Understanding Host–Pathogen Interactions in Brassica napus in the Omics Era




Abstract
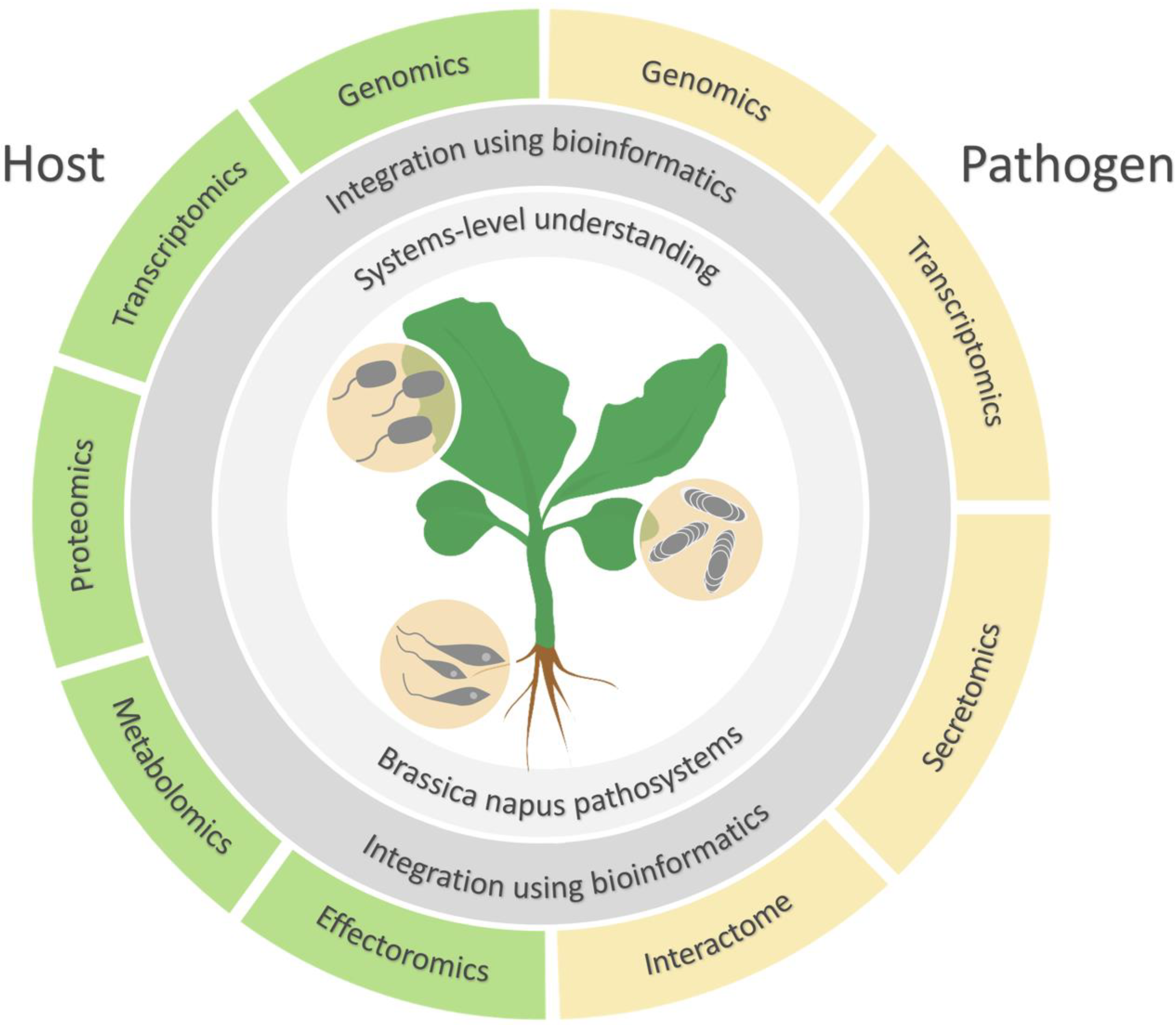
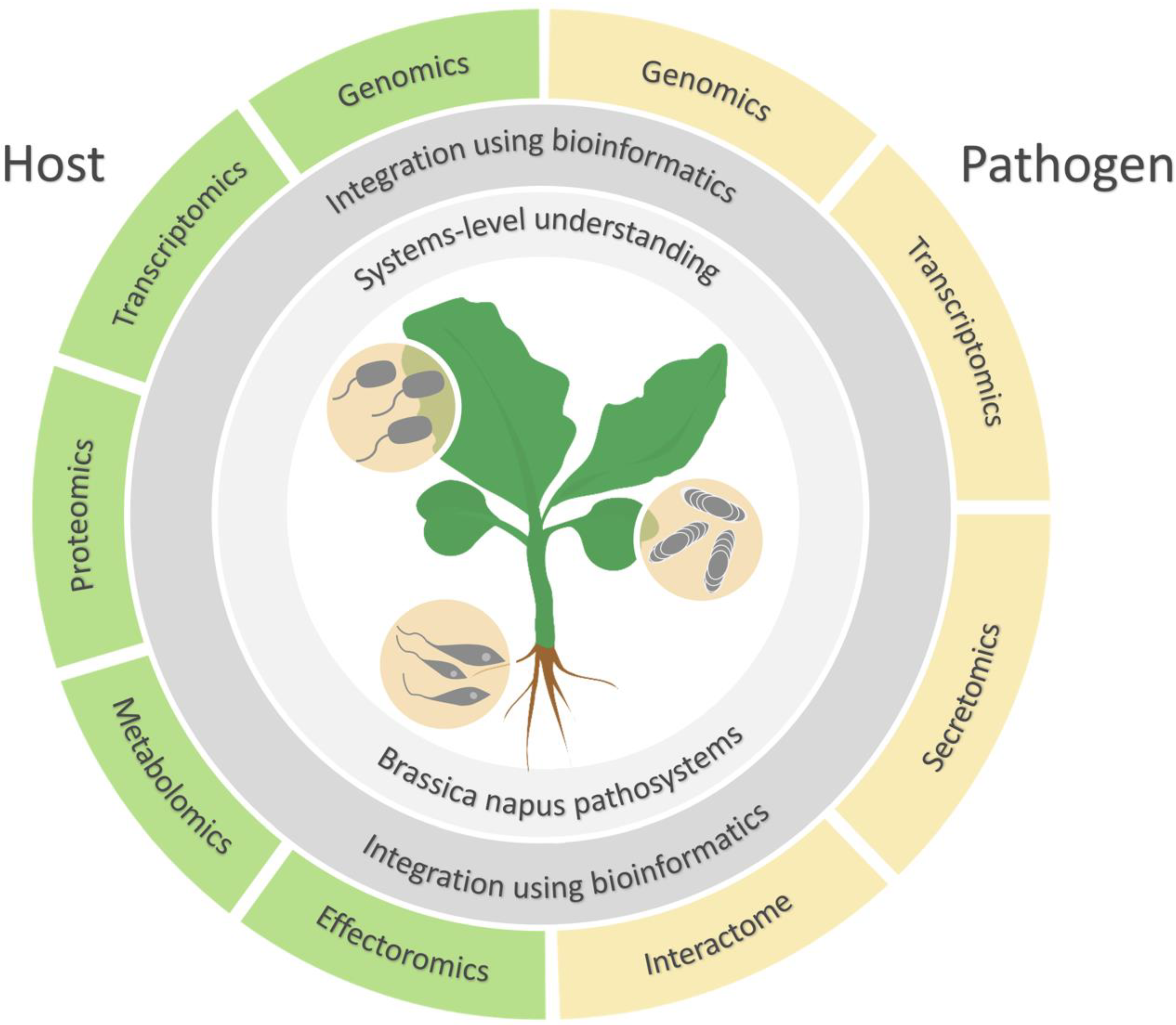
:1. Introduction
2. Application of Omics Technologies in Brassica Host Plants
2.1. High-Quality Genome Assemblies
2.2. Pangenomics

{kind=link}
{kind=link}
| Reference Genome | Approach | Major Findings Relevant to R Gene Study | Reference |
|---|---|---|---|
| Single genome | |||
| B. napus winter cultivar “Express 617” | PacBio, ONT, Illumina HiSeq, Optical mapping | Resolved break-point sequence at homoeologous exchange regions | Lee, et al. [33] |
| B. oleracea cultivar “C-8” | PacBio, Illumina HiSeq, transcriptomics | Cauliflower is the most recent var. to evolve within Brassica genus. It contains more repetitive sequences compared to other B. oleracea species | Sun, et al. [40] |
| B. nigra accession CGN7651 | ONT, Hi-C | Hotspot of ALE-type retroelement in the centromeric regions showed that these retroelements play an important role in the divergence of B. nigra centromere | Perumal, et al. [56] |
| B. rapa cultivar “Chiifu-401-42” | PacBio, Optical mapping, Hi-C | V3.0, improved repeat reads, defined locations of centromeres and annotated more genes in these difficult regions. Annotated higher number of TEs | Zhang, et al. [39] |
| Pangenome | |||
| Eight B. napus accessions of three ecotypes | Alignment of de novo assembled genomes against “ZS11” | PAV genes were highly represented by defence response gene | Song, et al. [51] |
| 33 non-synthetic and 20 synthetic B. napus accessions | Iterative mapping and assembly using improved “Darmor-bzh”(v8.1) from Bayer, Hurgobin, Golicz, Chan, Yuan, Lee, Renton, Meng, Li, Long, Zou, Bancroft, Chalhoub, King, Batley and Edwards [31] as reference | Homoeologous exchange-related PAV genes highly represented by defence, stress and auxin pathways | Hurgobin, et al. [44] |
| Nine B. oleracea subspecies and wild type comprising cabbage, kale, Brussels sprouts, kohlrabi, cauliflower, broccoli and B. macrocarpa | Iterative mapping and assembly using Chinese kale rapid cycling line (TO1000) as reference | 18.7% of genes showed PAV with annotation of disease resistance genes | Golicz, et al. [52] |
| Two B. rapa subspecies: turnip and rapid cycling | Alignment of de novo assembled genomes against “Chiifu” reference | Peroxidase genes that are involved in phenylpropanoid biosynthesis response pathway during biotic stress are unique in turnip, with evidence of copy number variation | Lin, et al. [57] |
2.3. Identification of Candidate QTLs/Genes Using NGS-Based SNP Methods
2.4. Identification of Candidate R Gene Using In Silico Methods
2.5. NGS-Based Bulked Segregant Analysis (BSA)
2.6. Resistance Gene Enrichment and Sequencing (RenSeq)
2.7. Effectoromics
2.8. Transcriptomics
2.9. Proteomics
3. Application of Omics Technologies in Brassica Pathogens
3.1. High-Quality Genome Assemblies
3.2. Transcriptomics of Virulence-Related Genes
3.3. Secretomics
3.4. Interactome
4. Application of Metabolomics and Systems Biology in the Brassica–Pathogen System
5. Future Perspectives
