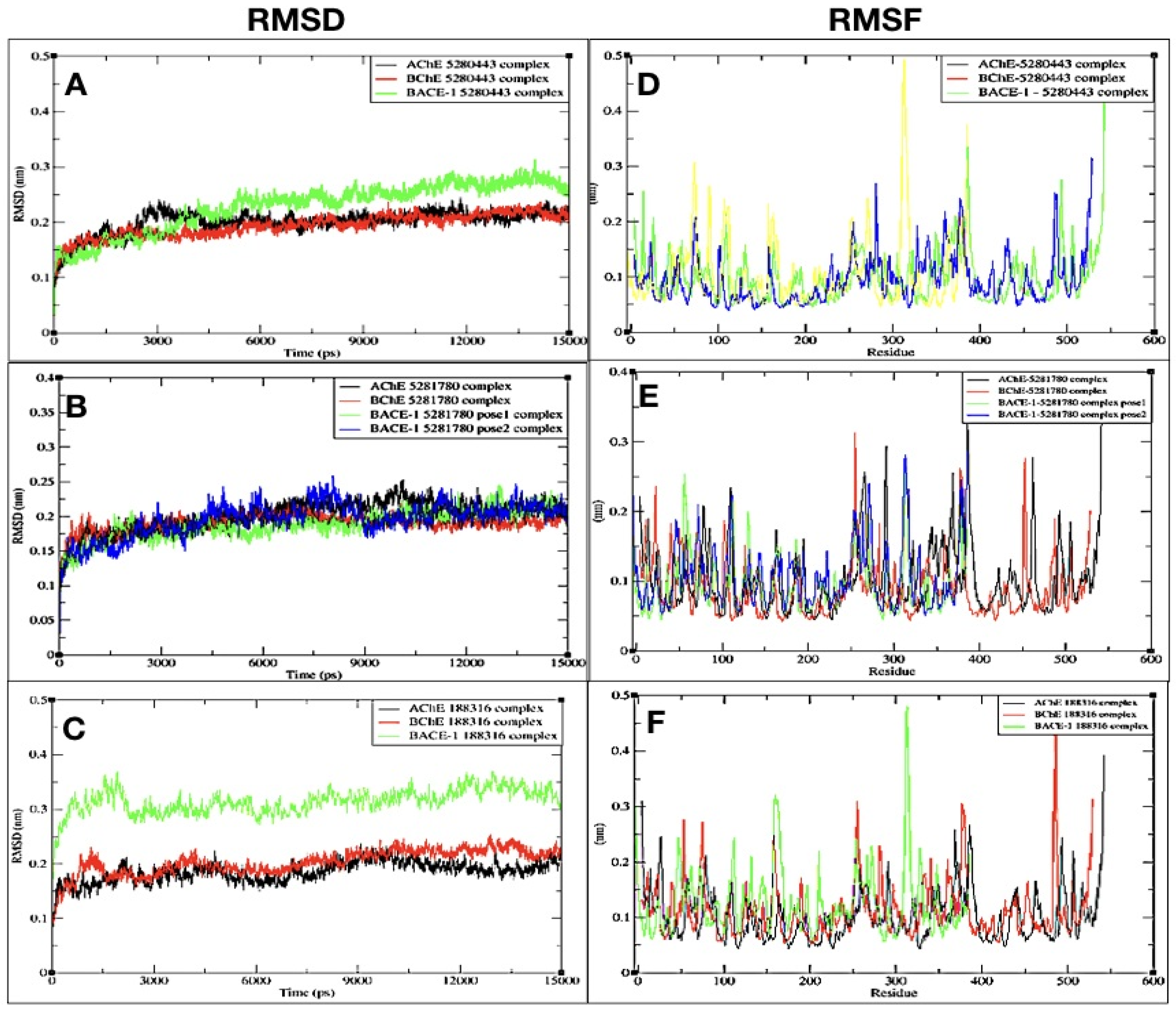
2.3. Molecular Dynamics (MD) and Simulations
2.3.1. RMSD
2.3.2. RMSF
2.3.3. Interacting H-Bond Analysis
2.4. Enzyme Inhibition
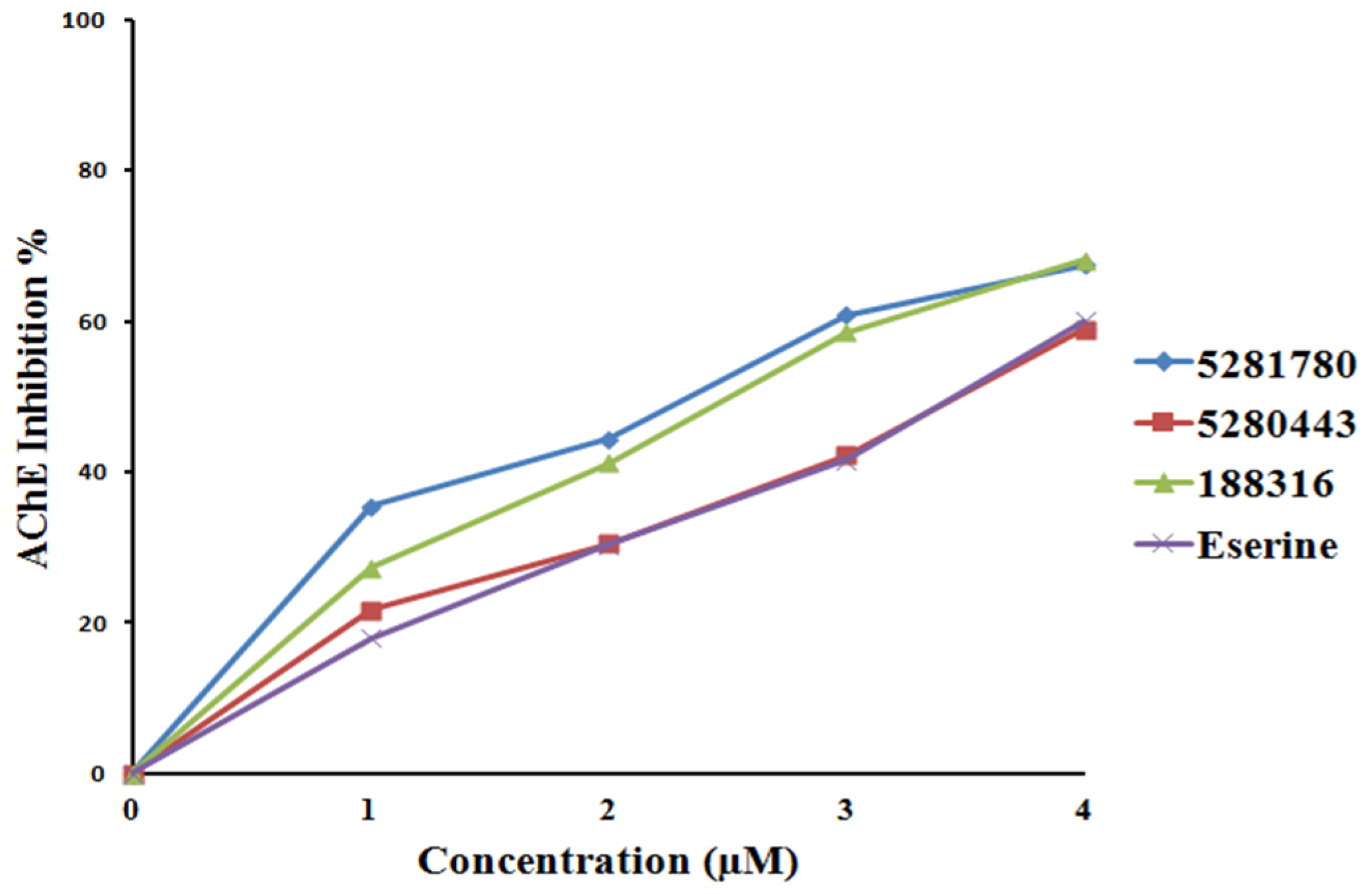
2.4.1. Cholinesterase Inhibition
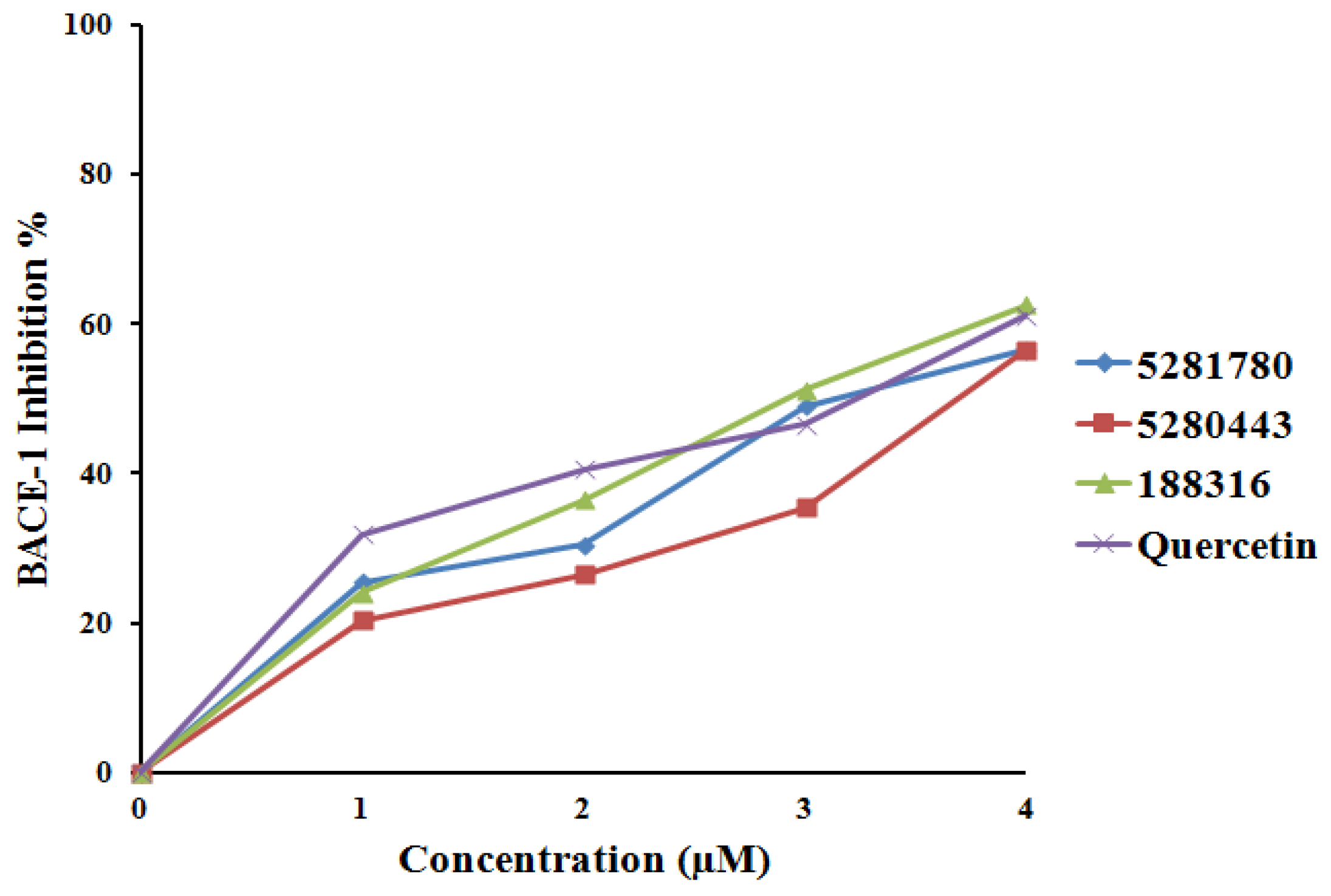
2.4.2. BACE-1 Inhibition
3. Materials and Methods
3.1. Protein Preparation
3.2. Ligand Preparation
3.3. Molecular Docking
3.4. ADME Prediction
3.5. Molecular Dynamics and Simulations
3.6. Enzyme Inhibition Assay of Isolated Flavonoids
3.6.1. In Vitro BACE-1 Activity Assay
3.6.2. In Vitro AChE and BChE Activity Assay
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Lemaire, H.G.; Unterbeck, A.; Salbaum, J.M.; Masters, C.L.; Grzeschik, K.H.; Multhaup, G.; Beyreuther, K.; Müller-Hill, B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 1987, 325, 733–736. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.W.; Thompson, R.; Zhang, H.; Xu, H. APP processing in Alzheimer’s disease. Mol. Brain 2011, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Cecilia, R.A.S.; Isabel, C.; Isabel, G. Alzheimer’s Disease Pathogenesis, Core Concepts, Shifting Paradigms and Therapeutic Targets; Intech Open: London, UK, 2011. [Google Scholar]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef] [PubMed]
- Rochette, M.J.; Murphy, M.P. Gamma secretase: Substrates and inhibitors. Mol. Neurobiol. 2002, 26, 81–95. [Google Scholar] [CrossRef]
- Tang, J.; Ghosh, A. Treating transgenic Alzheimer mice with a β-secretase inhibitor, what have we learned? Aging 2011, 3, 14–16. [Google Scholar] [CrossRef] [PubMed]
- Hussain, I.; Powell, D.; Howlett, D.R.; Tew, D.G.; Meek, T.D.; Chapman, C.; Gloger, I.S.; Murphy, K.E.; Southan, C.D.; Ryan, D.M.; et al. Identification of a novel aspartic protease (Asp 2) as beta-secretase. Mol. Cell. Neurosci. 1999, 14, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Anderson, J.P.; Barbour, R.; Basi, G.S.; Caccavello, R.; Davis, D.; Doan, M.; Dovey, H.; Frigon, N.; Hong, J.; et al. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature 1999, 402, 537–540. [Google Scholar] [CrossRef]
- Vassar, R.; Bennet, B.; Babu-khan, S.; Kahn, S.; Mendiaz, E.; Denis, P.; Teplow, D.; Ross, S.; Amarante, P.; Loeloff, R.; et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef]
- Yan, R.; Bienkowski, M.J.; Shuck, M.E.; Miao, H.; Tory, M.C.; Pauley, A.M.; Brashier, J.R.; Stratman, N.C.; Mathews, W.R.; Buhl, A.E.; et al. Membrane-anchored aspartyl protease with Alzheimer’s disease beta-secretase activity. Nature 1999, 402, 533–537. [Google Scholar] [CrossRef]
- Kennedy, M.E.; Stamford, A.W.; Chen, X.; Cox, K.; Cumming, J.N.; Dockendorf, M.F.; Egan, M.; Ereshefsky, L.; Hodgson, R.A.; Hyde, L.A.; et al. The BACE1 inhibitor verubecestat (MK-8931) reduces CNS β-amyloid in animal models and in Alzheimer’s disease patients. Sci. Transl. Med. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- May, P.C.; Dean, R.A.; Lowe, S.L.; Martenyi, F.; Sheehan, S.M.; Boggs, L.N.; Monk, S.A.; Mathes, B.M.; Mergott, D.J.; Watson, B.M.; et al. Robust central reduction of amyloid-β in humans with an orally available, non-peptidic β-secretase inhibitor. J. Neurosci. 2011, 31, 16507–16516. [Google Scholar] [CrossRef] [PubMed]
- May, P.C.; Willis, B.A.; Lowe, S.L.; Dean, R.A.; Monk, S.A.; Cocke, P.J.; Audia, J.E.; Boggs, L.N.; Borders, A.R.; Brier, R.A.; et al. Robust central reduction of amyloid-β in humans with an orally available, non-peptidic beta-secretase inhibitor. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Kato, S. Theoretical Perspectives on the Reaction Mechanism of Serine Proteases: The Reaction Free Energy Profiles of the Acylation Process. Process. J. Am. Chem. Soc. 2003, 125, 12035–12048. [Google Scholar] [CrossRef] [PubMed]