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kandel, S.L.; Joubert, P.M.; Doty, S.L. Bacterial endophyte colonization and distribution within plants. Microorganisms 2017, 5, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spanu, P.D.; Panstruga, R. Editorial: Biotrophic plant-microbe interactions. Front. Plant Sci. 2017, 8, 192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramovitch, R.B.; Anderson, J.C.; Martin, G.B. Bacterial elicitation and evasion of plant innate immunity. Nat. Rev. Mol. Cell Biol. 2006, 7, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Dodds, P.N.; Rathjen, J.P. Plant immunity: Towards an integrated view of plant–pathogen interactions. Nat. Rev. Genet. 2010, 11, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Lo Presti, L.; Lanver, D.; Schweizer, G.; Tanaka, S.; Liang, L.; Tollot, M.; Zuccaro, A.; Reissmann, S.; Kahmann, R. Fungal effectors and plant susceptibility. Annu. Rev. Plant Biol. 2015, 66, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.D.G.; Dangl, J.L. The plant immune system. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stotz, H.U.; Mitrousia, G.K.; de Wit, P.J.G.M.; Fitt, B.D.L. Effector-triggered defence against apoplastic fungal pathogens. Trends Plant Sci. 2014, 19, 491–500. [Google Scholar] [CrossRef] [Green Version]
- Cook, D.E.; Mesarich, C.H.; Thomma, B.P.H.J. Understanding plant immunity as a surveillance system to detect invasion. Annu. Rev. Phytopathol. 2015, 53, 541–563. [Google Scholar] [CrossRef]
- Van der Burgh, A.M.; Joosten, M.H.A.J. Plant immunity: Thinking outside and inside the box. Trends Plant Sci. 2019, 24, 587–601. [Google Scholar] [CrossRef]
- Kanyuka, K.; Rudd, J.J. Cell surface immune receptors: The guardians of the plant’s extracellular spaces. Curr. Opin. Plant Biol. 2019, 50, 1–8. [Google Scholar] [CrossRef]
- Hohmann, N.; Wolf, E.M.; Lysak, M.A.; Koch, M.A. A time-calibrated road map of Brassicaceae species radiation and evolutionary history. Plant Cell 2015, 27, 2770–2784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiefer, M.; Schmickl, R.; German, D.A.; Mandáková, T.; Lysak, M.A.; Al-Shehbaz, I.A.; Franzke, A.; Mummenhoff, K.; Stamatakis, A.; Koch, M.A. BrassiBase: Introduction to a novel knowledge database on Brassicaceae evolution. Plant Cell Physiol. 2014, 55, e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiessl, S.V.; Mason, A. Ancient and recent polyploid evolution in Brassica. In Brassica Improvement—Molecular, Genetics and Genomic Perspectives; Wani, S.H., Thakur, A.K., Jeshima Khan, Y., Eds.; Springer: Cham, Switzerland, 2020; pp. 49–66. [Google Scholar] [CrossRef]
- Friedt, W.; Tu, J.; Fu, T. Academic and economic importance of Brassica napus rapeseed. In The Brassica napus Genome; Liu, S., Snowdon, R., Chalhoub, B., Eds.; Springer International Publishing: Cham, Switzerland, 2018. [Google Scholar] [CrossRef]
- Cheng, F.; Wu, J.; Wang, X. Genome triplication drove the diversification of Brassica plants. Hortic. Res. 2014, 1, 14024. [Google Scholar] [CrossRef] [Green Version]
- Cheng, F.; Sun, R.; Hou, X.; Zheng, H.; Zhang, F.; Zhang, Y.; Liu, B.; Liang, J.; Zhuang, M.; Liu, Y.; et al. Subgenome parallel selection is associated with morphotype diversification and convergent crop domestication in Brassica rapa and Brassica oleracea. Nat. Genet. 2016, 48, 1218–1224. [Google Scholar] [CrossRef] [PubMed]
- Werner, C.; Snowdon, R. Genome-facilitated breeding of oilseed rape. In The Brassica napus Genome; Liu, S., Snowdon, R., Chalhoub, B., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 245–269. [Google Scholar] [CrossRef]
- FAO Stat. Food and Agriculture Organization of the United Nations—Statistics Division, Crop. Available online: http://www.fao.org/faostat/en/#data/QC (accessed on 31 January 2020).
- Varshney, R.K.; Roorkiwal, M.; Sorrells, M.E. Genomic selection for crop improvement: An introduction. In Genomic Selection for Crop Improvement: New Molecular Breeding Strategies for Crop Improvement; Varshney, R.K., Roorkiwal, M., Sorrells, M.E., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 1–6. [Google Scholar] [CrossRef] [Green Version]
- Anderson, R.; Bayer, P.E.; Edwards, D. Climate change and the need for agricultural adaptation. Curr. Opin. Plant Biol. 2020, 56, 197–202. [Google Scholar] [CrossRef]
- Kabbage, M.; Yarden, O.; Dickman, M.B. Pathogenic attributes of Sclerotinia sclerotiorum: Switching from a biotrophic to necrotrophic lifestyle. Plant Sci. 2015, 233, 53–60. [Google Scholar] [CrossRef]
- Neik, T.X.; Barbetti, M.J.; Batley, J. Current status and challenges in identifying disease resistance genes in Brassica napus. Front. Plant Sci. 2017, 8, 1788. [Google Scholar] [CrossRef]
- Lv, H.; Fang, Z.; Yang, L.; Zhang, Y.; Wang, Y. An update on the arsenal: Mining resistance genes for disease management of Brassica crops in the genomic era. Hortic. Res. 2020, 7, 34. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Fernando, W.G.D. Insights into fighting against blackleg disease of Brassica napus in Canada. Crop. Pasture Sci. 2017, 69, 40–47. [Google Scholar] [CrossRef]
- Hwang, S.-F.; Strelkov, S.E.; Peng, G.; Ahmed, H.; Zhou, Q.; Turnbull, G. Blackleg (Leptosphaeria maculans) severity and yield loss in canola in Alberta, Canada. Plants 2016, 5, 31. [Google Scholar] [CrossRef]
- Strehlow, B.; de Mol, F.; Struck, C. Risk potential of Clubroot disease on winter oilseed rape. Plant Dis. 2014, 99, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Cowling, W.; Banga, S.S.; You, M.P.; Tyagi, V.; Bharti, B.; Barbetti, M.J. Patterns of inheritance for cotyledon resistance against Sclerotinia sclerotiorum in Brassica napus. Euphytica 2020, 216, 79. [Google Scholar] [CrossRef]
- Al-Lami, H.F.D.; You, M.P.; Barbetti, M.J. Role of foliage component and host age on severity of Alternaria leaf spot (caused by Alternaria japonica and A. brassicae) in canola (Brassica napus) and mustard (B. juncea) and yield loss in canola. Crop. Pasture Sci. 2019, 70, 969–980. [Google Scholar] [CrossRef]
- Mohammed, A.E.; You, M.P.; Banga, S.S.; Barbetti, M.J. Resistances to downy mildew (Hyaloperonospora brassicae) in diverse Brassicaceae offer new disease management opportunities for oilseed and vegetable crucifer industries. Eur. J. Plant Pathol. 2019, 153, 915–929. [Google Scholar] [CrossRef]
- Bailey-Serres, J.; Parker, J.E.; Ainsworth, E.A.; Oldroyd, G.E.D.; Schroeder, J.I. Genetic strategies for improving crop yields. Nature 2019, 575, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Bayer, P.E.; Hurgobin, B.; Golicz, A.A.; Chan, C.K.K.; Yuan, Y.; Lee, H.; Renton, M.; Meng, J.; Li, R.; Long, Y.; et al. Assembly and comparison of two closely related Brassica napus genomes. Plant Biotechnol. J. 2017, 15, 1602–1610. [Google Scholar] [CrossRef] [Green Version]
- Sun, F.; Fan, G.; Hu, Q.; Zhou, Y.; Guan, M.; Tong, C.; Li, J.; Du, D.; Qi, C.; Jiang, L.; et al. The high-quality genome of Brassica napus cultivar ‘ZS11′ reveals the introgression history in semi-winter morphotype. Plant J. 2017, 92, 452–468. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Chawla, H.S.; Obermeier, C.; Dreyer, F.; Abbadi, A.; Snowdon, R. Chromosome-scale assembly of winter oilseed rape Brassica napus. Front. Plant Sci. 2020, 11, 496. [Google Scholar] [CrossRef]
- Roberts, R.J.; Carneiro, M.O.; Schatz, M.C. The advantages of SMRT sequencing. Genome Biol. 2013, 14, 405. [Google Scholar] [CrossRef]
- Jain, M.; Olsen, H.E.; Paten, B.; Akeson, M. The Oxford Nanopore MinION: Delivery of nanopore sequencing to the genomics community. Genome Biol. 2016, 17, 239. [Google Scholar] [CrossRef] [Green Version]
- Howe, K.; Wood, J.M.D. Using optical mapping data for the improvement of vertebrate genome assemblies. GigaScience 2015, 4, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Lin, F.; An, D.; Wang, W.; Huang, R. Genome sequencing and assembly by long reads in plants. Genes 2017, 9, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Berkum, N.L.; Lieberman-Aiden, E.; Williams, L.; Imakaev, M.; Gnirke, A.; Mirny, L.A.; Dekker, J.; Lander, E.S. Hi-C: A method to study the three-dimensional architecture of genomes. J. Vis. Exp. 2010, 6, 1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Cai, X.; Wu, J.; Liu, M.; Grob, S.; Cheng, F.; Liang, J.; Cai, C.; Liu, Z.; Liu, B.; et al. Improved Brassica rapa reference genome by single-molecule sequencing and chromosome conformation capture technologies. Hortic. Res. 2018, 5, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.; Wang, C.; Zhang, X.; Zhang, W.; Jiang, H.; Yao, X.; Liu, L.; Wen, Z.; Niu, G.; Shan, X. Draft genome sequence of cauliflower (Brassica oleracea L. var. botrytis) provides new insights into the C genome in Brassica species. Hortic. Res. 2019, 6, 82. [Google Scholar] [CrossRef] [Green Version]
- Rousseau-Gueutin, M.; Belser, C.; Silva, C.D.; Richard, G.; Istace, B.; Cruaud, C.; Falentin, C.; Boideau, F.; Boutte, J.; Delourme, R.; et al. Long-reads assembly of the Brassica napus; reference genome, Darmor-bzh. bioRxiv 2020. [Google Scholar] [CrossRef]
- Schiessl, S.-V.; Katche, E.; Ihien, E.; Chawla, H.S.; Mason, A.S. The role of genomic structural variation in the genetic improvement of polyploid crops. Crop J. 2019, 7, 127–140. [Google Scholar] [CrossRef]
- Bayer, P.E.; Golicz, A.A.; Tirnaz, S.; Chan, C.-K.K.; Edwards, D.; Batley, J. Variation in abundance of predicted resistance genes in the Brassica oleracea pangenome. Plant Biotechnol. J. 2019, 17, 789–800. [Google Scholar] [CrossRef] [Green Version]