- Hedstrom, L. Serine protease mechanism and specificity. Chem. Rev. 2002, 102, 4501–4523. [Google Scholar] [CrossRef]
- Topf, M.; Richards, W.G. Theoretical studies on the deacylation step of serine protease catalysis in the gas phase, in solution, and in elastase. J. Am. Chem. Soc. 2004, 126, 14631–14641. [Google Scholar] [CrossRef] [PubMed]
- Quinn, D.M. Acetylcholinesterase: Enzyme Structure, Reaction Dynamics, and Virtual Transition Stats. Chem. Rev. 1987, 87, 955–979. [Google Scholar] [CrossRef]
- Inestrosa, N.C.; Sagal, J.P.; Colombres, M. Acetylcholinesterase interaction with Alzheimer amyloid beta. Subcell. Biochem. 2005, 38, 299–317. [Google Scholar]
- Inestrosa, N.C.; Dinamarc, M.C.; Alvarez, A. Amyloid-cholinesterase interactions. Implications for Alzheimer’s disease. FEBS J. 2008, 275, 625–632. [Google Scholar] [CrossRef]
- Huang, Y.; Mucke, L. Alzheimer mechanisms and therapeutic strategies. Cell 2012, 148, 1204–1222. [Google Scholar] [CrossRef]
- Herrmann, N.; Chau, S.A.; Kircanski, I.; Lanctot, K.L. Current and emerging drug treatment options for Alzheimer’s disease: A systematic review. Drugs 2011, 71, 2031–2065. [Google Scholar] [CrossRef] [PubMed]
- Danysz, W.; Parsons, C. Alzheimer’s disease beta-amyloid, glutamate, NMDA receptors and memantine-searching for the connections. Br. J. Pharmacol. 2012, 16, 324–352. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xie, J.; Bao, D.; Liu, S.; Zhou, Q. Total synthesis of (-)-galanthamine and (-)-lycoramine via catalytic asymmetric hydrogenation and intramolecular reductive heck cyclization. Org. Lett. 2012, 14, 2714–2717. [Google Scholar] [CrossRef] [PubMed]
- Nikiforuk, A.; Potasiewicz, A.; Kos, T.; Popik, P. The combination of memantine and galantamine improves cognition in rats: The synergistic role of the α7 nicotinic acetylcholine and NMDA receptors. Behav. Brain Res. 2016, 313, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Peters, O.; Fuentes, M.; Joachim, L.K.; Jessen, F.; Luckhaus, C.; Kornhuber, J.; Pantel, J.; Hüll, M.; Schmidtke, K.; Rüther, E.; et al. Combined treatment with memantine and galantamine-CR compared with galantamine-CR only in antidementia drug naïve patients with mild-to-moderate Alzheimer’s disease. Transl. Res. Clin. Interv. 2015, 1, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Kaye, A.D.; Liu, H.; Fox, C.; Baluch, A.; Sutker, P.B. Psychiatric and behavioral disorders A2. In Anesthesia and Uncommon Diseases, 6th ed.; Fleisher, L.A., Ed.; Elsevier: Philadelphia, PA, USA, 2012; pp. 444–469. [Google Scholar]
- Darvesh, S.; Hopkins, D.; Geula, C. Neurobiology of Butyrylcholinesterase. Nat. Rev. Neurosci. 2003, 4, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Turner, A.; Hartman, L.; Nakayama, K.; Acey, R. The Role of Butyrylcholinesterase in Beta-Amyloid Formation in Neuroblastoma Cells. FASEB J. 2015, 29, 1. [Google Scholar]
- Matsuoka, S.; Inoue, M. Application of REDOR NMR in natural product chemistry. Chem. Commun. 2009, 5664–5675. [Google Scholar] [CrossRef]
- Kingston, D.G.I. Modern Natural Products Drug Discovery and Its Relevance to Biodiversity Conservation. J. Nat. Prod. 2011, 74, 496–511. [Google Scholar] [CrossRef]
- Maione, F.; Russo, R.; Khan, H.; Mascolo, N. Medicinal plants with anti-inflammatory activities. Nat. Prod. Res. 2016, 30, 1343–1352. [Google Scholar] [CrossRef]
- Mishra, B.B.; Tiwari, V.K. Natural products: An evolving role in future drug discovery. Eur. J. Med. Chem. 2011, 46, 4769–4807. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.; Amin, S. ACE Inhibition of Plant Alkaloids Targeted Approach for Selective Inhibition. Mini Rev. Org. Chem. 2017, 14, 85–89. [Google Scholar] [CrossRef]