- Hurgobin, B.; Golicz, A.A.; Bayer, P.E.; Chan, C.-K.K.; Tirnaz, S.; Dolatabadian, A.; Schiessl, S.V.; Samans, B.; Montenegro, J.D.; Parkin, I.A.P.; et al. Homoeologous exchange is a major cause of gene presence/absence variation in the amphidiploid Brassica napus. Plant Biotechnol. J. 2018, 16, 1265–1274. [Google Scholar] [CrossRef] [Green Version]
- Dolatabadian, A.; Bayer, P.E.; Tirnaz, S.; Hurgobin, B.; Edwards, D.; Batley, J. Characterization of disease resistance genes in the Brassica napus pangenome reveals significant structural variation. Plant Biotechnol. J. 2019, 18, 969–982. [Google Scholar] [CrossRef] [Green Version]
- Belser, C.; Istace, B.; Denis, E.; Dubarry, M.; Baurens, F.-C.; Falentin, C.; Genete, M.; Berrabah, W.; Chèvre, A.-M.; Delourme, R.; et al. Chromosome-scale assemblies of plant genomes using nanopore long reads and optical maps. Nat. Plants 2018, 4, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Parkin, I.A.P.; Koh, C.; Tang, H.; Robinson, S.J.; Kagale, S.; Clarke, W.E.; Town, C.D.; Nixon, J.; Krishnakumar, V.; Bidwell, S.L.; et al. Transcriptome and methylome profiling reveals relics of genome dominance in the mesopolyploid Brassica oleracea. Genome Biol. 2014, 15, R77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tirnaz, S.; Edwards, D.; Batley, J. The importance of plant pan-genomes in breeding. In Quantitative Genetics, Genomics and Plant Breeding, 2nd ed.; Kang, M.S., Ed.; CABI: Boston, MA, USA, 2020; pp. 27–32. [Google Scholar] [CrossRef]
- Tao, Y.; Zhao, X.; Mace, E.; Henry, R.; Jordan, D. Exploring and exploiting pan-genomics for crop improvement. Mol. Plant 2019, 12, 156–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayer, P.E.; Golicz, A.A.; Scheben, A.; Batley, J.; Edwards, D. Plant pan-genomes are the new reference. Nat. Plants 2020, 6, 914–920. [Google Scholar] [CrossRef]
- Song, J.-M.; Guan, Z.; Hu, J.; Guo, C.; Yang, Z.; Wang, S.; Liu, D.; Wang, B.; Lu, S.; Zhou, R.; et al. Eight high-quality genomes reveal pan-genome architecture and ecotype differentiation of Brassica napus. Nat. Plants 2020, 6, 34–45. [Google Scholar] [CrossRef]
- Golicz, A.A.; Bayer, P.E.; Barker, G.C.; Edger, P.P.; Kim, H.; Martinez, P.A.; Chan, C.K.K.; Severn-Ellis, A.; McCombie, W.R.; Parkin, I.A.P.; et al. The pangenome of an agronomically important crop plant Brassica oleracea. Nat. Commun. 2016, 7, 13390. [Google Scholar] [CrossRef]
- Amsbury, S. Sensing attack: The role of wall-associated kinases in plant pathogen responses. Plant Physiol. 2020, 183, 1420. [Google Scholar] [CrossRef]
- Tirnaz, S.; Bayer, P.; Inturrisi, F.; Zhang, F.; Yang, H.; Dolatabadian, A.; Neik, T.X.; Severn-Ellis, A.; Patel, D.; Ibrahim, M.I.; et al. Resistance gene analogs in the Brassicaceae: Identification, characterization, distribution, and evolution. Plant Physiol. 2020, 184, 909–922. [Google Scholar] [CrossRef]
- Khan, A.W.; Garg, V.; Roorkiwal, M.; Golicz, A.A.; Edwards, D.; Varshney, R.K. Super-pangenome by integrating the wild side of a species for accelerated crop improvement. Trends Plant Sci. 2020, 25, 148–158. [Google Scholar] [CrossRef] [Green Version]
- Perumal, S.; Koh, C.S.; Jin, L.; Buchwaldt, M.; Higgins, E.E.; Zheng, C.; Sankoff, D.; Robinson, S.J.; Kagale, S.; Navabi, Z.-K.; et al. A high-contiguity Brassica nigra genome localizes active centromeres and defines the ancestral Brassica genome. Nat. Plants 2020, 6, 929–941. [Google Scholar] [CrossRef]
- Lin, K.; Zhang, N.; Severing, E.I.; Nijveen, H.; Cheng, F.; Visser, R.G.F.; Wang, X.; de Ridder, D.; Bonnema, G. Beyond genomic variation—Comparison and functional annotation of three Brassica rapa genomes: A turnip, a rapid cycling and a Chinese cabbage. BMC Genom. 2014, 15, 250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delourme, R.; Laperche, A.; Bouchet, A.-S.; Jubault, M.; Paillard, S.; Manzanares-Dauleux, M.-J.; Nesi, N. Genes and quantitative trait loci mapping for major agronomic traits in Brassica napus L. In The Brassica napus Genome; Liu, S., Snowdon, R., Chalhoub, B., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 41–85. [Google Scholar] [CrossRef]
- Nadeem, M.A.; Nawaz, M.A.; Shahid, M.Q.; Doğan, Y.; Comertpay, G.; Yıldız, M.; Hatipoğlu, R.; Ahmad, F.; Alsaleh, A.; Labhane, N.; et al. DNA molecular markers in plant breeding: Current status and recent advancements in genomic selection and genome editing. Biotechnol. Biotechnol. Equip. 2018, 32, 261–285. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, K.L.; Grondin, A.; Courtois, B.; Gantet, P. Next-generation sequencing accelerates crop gene discovery. Trends Plant Sci. 2019, 24, 263–274. [Google Scholar] [CrossRef]
- Singh, B.D.; Singh, A.K. High-throughput SNP genotyping. In Marker-Assisted Plant Breeding: Principles and Practices; Singh, B.D., Singh, A.K., Eds.; Springer India: New Delhi, India, 2015; pp. 367–400. [Google Scholar] [CrossRef]
- Mammadov, J.; Aggarwal, R.; Buyyarapu, R.; Kumpatla, S. SNP markers and their impact on plant breeding. Int. J. Plant Genom. 2012, 2012, 728398. [Google Scholar] [CrossRef] [PubMed]
- Scheben, A.; Batley, J.; Edwards, D. Genotyping-by-sequencing approaches to characterize crop genomes: Choosing the right tool for the right application. Plant Biotechnol. J. 2017, 15, 149–161. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Wang, H.; Zhong, W.; Bai, J.; Liu, P.; He, Y. QTL mapping of leafy heads by genome resequencing in the RIL population of Brassica rapa. PLoS ONE 2013, 8, e76059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple Genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [Green Version]
- Chung, Y.S.; Choi, S.C.; Jun, T.-H.; Kim, C. Genotyping-by-sequencing: A promising tool for plant genetics research and breeding. Hortic. Environ. Biotechnol. 2017, 58, 425–431. [Google Scholar] [CrossRef]
- Ott, A.; Liu, S.; Schnable, J.C.; Yeh, C.-T.E.; Wang, K.-S.; Schnable, P.S. tGBS® genotyping-by-sequencing enables reliable genotyping of heterozygous loci. Nucleic Acids Res. 2017, 45, e178. [Google Scholar] [CrossRef] [Green Version]
- Clarke, W.E.; Higgins, E.E.; Plieske, J.; Wieseke, R.; Sidebottom, C.; Khedikar, Y.; Batley, J.; Edwards, D.; Meng, J.; Li, R.; et al. A high-density SNP genotyping array for Brassica napus and its ancestral diploid species based on optimised selection of single-locus markers in the allotetraploid genome. Theor. Appl. Genet. 2016, 129, 1887–1899. [Google Scholar] [CrossRef] [Green Version]
- You, Q.; Yang, X.; Peng, Z.; Xu, L.; Wang, J. Development and applications of a high throughput genotyping tool for polyploid crops: Single Nucleotide Polymorphism (SNP) array. Front. Plant Sci. 2018, 9, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason, A.S.; Zhang, J.; Tollenaere, R.; Vasquez Teuber, P.; Dalton-Morgan, J.; Hu, L.; Yan, G.; Edwards, D.; Redden, R.; Batley, J. High-throughput genotyping for species identification and diversity assessment in germplasm collections. Mol. Ecol. Resour. 2015, 15, 1091–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheben, A.; Verpaalen, B.; Lawley, C.T.; Chan, C.-K.K.; Bayer, P.E.; Batley, J.; Edwards, D. CropSNPdb: A database of SNP array data for Brassica crops and hexaploid bread wheat. Plant J. 2019, 98, 142–152. [Google Scholar] [CrossRef]
- Raman, H.; Raman, R.; Coombes, N.; Song, J.; Diffey, S.; Kilian, A.; Lindbeck, K.; Barbulescu, D.M.; Batley, J.; Edwards, D.; et al. Genome-wide association study identifies new loci for resistance to Leptosphaeria maculans in canola. Front. Plant Sci. 2016, 7, 1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, F.; Zhang, X.; Liu, F.; Peng, G.; Yu, F.; Fernando, D. Identification of resistance loci in Chinese and Canadian canola/rapeseed varieties against Leptosphaeria maculans based on Genome-Wide Association Studies. BMC Genom. 2020, 21, 501. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhao, Q.; Liu, S.; Shahid, M.; Lan, L.; Cai, G.; Zhang, C.; Fan, C.; Wang, Y.; Zhou, Y. Genome-wide association study identifies new loci for resistance to Sclerotinia Stem Rot in Brassica napus. Front. Plant Sci. 2016, 7, 1418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atri, C.; Akhatar, J.; Gupta, M.; Gupta, N.; Goyal, A.; Rana, K.; Kaur, R.; Mittal, M.; Sharma, A.; Singh, M.P.; et al. Molecular and genetic analysis of defensive responses of Brassica juncea—B. fruticulosa introgression lines to Sclerotinia infection. Sci. Rep. 2019, 9, 17089. [Google Scholar] [CrossRef]
- Rana, K.; Atri, C.; Akhatar, J.; Kaur, R.; Goyal, A.; Singh Mohini, P.; Kumar, N.; Sharma, A.; Sandhu Prabhjodh, S.; Kaur, G.; et al. Detection of first marker trait associations for resistance against Sclerotinia sclerotiorum in Brassica juncea–Erucastrum cardaminoides introgression lines. Front. Plant Sci. 2019, 10, 1015. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Luo, Y.; Chen, B.; Xu, K.; Zhang, F.; Li, H.; Huang, Q.; Xiao, X.; Zhang, T.; Hu, J.; et al. A genome-wide association study reveals new loci for resistance to clubroot disease in Brassica napus. Front. Plant Sci. 2016, 7, 1483. [Google Scholar] [CrossRef] [Green Version]
- Peng, L.; Zhou, L.; Li, Q.; Wei, D.; Ren, X.; Song, H.; Mei, J.; Si, J.; Qian, W. Identification of quantitative trait loci for Clubroot resistance in Brassica oleracea with the use of Brassica SNP microarray. Front. Plant Sci. 2018, 9, 822. [Google Scholar] [CrossRef]
- Laila, R.; Park, J.-I.; Robin, A.H.K.; Natarajan, S.; Vijayakumar, H.; Shirasawa, K.; Isobe, S.; Kim, H.-T.; Nou, I.-S. Mapping of a novel clubroot resistance QTL using ddRAD-seq in Chinese cabbage (Brassica rapa L.). BMC Plant Biol. 2019, 19, 13. [Google Scholar] [CrossRef] [PubMed]
- Mei, J.; Shao, C.; Yang, R.; Feng, Y.; Gao, Y.; Ding, Y.; Li, J.; Qian, W. Introgression and pyramiding of genetic loci from wild Brassica oleracea into B. napus for improving Sclerotinia resistance of rapeseed. Theor. Appl. Genet. 2020, 133, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- Gabur, I.; Chawla, H.S.; Lopisso, D.T.; von Tiedemann, A.; Snowdon, R.J.; Obermeier, C. Gene presence-absence variation associates with quantitative Verticillium longisporum disease resistance in Brassica napus. Sci. Rep. 2020, 10, 4131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Liang, Z.; Yan, T.; Xu, Y.; Xuan, L.; Tang, J.; Zhou, G.; Lohwasser, U.; Hua, S.; Wang, H.; et al. Whole-genome resequencing of a worldwide collection of rapeseed accessions reveals the genetic basis of ecotype divergence. Mol. Plant 2019, 12, 30–43. [Google Scholar] [CrossRef] [Green Version]
- Lu, K.; Wei, L.; Li, X.; Wang, Y.; Wu, J.; Liu, M.; Zhang, C.; Chen, Z.; Xiao, Z.; Jian, H.; et al. Whole-genome resequencing reveals Brassica napus origin and genetic loci involved in its improvement. Nat. Commun. 2019, 10, 1154. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Izzah, N.K.; Jayakodi, M.; Perumal, S.; Joh, H.J.; Lee, H.J.; Lee, S.-C.; Park, J.Y.; Yang, K.-W.; Nou, I.-S.; et al. Genome-wide SNP identification and QTL mapping for black rot resistance in cabbage. BMC Plant Biol. 2015, 15, 32. [Google Scholar] [CrossRef] [Green Version]
- Farid, M.; Yang, R.-C.; Kebede, B.; Rahman, H. Evaluation of Brassica oleracea accessions for resistance to Plasmodiophora brassicae and identification of genomic regions associated with resistance. Genome 2019, 63, 91–101. [Google Scholar] [CrossRef]