- Khan, H. Brilliant future of phytomedicines in the light of latest technological developments. J. Phytopharmacol. 2015, 4, 58–60. [Google Scholar]
- Feng, Y.; Wang, X. Antioxidant Therapies for Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2012, 2012, 472–932. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.A.; Arvanitakis, Z.; Bang, W.; Bennett, D.A. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 2007, 69, 2197–2204. [Google Scholar] [CrossRef] [PubMed]
- Dolan, H.; Troncoso, J.; Crain, B.; Resnick, S.; Zonderman, A.; O’Brien, R. Atherosclerosis, dementia, and Alzheimer disease in the Baltimore Longitudinal Study of aging cohort. Ann. Neurol. 2010, 68, 231–240. [Google Scholar] [PubMed]
- Tang, W.; Eisenbrand, G. Chinese Drugs of Plant Origin, Chemistry, Pharmacology and Use in Traditional and Modern Medicine; Springer: Heidelberg/Berlin, Germany, 1992. [Google Scholar]
- Gamble, J.S. Flora of the Presidency of Madras; Botanical Survey of India: Calcutta, India, 1956. [Google Scholar]
- Lim, J.C.W.; Chan, T.K.; Ng, D.S.W.; Sagineedu, S.R.; Stanslas, J.; Wong, W.S.F. Andrographolide and its analogues: Verstaile bioactive molecules for combating inflammation and cancer. Clin. Exp. Pharmacol. Physiol. 2012, 39, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Prachayasittukul, S.; Suphapong, S.; Worachartcheewan, A.; Lawung, R.; Ruchirawat, S.; Prachayasittukul, V. Bioactive metabolites from Spilanthes acmella Murr. Molecules 2009, 14, 850–867. [Google Scholar] [CrossRef]
- Thakur, H.R.; Bhamare, M.R. Phytochemical and Antimicrobial activity of Spilanthes acmella. Adv. Pharmacol. Sci. 2015, 4, 723–733. [Google Scholar]
- Sana, H.; Rani, A.S.; Sulakshana, G. Determination of Antioxidant Potential in Spilanthes acmella using DPPH assay. Int. J. Curr. Microbiol. Appl. Sci. 2014, 3, 219–223. [Google Scholar]
- Urankar, M.; Desai, A.; Bhat, R. Review on medicinal herb genus Spilanthes and Applications in oral hygiene. Univ. J. Pharm. 2013, 2, 25–33. [Google Scholar]
- Suwanjang, W.; Khongniam, B.; Srisung, S.; Prachayasittikul, S.; Prachayasittikul, S. Neuroprotective effect of Spilanthes acmella Murr on pesticide-induced neuronal cells death. Asian Pacific J. Trop. Med. 2016, 5, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Rao, Y.K.; Vimalamma, G.; Rao, C.V.; Tzeng, Y. Flavonoids and andrographolides from Andrographis paniculata. Phytochemistry 2004, 65, 2317–2321. [Google Scholar] [CrossRef]
- Gulam, N.N.; Wani, T.A.; Shrivastava, M.; Wani, A.; Shah, S.N. Spilanthes acmella an endangered medicinal plant—Its Traditional, Phytochemical and Therapeutic properties—An overview. Int. J. Adv. Res. 2016, 4, 627–639. [Google Scholar]
- Ncube, B.; Nair, J.J.; Rárová, L.; Strnad, M.; Finnie, J.F.; Van Staden, J. Seasonal pharmacological properties and alkaloid content in Cyrtanthus contractus N.E. Br. S. Afr. J. Bot. 2015, 97, 69–76. [Google Scholar] [CrossRef]
- Stafford, G.I.; Pedersen, M.E.; Van Staden, J.; Jäger, A.K. Review on plants with CNS effects used in traditional South African medicine against mental diseases. J. Ethnopharmacol. 2008, 119, 513–537. [Google Scholar] [CrossRef] [PubMed]
- Uysal, S.; Zengina, G.; Mahomoodally, M.F.; Yilmaz, M.A.; Aktumsek, A. Chemical profile, antioxidant properties and enzyme inhibitory effects of the root extracts of selected Potentilla species. S. Afr. J. Bot. 2019, 120, 124–128. [Google Scholar] [CrossRef]
- Sinha, S.; Kumar, B.; Luqman, S.; Singh, D.K. Neuroprotective potential of Cucurbita maxima Duchesne ex Poir, Caeselpenia bunduc (L.) Roxb and Bombax ceiba Linn extracts. S. Afr. J. Bot. 2019, 120, 319–325. [Google Scholar] [CrossRef]