- Wei, D.; Cui, Y.; He, Y.; Xiong, Q.; Qian, L.; Tong, C.; Lu, G.; Ding, Y.; Li, J.; Jung, C.; et al. A genome-wide survey with different rapeseed ecotypes uncovers footprints of domestication and breeding. J. Exp. Bot. 2017, 68, 4791–4801. [Google Scholar] [CrossRef] [Green Version]
- Larkan, N.J.; Yu, F.; Lydiate, D.J.; Rimmer, S.R.; Borhan, M.H. Single R gene introgression lines for accurate dissection of the Brassica-Leptosphaeria pathosystem. Front. Plant Sci. 2016, 7, 1771. [Google Scholar] [CrossRef] [Green Version]
- Ferdous, M.J.; Hossain, M.R.; Park, J.-I.; Robin, A.H.K.; Natarajan, S.; Jesse, D.M.I.; Jung, H.-J.; Kim, H.-T.; Nou, I.-S. In-silico identification and differential expressions of LepR4-syntenic disease resistance related domain containing genes against Blackleg causal fungus Leptosphaeria maculans in Brassica oleracea. Gene Rep. 2020, 19, 100598. [Google Scholar] [CrossRef]
- Alamery, S.; Tirnaz, S.; Bayer, P.; Tollenaere, R.; Chaloub, B.; Edwards, D.; Batley, J. Genome-wide identification and comparative analysis of NBS-LRR resistance genes in Brassica napus. Crop. Pasture Sci. 2017, 69, 79–93. [Google Scholar] [CrossRef]
- Fu, Y.; Zhang, Y.; Mason, A.S.; Lin, B.; Zhang, D.; Yu, H.; Fu, D. NBS-Encoding Genes in Brassica napus Evolved Rapidly After Allopolyploidization and Co-localize With Known Disease Resistance Loci. Front. Plant Sci. 2019, 10, 26. [Google Scholar] [CrossRef] [PubMed]
- Graham-Taylor, C.; Kamphuis, L.G.; Derbyshire, M.C. A detailed in silico analysis of secondary metabolite biosynthesis clusters in the genome of the broad host range plant pathogenic fungus Sclerotinia sclerotiorum. BMC Genom. 2020, 21, 7. [Google Scholar] [CrossRef] [Green Version]
- Stotz, H.U.; Harvey, P.J.; Haddadi, P.; Mashanova, A.; Kukol, A.; Larkan, N.J.; Borhan, M.H.; Fitt, B.D.L. Genomic evidence for genes encoding leucine-rich repeat receptors linked to resistance against the eukaryotic extra- and intracellular Brassica napus pathogens Leptosphaeria maculans and Plasmodiophora brassicae. PLoS ONE 2018, 13, e0198201. [Google Scholar] [CrossRef] [PubMed]
- Takagi, H.; Abe, A.; Yoshida, K.; Kosugi, S.; Natsume, S.; Mitsuoka, C.; Uemura, A.; Utsushi, H.; Tamiru, M.; Takuno, S.; et al. QTL-seq: Rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J. 2013, 74, 174–183. [Google Scholar] [CrossRef]
- Liu, S.; Yeh, C.-T.; Tang, H.M.; Nettleton, D.; Schnable, P.S. Gene mapping via bulked segregant RNA-Seq (BSR-Seq). PLoS ONE 2012, 7, e36406. [Google Scholar] [CrossRef] [Green Version]
- Michelmore, R.W.; Paran, I.; Kesseli, R.V. Identification of markers linked to disease-resistance genes by bulked segregant analysis: A rapid method to detect markers. Proc. Natl. Acad. Sci. USA 1991, 88. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Zhai, W.; Li, X.; Zhu, Y. Two QTLs controlling Clubroot resistance identified from Bulked Segregant Sequencing in Pakchoi (Brassica campestris ssp. chinensis Makino). Sci. Rep. 2019, 9, 9228. [Google Scholar] [CrossRef] [Green Version]
- Fu, F.; Liu, X.; Wang, R.; Zhai, C.; Peng, G.; Yu, F.; Fernando, W.G.D. Fine mapping of Brassica napus Blackleg resistance gene Rlm1 through bulked segregant RNA sequencing. Sci. Rep. 2019, 9, 14600. [Google Scholar] [CrossRef] [Green Version]
- Dakouri, A.; Zhang, X.; Peng, G.; Falk, K.C.; Gossen, B.D.; Strelkov, S.E.; Yu, F. Analysis of genome-wide variants through bulked segregant RNA sequencing reveals a major gene for resistance to Plasmodiophora brassicae in Brassica oleracea. Sci. Rep. 2018, 8, 17657. [Google Scholar] [CrossRef]
- Huang, Z.; Peng, G.; Liu, X.; Deora, A.; Falk, K.C.; Gossen, B.D.; McDonald, M.R.; Yu, F. Fine mapping of a Clubroot resistance gene in Chinese cabbage using SNP markers identified from bulked segregant RNA sequencing. Front. Plant Sci. 2017, 8, 1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, A.; Lamara, M.; Wei, Y.; Hu, H.; Parkin, I.A.P.; Gossen, B.D.; Peng, G.; Yu, F. Clubroot resistance gene Rcr6 in Brassica nigra resides in a genomic region homologous to chromosome A08 in B. rapa. BMC Plant Biol. 2019, 19, 224. [Google Scholar] [CrossRef] [PubMed]
- Pang, W.; Fu, P.; Li, X.; Zhan, Z.; Yu, S.; Piao, Z. Identification and mapping of the Clubroot resistance gene CRd in Chinese Cabbage (Brassica rapa ssp. pekinensis). Front. Plant Sci. 2018, 9, 653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jupe, F.; Witek, K.; Verweij, W.; Śliwka, J.; Pritchard, L.; Etherington, G.J.; Maclean, D.; Cock, P.J.; Leggett, R.M.; Bryan, G.J.; et al. Resistance gene enrichment sequencing (RenSeq) enables reannotation of the NB-LRR gene family from sequenced plant genomes and rapid mapping of resistance loci in segregating populations. Plant J. 2013, 76, 530–544. [Google Scholar] [CrossRef] [Green Version]
- Cevik, V.; Boutrot, F.; Apel, W.; Robert-Seilaniantz, A.; Furzer, O.J.; Redkar, A.; Castel, B.; Kover, P.X.; Prince, D.C.; Holub, E.B.; et al. Transgressive segregation reveals mechanisms of Arabidopsis; immunity to Brassica-infecting races of white rust (Albugo candida). Proc. Natl. Acad. Sci. USA 2019, 116, 2767. [Google Scholar] [CrossRef] [Green Version]
- Van de Weyer, A.-L.; Monteiro, F.; Furzer, O.J.; Nishimura, M.T.; Cevik, V.; Witek, K.; Jones, J.D.G.; Dangl, J.L.; Weigel, D.; Bemm, F. A species-wide inventory of NLR genes and alleles in Arabidopsis thaliana. Cell 2019, 178, 1260–1272.e14. [Google Scholar] [CrossRef] [Green Version]
- Domazakis, E.; Lin, X.; Aguilera-Galvez, C.; Wouters, D.; Bijsterbosch, G.; Wolters, P.J.; Vleeshouwers, V.G.A.A. Effectoromics-based identification of cell surface receptors in potato. In Plant Pattern Recognition Receptors—Methods in Molecular Biology; Shan, L., He, P., Eds.; Humana Press: New York, NY, USA, 2017; Volume 1578. [Google Scholar]