- Akata, G.; Zenginb, C.M.N.; Picot, M.F. Enzyme inhibitory and antioxidant properties of six mushroom species from the Agaricaceae family. S. Afr. J. Bot. 2019, 120, 95–99. [Google Scholar] [CrossRef]
- Ozarowski, M.; Mikolajczak, P.L.; Piasecka, A.; Kujawski, R.; Bartkowiak-Wieczorek, J.; Bogacz, A.; Szulc, M.; Kaminska, E.; Kujawska, M.; Gryszczynska, A.; et al. Effect of Salvia miltiorrhiza root extract on brain acetylcholinesterase and butyrylcholinesterase activities, their mRNA levels and memory evaluation in rats. Physiol. Behav. 2017, 173, 223–230. [Google Scholar] [CrossRef]
- Ahmad, A.; Ramasamy, K.; Majeed, A.B.A.; Mani, V. Enhancement of β-secretase inhibition and antioxidant activities of tempeh, a fermented soybean cake through enrichment of bioactive aglycones. Pharm. Biol. 2015, 53, 758–766. [Google Scholar] [CrossRef] [PubMed]
- Aliaga, K.J.; Bescós, P.B.; Benedi, J.; Aragón, S.M. Quercetin and rutin exhibit antiamyloidogenic and fibril-disaggregating effects in vitro and potent antioxidant activity in APPswe cells. Life Sci. 2011, 89, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, Y.; Su, Y.; Zhou, W.; Yang, S.; Zhang, R.; Zhao, M.; Li, Y.; Zhang, Z.; Zhan, D.; et al. Rutin inhibits β-amyloid aggregation and cytotoxicity, attenuates oxidative stress, and decreases the production of nitric oxide and proinflammatory cytokines. Neurotoxicology 2012, 33, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, A.; D’Abrosca, B.; Pacifico, S.; Mastellone, C.; Piccolella, S.; Monaco, P. Isolation and Structure Elucidation of Antioxidant Polyphenols from Quince (Cydonia vulgaris) Peels. J. Agric. Food Chem. 2008, 56, 2660–2667. [Google Scholar] [CrossRef] [PubMed]
- Hoper, L.; Cassidy, A. A review of the health care potential of bioactive compounds. J. Sci. Food Agric. 2006, 86, 1805–1813. [Google Scholar] [CrossRef]
- Pu, F.; Ren, X.L.; Zhang, X.P. Phenolic compounds and antioxidant activity in fruits of six Diospyros kaki genotypes. Eur. Food. Res. Technol. 2013, 237, 923–932. [Google Scholar] [CrossRef]
- Bolognesi, M.L. Polypharmacology in a single drug: Multitarget drugs. Curr. Med. Chem. 2013, 20, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, M.L.; Cavalli, A. Multitarget drug discovery and polypharmacology. Chem. Med. Chem. 2016, 11, 1190–1192. [Google Scholar] [CrossRef] [PubMed]
- Morphy, R.; Kay, C.; Rankovic, Z. From magic bullets to designed multiple ligands. Drug Discov. Today 2004, 9, 641–651. [Google Scholar] [CrossRef]
- Youdim, M.B.H.; Buccafusco, J.J. Multi-functional drugs for various CNS targets in the treatment of neurodegenerative disorders. Trends Pharmacol. Sci. 2005, 26, 27–35. [Google Scholar] [CrossRef]
- Roth, B.L.; Sheffler, D.J.; Kroeze, W.K. Magic shotguns versus magic bullets: Selectively non-selective drugs for mood disorders and schizophrenia. Nat. Rev. Drug Discov. 2004, 3, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nokolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Trans. Med. 2018, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.S.; Chen, H.J.; Damu, A.G.; Kuo, P.C.; Su, C.R.; Lee, E.J.; Teng, C.M. Flavonoids and ent-labdane diterpenoids from Andrographis paniculata and their antiplatelet aggregatory and vasorelaxing effects. J. Asian Nat. Prod. Res. 2008, 10, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Radic, Z.; Pickering, N.A.; Vellom, D.C.; Camp, S.; Taylor, P. Three distinct domains in the cholinesterase molecule confer selectivity for acetyl- and butyrylcholinesterase inhibitors. Biochemistry 1993, 32, 12074–12084. [Google Scholar] [CrossRef] [PubMed]
- Radic, Z.; Quinn, D.M.; Vellom, D.C.; Camp, S.; Taylor, P. Allosteric control of acetylcholinesterase catalysis by fasciculin. J. Biol. Chem. 1995, 270, 20391–20399. [Google Scholar] [CrossRef] [PubMed]
- Vellom, D.C.; Radic, Z.; Li, Y.; Pickering, N.A.; Camp, S.; Taylor, P. Amino acid residues controlling acetylcholinesterase and butyryl- cholinesterase specificity. Biochemistry 1993, 32, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Kryger, G.; Silman, I.; Sussman, J.L. Three-dimensional structure of a complex of E2020 with acetylcholinesterase from Torpedo califor-nica. J. Physiol. Paris 1998, 92, 191–194. [Google Scholar] [CrossRef]
- Felder, C.E.; Harel, M.; Silman, I.; Sussman, J.L. Structure of a complex of the potent and specific inhibitor BW284C51 with Torpedo californica acetylcholinesterase. Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 1765–1771. [Google Scholar] [CrossRef]
- Ashani, Y.; Peggins, J.O.; Doctor, B.P. Mechanism of inhibition of cho-linesterases by Huperzine A. Biochem. Biophys. Res. Commun. 1992, 184, 719–726. [Google Scholar] [CrossRef]
- Davis, A.; Teague, S. Hydrogen bonding, hydrophobic interactions and failure of the rigid receptor hypothesis. Angew. Chem. Int. Ed. 1999, 38, 736–749. [Google Scholar] [CrossRef]
- Varma, A.; Patil, R.; Das, S.; Stanley, A.; Yadav, L.; Akulapalli, S. Optimized hydrophobic interactions and hydrogen bonding at the target ligand interface leads pathway of Drug designing. PLoS ONE 2010, 5, e12029. [Google Scholar]
- Liu, S.; Fu, R.; Cheng, X.; Chen, S.; Zhou, L. Exploring the binding of BACE-1 inhibitors using comparative binding energy analysis(COMBINE). BMC Struct. Biol. 2012, 12, 21. [Google Scholar] [CrossRef] [PubMed]
- Bourne, Y.; Kolb, H.C.; Radic, Z.; Sharpless, K.B.; Taylor, P.; Marchot, P. Freeze-frame inhibitor captures acetylcholinesterase in a unique conformation. Proc. Natl. Acad. Sci. USA 2004, 101, 1449–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darvesh, S.; Macdonald, I.R.; Martin, E.; Rosenberry, T.L. Probing the Peripheral Site of Human Butyrylcholinesterase. Biochemistry 2012, 51, 7046–7053. [Google Scholar]
- Mallender, W.D.; Szegletes, T.; Rosenberry, T.L. Acetylthiocholine binds to Asp74 at the peripheral site of human acetylcholinesterase as the first step in the catalytic pathway. Biochemistry 2000, 39, 7753–7763. [Google Scholar] [CrossRef]
- Nachmansohn, D.; Wilson, I.B. The enzymic hydrolysis and synthesis of acetylcholine. Adv. Enzymol. Relat. Subj. Biochem. 1951, 12, 259–339. [Google Scholar] [PubMed]
- Masson, P.; Froment, M.T.; Bartels, C.F.; Lockridge, O. Asp70 in the peripheral anionic site of human butyrylcholines-terase. Eur. J. Biochem. 1996, 235, 36–48. [Google Scholar] [CrossRef]
- Johnson, G.; Moore, S.W. The Peripheral Anionic Site of Acetylcholinesterase: Structure, Functions and Potential Role in Rational Drug Design. Curr. Pharm. Des. 2006, 12, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Radic, Z.; Duran, R.; Vellom, D.C.; Li, Y.; Cervenansky, C.; Taylor, P. Site of fasciculin interaction with acetylcholinesterase. J. Biol. Chem. 1994, 269, 11233–11239. [Google Scholar]
- Jung, H.A.; Shrestha, S.; Seong, S.H.; Paudel, P.; Choi, J.S. Structure Related Inhibition of Enzyme Systems in Cholinesterases and BACE1 In vitro by Naturally occurring Naphthopyrone and Its Glycosides Isolated from Cassia obtusifolia. Molecules 2018, 23, 69. [Google Scholar] [CrossRef]
- Matheus, F.; Vargas, J.A.R.; Lopez, A.G.; Piñol, M.C. Molecular docking study on the interaction between 2-substituted-4,5-difuryl Imidazoles with different Protein Target for antileishmanial activity. J. Appl. Pharm. Sci. 2018, 8, 14–22. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- QikProp, Version 3.5; Schrodinger, LLC: New York, NY, USA, 2015.