- Rodriguez-Moreno, L.; Song, Y.; Thomma, B.P.H.J. Transfer and engineering of immune receptors to improve recognition capacities in crops. Curr. Opin. Plant Biol. 2017, 38, 42–49. [Google Scholar] [CrossRef]
- Gu, B.; Cao, X.; Zhou, X.; Chen, Z.; Wang, Q.; Liu, W.; Chen, Q.; Zhao, H. The histological, effectoromic, and transcriptomic analyses of Solanum pinnatisectum reveal an upregulation of multiple NBS-LRR genes suppressing Phytophthora infestans infection. Int. J. Mol. Sci. 2020, 21, 3211. [Google Scholar] [CrossRef]
- Aguilera-Galvez, C.; Champouret, N.; Rietman, H.; Lin, X.; Wouters, D.; Chu, Z.; Jones, J.D.G.; Vossen, J.H.; Visser, R.G.F.; Wolters, P.J.; et al. Two different R gene loci co-evolved with Avr2 of Phytophthora infestans and confer distinct resistance specificities in potato. Stud. Mycol. 2018, 89, 105–115. [Google Scholar] [CrossRef]
- Wani, Z.A.; Ashraf, N. Transcriptomic studies revealing enigma of plant-pathogen interaction. In Molecular Aspects of Plant-Pathogen Interaction; Singh, A., Singh, I.K., Eds.; Springer: Singapore, 2018; pp. 219–238. [Google Scholar] [CrossRef]
- Wang, B.; Kumar, V.; Olson, A.; Ware, D. Reviving the transcriptome studies: An insight into the emergence of single-molecule transcriptome sequencing. Front. Genet. 2019, 10, 384. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10. [Google Scholar] [CrossRef] [PubMed]
- Metzker, M.L. Sequencing technologies—The next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westermann, A.J.; Gorski, S.A.; Vogel, J. Dual RNA-seq of pathogen and host. Nat. Rev. Microbiol. 2012, 10, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Harper, A.L.; Trick, M.; Higgins, J.; Fraser, F.; Clissold, L.; Wells, R.; Hattori, C.; Werner, P.; Bancroft, I. Associative transcriptomics of traits in the polyploid crop species Brassica napus. Nat. Biotechnol. 2012, 30, 798–802. [Google Scholar] [CrossRef]
- Adams, K.L.; Cronn, R.; Percifield, R.; Wendel, J.F. Genes duplicated by polyploidy show unequal contributions to the transcriptome and organ-specific reciprocal silencing. Proc. Natl. Acad. Sci. USA 2003, 100, 4649. [Google Scholar] [CrossRef] [Green Version]
- Larkan, N.J.; Ma, L.; Borhan, M.H. The Brassica napus receptor-like protein RLM2 is encoded by a second allele of the LepR3/Rlm2 Blackleg resistance locus. Plant Biotechnol. J. 2015, 13, 983–992. [Google Scholar] [CrossRef]
- Larkan, N.J.; Ma, L.; Haddadi, P.; Buchwaldt, M.; Parkin, I.A.P.; Djavaheri, M.; Borhan, M.H. The Brassica napus Wall-Associated Kinase-Like (WAKL) gene Rlm9 provides race-specific Blackleg resistance. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Xu, W.; Hirani, A.H.; Liu, Z.; Tuan, P.A.; Ayele, B.T.; Daayf, F.; McVetty, P.B.E.; Duncan, R.W.; Li, G. Transcriptional insight into Brassica napus resistance genes LepR3 and Rlm2-mediated defense response against the Leptosphaeria maculans infection. Front. Plant Sci. 2019, 10, 823. [Google Scholar] [CrossRef]
- Dmochowska-Boguta, M.; Kloc, Y.; Zielezinski, A.; Werecki, P.; Nadolska-Orczyk, A.; Karlowski, W.M.; Orczyk, W. TaWAK6 encoding wall-associated kinase is involved in wheat resistance to leaf rust similar to adult plant resistance. PLoS ONE 2020, 15, e0227713. [Google Scholar] [CrossRef] [Green Version]
- Brutus, A.; Sicilia, F.; Macone, A.; Cervone, F.; De Lorenzo, G. A domain swap approach reveals a role of the plant wall-associated kinase 1 (WAK1) as a receptor of oligogalacturonides. Proc. Natl. Acad. Sci. USA 2010, 107, 9452. [Google Scholar] [CrossRef] [Green Version]
- Haddadi, P.; Larkan, N.J.; Borhan, M.H. Dissecting R gene and host genetic background effect on the Brassica napus defense response to Leptosphaeria maculans. Sci. Rep. 2019, 9, 6947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haddadi, P.; Ma, L.; Wang, H.; Borhan, M.H. Genome-wide transcriptomic analyses provide insights into the lifestyle transition and effector repertoire of Leptosphaeria maculans during the colonization of Brassica napus seedlings. Mol. Plant Pathol. 2016, 17, 1196–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, M.G.; Zhang, X.; Walker, P.L.; Wan, J.C.; Millar, J.L.; Khan, D.; Granger, M.J.; Cavers, J.D.; Chan, A.C.; Fernando, D.W.G.; et al. Transcriptome analysis of the Brassica napus–Leptosphaeria maculans pathosystem identifies receptor, signaling and structural genes underlying plant resistance. Plant J. 2017, 90, 573–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Djavaheri, M.; Wang, H.; Larkan, N.J.; Haddadi, P.; Beynon, E.; Gropp, G.; Borhan, M.H. Leptosphaeria maculans effector protein AvrLm1 modulates plant immunity by enhancing MAP kinase 9 phosphorylation. iScience 2018, 3, 177–191. [Google Scholar] [CrossRef] [Green Version]
- Jammes, F.; Yang, X.; Xiao, S.; Kwak, J.M. Two Arabidopsis guard cell-preferential MAPK genes, MPK9 and MPK12, function in biotic stress response. Plant Signal. Behav. 2011, 6, 1875–1877. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Kim, Y.J.; Kim, M.-H.; Kwak, J.M. MAPK cascades in guard cell sgnal transduction. Front. Plant Sci. 2016, 7, 80. [Google Scholar] [CrossRef] [Green Version]
- Ueno, H.; Matsumoto, E.; Aruga, D.; Kitagawa, S.; Matsumura, H.; Hayashida, N. Molecular characterization of the CRa gene conferring clubroot resistance in Brassica rapa. Plant Mol. Biol. 2012, 80, 621–629. [Google Scholar] [CrossRef] [Green Version]
- Hatakeyama, K.; Suwabe, K.; Tomita, R.N.; Kato, T.; Nunome, T.; Fukuoka, H.; Matsumoto, S. Identification and characterization of Crr1a, a gene for resistance to clubroot disease (Plasmodiophora brassicae Woronin) in Brassica rapa L. PLoS ONE 2013, 8, e54745. [Google Scholar] [CrossRef]