- Ghose, A.K.; Herbertz, T.; Hudkins, R.L.; Dorsey, B.D.; Mallamo, J.P. Knowledge-Based, Central Nervous System (CNS) Lead Selection and Lead Optimization for CNS Drug Discovery. ACS Chem. Neurosci. 2012, 3, 50–68. [Google Scholar] [CrossRef]
- Gerald, M.; Petita, J.; Meuricea, N.; Kaisera, C. Softening the Rule of Five—where to draw the line? Bioorg. Med. Chem. 2012, 20, 5343–5351. [Google Scholar]
- Available online: http://www.bccancer.bc.ca/health-professionals/clinical-resources/cancer-drug-manual (accessed on 6 April 2019).
- Ahmed, M.; Hafsa, A. Designing second generation anti-alzheimer compounds as inhibitors of human acetylcholinesterase: Computational screening of synthetic molecules and dietary phytochemicals. PLoS ONE 2015, 10, e0136509. [Google Scholar] [CrossRef]
- Adcock, S.A.; McCammon, J.A. Molecular dynamics: Survey of methods for simulating the activity of protein. Chem. Rev. 2006, 106, 1589–1615. [Google Scholar] [CrossRef]
- Cardone, A.; Hassan, S.A.; Albers, R.W.; Sriram, R.D.; Pant, H.C. Structural and dynamic determinants of ligand binding and regulation of cyclin-dependent kinase 5 by pathological activator p25 and inhibitory peptide CIP. J. Mole. Biol. 2010, 401, 478–492. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. Model. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Martínez, L. Automatic Identification of Mobile and Rigid Substructures in Molecular Dynamics Simulations and Fractional Structural Fluctuation Analysis. PLoS ONE 2015, 10, e0119264. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.B.; Jeong, S.K.; Lee, Y.S.; Lee, S.O.; Kang, I.J.; Lim, S.S. Inhibitory activity of caffeoylquinic acids from the aerial parts of Artemisia princes on rat lens aldose reductase and on the formation of advanced glycation end product. J. Korean Soc. Appl. Biol. Chem. 2009, 52, 655–662. [Google Scholar] [CrossRef]
- Shimozono, H.; Kobori, M.; Shinmoto, H.; Tsushida, T. Suppression of the Melanogenesis of Mouse Melanoma B 16 Cells by Sweet Potato Extract. Nippon Shokuhin Kagaku Kaishi 1996, 43, 313–317. [Google Scholar] [CrossRef]
- Yagasaki, K.; Miura, Y.; Okauchi, R.; Furuse, T. Inhibitory effects of chlorogenic acid and its related compounds on the invasion of hepatoma cells in culture. Cytochemistry 2000, 33, 229–235. [Google Scholar]
- Liu, H.; Zhang, X.; Wu, C.; Wu, H.; Guo, P.; Xu, X. Anti-hyperlipidemic caffeoylquinic acids from the fruits of Pandanus tectorius Soland. J. Appl. Pharm. Sci. 2013, 3, 016–019. [Google Scholar]
- Kim, K.H.; Kim, Y.H.; Lee, K.R. Isolation of quinic acid derivatives and flavonoids from the aerial parts of Lactuca indica L. and their hepatoprotective activity in vitro. Bioorg. Med. Chem. Lett. 2007, 17, 6739–6743. [Google Scholar] [CrossRef] [PubMed]
- Hyun, Y.J.; Piaoa, M.J.; Kanga, K.A.; Ryua, Y.S.; Zhena, A.O.; Choa, S.J.; Kanga, H.K.; Koha, Y.S.; Ahn, M.J.; Kim, T.H.; et al. 3,4-Dicaffeoylquinic acid protects human keratinocytes against environmental oxidative damage. J. Funct. Foods 2019, 52, 430–441. [Google Scholar] [CrossRef]
- Hadi, A.H.A.; Fadaeinasab, M.; Kia, Y.; Basiri, A.; Murugaiyah, V. Cholinesterase enzymes inhibitors from the leaves of Rauvolfia Reflexa and their Molecular Docking Study. Molecules 2013, 18, 3779–3788. [Google Scholar]
- Wei, H.; Tye, L.; Bresnick, E.; Birt, D.F. Inhibitory effect of apigenin, a plant flavonoid, on epidermal ornithine decarboxylase and skin tumor promotion in mice. Cancer Res. 1990, 50, 499–502. [Google Scholar]
- Verbeek, R.; Plomp, A.C.; Van tol, E.A.F.; Van Noort, J.M. The flavones luteolin and apigenin inhibit in vitro antigen-specific proliferation and interferon-gamma production by murine and human autoimmune T cells. Biochem. Pharmacol. 2004, 68, 621–629. [Google Scholar] [CrossRef]
- Johnson, J.L.; Rupasinghe, S.G.; Stefani, F.; Schuler, M.A.; De Mejia, E.G. Citrus flavonoids luteolin, apigenin, and quercetin inhibit glycogen synthase kinase-3β enzymatic activity by lowering the interaction energy within the binding cavity. J. Med. Food. 2011, 14, 325–333. [Google Scholar] [CrossRef]
- Shoubaky, G.A.E.; Abdel-Daim, M.M.; Mansour, M.H.; Salem, E.A. Isolation and Identification of a Flavone Apigenin from Marine Red Alga Acanthophora spicifera with Antinociceptive and Anti-Inflammatory Activities. J. Exp. Neurosci. 2016, 10, 21–29. [Google Scholar] [CrossRef]
- Yan, X.; Qi, M.; Li, P.; Zhan, Y.; Shao, H. Apigenin in cancer therapy: Anti-cancer effects and mechanisms of action. Cell Biosci. 2017, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, A.; Tel-Cayan, G.; Öztürk, M.; Nadeem, S.; Duru, M.E.; Anis, I.; Ng, S.W.; Shah, M.R. Biologically active flavonoids from Dodonaea viscosa and their structure–activity relationships. Ind. Crops Prod. 2015, 78, 66–72. [Google Scholar] [CrossRef]
- Chandrasekaran, C.V.; Thiyagarajan, P.; Deepak, H.B.; Agarwal, A. In vitro modulation of LPS/calcimycin induced inflammatory and allergic mediators by pure compounds of Andrographis paniculata (King of bitters) extract. Int. Immunopharmacol. 2011, 11, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Larik, F.A.; Shah, M.S.; Saeed, A.; Shah, H.S.; Channar, P.A.; Bolte, M.; Iqbal, J. New cholinesterase inhibitors for Alzheimer’s disease: Structure activity relationship, kinetics and molecular docking studies of 1-butanoyl-3-arylthiourea derivatives. Int. J. Biol. Macromol. 2018, 116, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Kostelnik, A.; Pohanka, M. Inhibition of Acetylcholinesterase and Butyrylcholinesterase by a Plant Secondary Metabolite Boldine. Bio. Med. Res. Int. 2018, 2018, 9634349. [Google Scholar] [CrossRef] [PubMed]
- Dajas, F. Life or death: Neuroprotective and anticancer effects of quercetin. J. Ethnopharmacol. 2012, 143, 383–396. [Google Scholar] [CrossRef]
- Bahar, E.; Kim, J.Y.; Yoon, H. Quercetin attenuates manganese-induced neuroinflammation by alleviating oxidative stress through regulation of apoptosis, iNOS/NF-κB and HO-1/Nrf2 pathways. Int. J. Mol. Sci. 2017, 18, 1989. [Google Scholar] [CrossRef]
- Mukherjee, P.K.; Kumar, V.; Houghton, P.J. Acetylcholinesterase inhibitors from plants. Phytother. Res. 2007, 21, 1142–1145. [Google Scholar] [CrossRef]
- Greig, N.H.; Utsuki, T.; Ingram, D.K.; Wang, Y.; Pepeu, G.; Scali, C.; Yu, Q.S.; Mamczarz, J.; Holloway, H.W.; Giordano, T.; et al. Selective butyrylcholinesterase inhibition elevates brain acetylcholine, augments learning and lowers Alzheimer β-amyloid peptide in rodent. Proc. Natl. Acad. Sci. USA 2005, 102, 17213–17218. [Google Scholar] [CrossRef]
- Furukawa-Hibi, Y.; Alkam, T.; Nitta, A.; Matsuyama, A.; Mizoguchi, H.; Suzuki, K.; Moussaoui, S.; Yu, Q.S.; Greig, N.H.; Nagai, T.; et al. Butyrylcholinesterase inhibitors ameliorate cognitive dysfunction induced by amyloid-β peptide in mice. Behav. Brain Res. 2011, 225, 222–229. [Google Scholar] [CrossRef]
- Peters, F.; Salihoglu, H.; Rodrigues, E.; Herzog, E.; Blume, T.; Filser, S.; Dorostkar, M.; Shimshek, D.R.; Brose, N.; Neumann, U.; et al. BACE1 inhibition more effectively suppresses initiation than progression of β-amyloid pathology. Acta Neuropathol. 2018, 135, 695–710. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Choi, S.M.; Kim, B.C.; Cho, Y.H.; Choi, K.H.; Chang, J.; Park, M.S.; Cho, K.H.; Kim, J.K. Effects of Flavonoid Compounds on β-amyloid-peptide-induced Neuronal Death in Cultured Mouse Cortical Neurons. Chonnam. Med. J. 2014, 50, 45–51. [Google Scholar]
- Butler, M.S. The role of natural product chemistry in drug discovery. J. Nat. Prod. 2004, 67, 2141–2153. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.N.; Chi, C.W.; Lin, Y.L.; Chen, C.F.; Shiao, Y.J. The neuroprotective effects of phytoestrogens on amyloid beta protein-induced toxicity are mediated by abrogating the activation of caspase cascade in rat cortical neurons. J. Biol. Chem. 2001, 276, 5287–5295. [Google Scholar] [CrossRef]
- Dajas, F.; Rivera, F.; Blasina, F.; Arredondo, F.; Echeverry, C.; Lafon, L.; Morquio, A.; Heizen, H. Cell culture protection and in vivo neuroprotective capacity of flavonoids. Neurotox. Res. 2003, 5, 425–432. [Google Scholar] [CrossRef]
- Mandel, S.; Youdim, M.B. Catechin polyphenols: Neurodegeneration and neuroprotection in neurodegenerative diseases. Free Radic. Biol. Med. 2004, 37, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Simonyi, A.; Wang, Q.; Miller, R.L.; Yusof, M.; Shelat, P.B.; Sun, A.Y.; Sun, G.Y. Polyphenols in cerebral ischemia. Mol. Neurobiol. 2005, 31, 135–147. [Google Scholar] [CrossRef]
- Borowiec, K.; Szwajgier, D.; Pustelniak, K. The Neuroprotective Effects of Phenolic Acids: Molecular Mechanism of Action. Nutrients 2017, 9, 477. [Google Scholar] [Green Version]
- Wang, Y.; Xiao, J.; Suzek, T.; Zhang, J.; Wang, J.; Bryant, S. PubChem: A public information system for analysing bioactivities of small molecules. Nucleic Acids Res. 2009, 2009, 1–11. [Google Scholar] [CrossRef]
- Degtyarenko, K.; Matos, P.; Ennis, M.; Hasting, J.; Zbinden, M.; McNaught, A.; Alcantara, R.; Darsow, M.; Guedj, M.; Ashburner, M. CHEBI: A database and ontology for chemical entities of biological interest. Nucleic Acids. Res. 2008, 36, D344–D350. [Google Scholar] [CrossRef]
- Protein Preparation Wizard 2015-1, Epik Version 2.4; Impact Version 5.9; Prime Version 3.2; Schrödinger, LLC: New York, NY, USA, 2015.
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W.J. Comput. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, G.; Friesner, R.; Tirado-Rives, J.; Jorgensen, W. Evaluation and reparametrization of the OPLS-aA forcefield for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- LigPrep, Version 3.3; Schrodinger, LLC: New York, NY, USA, 2015.
- Glide, Version 6.6; Schrodinger, LLC: New York, NY, USA, 2015.
- Bekker, H.; Berendsen, H.J.C.; Dijkstra, E.J.; Achterop, S.; Vondrumen, R.; Van Der Spoel, D.; Sijbers, A.; Keegstra, H.; Renardus, M.K.R. Physics Computing 92; DeGroot, R.A., Nadrchal, J., Eds.; World Scientific Publishing: Singapore, 1993; pp. 252–256. [Google Scholar]
- Van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comp. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Pall, S.; Smith, J.C.; Hess, B.; Lindah, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Schuttelkopf, W.; Van Aalten, D.M.F. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef]
- Citron, M. Beta-secretase as a target for the treatment of Alzheimer’s disease. J. Neurosci. Res. 2002, 70, 373–379. [Google Scholar] [CrossRef]
- Hong, L.; Turner, R.T.; Koelsch, G.; Ghosh, A.K.; Tang, J. Memapsin 2 (beta-secretase) as a therapeutic target. Biochem. Soc. Trans. 2002, 30, 530–534. [Google Scholar] [CrossRef]
- Roberds, S.L.; Anderson, J.; Basi, G.; Bienkowski, M.J.; Branstetter, D.G.; Chen, K.S.; Freedman, S.B.; Frigon, N.L.; Games, D.; Hu, K.; et al. BACE knockout mice are healthy despite lacking the primary beta-secretase activity in brain: Implications for Alzheimer’s disease therapeutics. Hum. Mol. Genet. 2001, 10, 1317–1324. [Google Scholar] [CrossRef]
- Jung, H.A.; Min, B.S.; Yokozawa, T.; Lee, J.H.; Kim, Y.S.; Choi, J.S. Anti-Alzheimer and antioxidant activities of Coptidis Rhizoma alkaloids. Biol. Pharm. Bull. 2009, 32, 1433–1438. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V.J.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | AChE Docking Score (kcal/mol) | AChE XP Score (kcal/mol) | BChE Docking Score (kcal/mol) | BChE XP Score (kcal/mol) | BACE−1 Docking Score (kcal/mol) | BACE−1 XP Score (kcal/mol) |
|---|---|---|---|---|---|---|
| 5281780 | −9.770 | −9.770 | −11.946 | −11.946 | −7.648 (pose1) −7.108 (pose2) | −7.648 (pose1) −7.108 (pose2) |
| 5280443 | −7.071 | −7.088 | −9.038 | −9.021 | −7.422 | −7.422 |
| 42608095 | −6.458 | −6.458 | −6.915 | −6.915 | −4.161 | −4.161 |
| 188316 | −6.837 | −6.837 | −7.370 | −7.364 | −4.872 | −4.879 |
| 8468 | −5.303 | −5.303 | −5.114 | −5.114 | −2.966 | −2.966 |
| 5280460 | −4.905 | −4.905 | −5.835 | −5.835 | −4.424 | −4.431 |
| 445858 | −5.909 | −5.909 | −5.646 | −5.646 | −4.532 | −4.532 |
| 68105 | −7.031 | −7.031 | −7.082 | −7.082 | −3.982 | −3.982 |
| Compound | Molecular Weight | Donar HB | AcceptHB | QPlogP o/w | QPlogBB | Human Oral Absorption | Percent of Human Oral Absorption | Rule of Five | Rule of Three |
|---|---|---|---|---|---|---|---|---|---|
| 5281780 | 516.457 | 7 | 11.45 | 0.926 | −5.376 | 1 | 0 | 3 | 1 |
| 5280443 | 270.441 | 2 | 3.75 | 1.624 | −1.411 | 3 | 74 | 0 | 0 |
| 42608095 | 300.310 | 0 | 4 | 3.462 | −0.556 | 3 | 100 | 0 | 0 |
| 188316 | 298.295 | 0 | 3.75 | 3.165 | −0.430 | 3 | 100 | 0 | 0 |
| 8468 | 168.149 | 2 | 3.5 | 1.058 | −0.779 | 2 | 68 | 0 | 0 |
| 5280460 | 192.171 | 1 | 4 | 0.891 | −0.474 | 3 | 84 | 0 | 0 |
| 445858 | 194.187 | 0 | 3.5 | 1.398 | −1.062 | 3 | 69 | 0 | 0 |
| 68105 | 416.729 | 1 | 1.7 | 7.498 | −0.334 | 1 | 100 | 1 | 1 |
| Sr.No. | AChE_RMSD | BChE_RMSD | BACE-1_RMSD | |||
|---|---|---|---|---|---|---|
| Stable From (ns) | Average | Stable From (ns) | Average | Stable From (ns) | Average | |
| 5281780 (pose1) | 12 | 0.191 | 13.5 | 0.176 | 8 | 0.175 |
| 5281780 (pose2) | - | - | - | - | 12 | 0.190 |
| 5280443 | 10 | 0.180 | 5 | 0.170 | 11 | 0.220 |
| 188316 | 12 | 0.160 | 12 | 0.185 | 09 | 0.298 |
| Sr. No. | Compound | MeanIC50-AChE (μM) | Mean IC50-BChE (μM) | Mean IC50-BACE-1(μM) |
|---|---|---|---|---|
| 1 | 5281780 | 2.14 ± 0.04 | 1.44 ± 0.02 | 3.31 ± 0.12 |
| 2 | 5280443 | 3.42 ± 0.02 | 1.97 ± 0.01 | 3.79 ± 0.26 |
| 3 | 188316 | 2.46 ± 0.03 | 1.46 ± 0.02 | 2.91 ± 0.04 |
| 4 | Eserine | 3.39 ± 0.22 | 2.88 ± 0.01 | - |
| 5 | Quercetin | - | - | 2.91 ± 0.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panche, A.N.; Chandra, S.; Diwan, A. Multi-Target β-Protease Inhibitors from Andrographis paniculata: In Silico and In Vitro Studies. Plants 2019, 8, 231. https://doi.org/10.3390/plants8070231
Panche AN, Chandra S, Diwan A. Multi-Target β-Protease Inhibitors from Andrographis paniculata: In Silico and In Vitro Studies. Plants. 2019; 8(7):231. https://doi.org/10.3390/plants8070231
Chicago/Turabian StylePanche, Archana N, Sheela Chandra, and AD Diwan. 2019. "Multi-Target β-Protease Inhibitors from Andrographis paniculata: In Silico and In Vitro Studies" Plants 8, no. 7: 231. https://doi.org/10.3390/plants8070231