- Shah, N.; Li, Q.; Xu, Q.; Liu, J.; Huang, F.; Zhan, Z.; Qin, P.; Zhou, X.; Yu, W.; Zhu, L.; et al. CRb and PbBa8.1 synergically increases resistant genes expression upon infection of Plasmodiophora brassicae in Brassica napus. Genes 2020, 11, 202. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Zhang, S. MAPK cascades in plant disease resistance signaling. Ann. Rev. Phytopathol. 2013, 51, 245–266. [Google Scholar] [CrossRef]
- Chen, J.; Pang, W.; Chen, B.; Zhang, C.; Piao, Z. Transcriptome analysis of Brassica rapa Near-Isogenic Lines carrying Clubroot-resistant and –susceptible alleles in response to Plasmodiophora brassicae during early Infection. Front. Plant Sci. 2016, 6, 1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, R.; Wang, Y.; Wang, X.; Liu, Y.; Shen, X.; Feng, H. Proteomic analysis of the interaction between Plasmodiophora brassicae and Chinese cabbage (Brassica rapa L. ssp. Pekinensis) at the initial infection stage. Sci. Hortic. 2018, 233, 386–393. [Google Scholar] [CrossRef]
- Ji, R.; Gao, S.; Bi, Q.; Wang, Y.; Lv, M.; Ge, W.; Feng, H. The salicylic acid signaling pathway plays an important role in the resistant process of Brassica rapa L. ssp. pekinensis to Plasmodiophora brassicae Woronin. J. Plant Growth Regul. 2020, 1–18. [Google Scholar] [CrossRef]
- Chu, M.; Song, T.; Falk, K.C.; Zhang, X.; Liu, X.; Chang, A.; Lahlali, R.; McGregor, L.; Gossen, B.D.; Yu, F.; et al. Fine mapping of Rcr1 and analyses of its effect on transcriptome patterns during infection by Plasmodiophora brassicae. BMC Genom. 2014, 15, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Galindo-González, L.; Manolii, V.; Hwang, S.-F.; Strelkov, S.E. Response of Brassica napus to Plasmodiophora brassicae involves salicylic acid-mediated immunity: An RNA-seq-based study. Front. Plant Sci. 2020, 11, 1025. [Google Scholar] [CrossRef]
- Qasim, M.U.; Zhao, Q.; Shahid, M.; Samad, R.A.; Ahmar, S.; Wu, J.; Fan, C.; Zhou, Y. Identification of QTLs containing resistance genes for Sclerotinia Stem Rot in Brassica napus using comparative transcriptomic studies. Front. Plant Sci. 2020, 11, 776. [Google Scholar] [CrossRef]
- Wu, J.; Zhao, Q.; Yang, Q.; Liu, H.; Li, Q.; Yi, X.; Cheng, Y.; Guo, L.; Fan, C.; Zhou, Y. Comparative transcriptomic analysis uncovers the complex genetic network for resistance to Sclerotinia sclerotiorum in Brassica napus. Sci. Rep. 2016, 6, 19007. [Google Scholar] [CrossRef] [Green Version]
- Girard, I.J.; Tong, C.; Becker, M.G.; Mao, X.; Huang, J.; de Kievit, T.; Fernando, W.G.D.; Liu, S.; Belmonte, M.F. RNA sequencing of Brassica napus reveals cellular redox control of Sclerotinia infection. J. Exp. Bot. 2017, 68, 5079–5091. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Ma, L.-Y.; Li, X.; Zhao, F.-Y.; Sarwar, R.; Cao, J.; Li, Y.-L.; Ding, L.-N.; Zhu, K.-M.; Yang, Y.-H.; et al. Genome-wide identification of the NPR1-like gene family in Brassica napus and functional characterization of BnaNPR1 in resistance to Sclerotinia sclerotiorum. Plant Cell Rep. 2020, 39, 709–722. [Google Scholar] [CrossRef]
- Zhong, X.; Zhou, Q.; Cui, N.; Cai, D.; Tang, G. BvcZR3 and BvHs1pro-1 genes pyramiding enhanced beet cyst nematode (Heterodera schachtii Schm.) resistance in oilseed rape (Brassica napus L.). Int. J. Mol. Sci. 2019, 20, 1740. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Huai, D.; Yang, Q.; Cheng, Y.; Ma, M.; Kliebenstein, D.J.; Zhou, Y. Overexpression of three glucosinolate biosynthesis genes in Brassica napus identifies enhanced resistance to Sclerotinia sclerotiorum and Botrytis cinerea. PLoS ONE 2015, 10, e0140491. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Mei, J.; Chai, Y.; Yu, Y.; Shao, C.; Wu, Q.; Disi, J.O.; Li, Y.; Wan, H.; Qian, W. Simultaneous transcriptome analysis of host and pathogen highlights the interaction between Brassica oleracea and Sclerotinia sclerotiorum. Phytopathology 2018, 109, 542–550. [Google Scholar] [CrossRef] [Green Version]
- Mei, J.; Ding, Y.; Li, Y.; Tong, C.; Du, H.; Yu, Y.; Wan, H.; Xiong, Q.; Yu, J.; Liu, S.; et al. Transcriptomic comparison between Brassica oleracea and rice (Oryza sativa) reveals diverse modulations on cell death in response to Sclerotinia sclerotiorum. Sci. Rep. 2016, 6, 33706. [Google Scholar] [CrossRef]
- Zheng, H.; Zhang, Y.; Li, J.; He, L.; Wang, F.; Bi, Y.; Gao, J. Comparative transcriptome analysis between a resistant and a susceptible Chinese cabbage in response to Hyaloperonospora brassicae. Plant Signal. Behav. 2020, 15, 1777373. [Google Scholar] [CrossRef]
- Xiao, D.; Liu, S.-T.; Wei, Y.-P.; Zhou, D.-Y.; Hou, X.-L.; Li, Y.; Hu, C.-M. cDNA-AFLP analysis reveals differential gene expression in incompatible interaction between infected non-heading Chinese cabbage and Hyaloperonospora parasitica. Hortic. Res. 2016, 3, 16034. [Google Scholar] [CrossRef] [Green Version]
- Xing, M.; Lv, H.; Ma, J.; Xu, D.; Li, H.; Yang, L.; Kang, J.; Wang, X.; Fang, Z. Transcriptome profiling of resistance to Fusarium oxysporum f. sp. conglutinans in cabbage (Brassica oleracea) roots. PLoS ONE 2016, 11, e0148048. [Google Scholar] [CrossRef]
- Tortosa, M.; Cartea, M.E.; Velasco, P.; Soengas, P.; Rodriguez, V.M. Calcium-signaling proteins mediate the plant transcriptomic response during a well-established Xanthomonas campestris pv. campestris infection. Hortic. Res. 2019, 6, 103. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Wu, F.; Wang, S.; Lu, Y.; Chen, X.; Wang, Y.; Gu, A.; Zhao, J.; Shen, S. Comparative transcriptome analysis reveals defense responses against soft rot in Chinese cabbage. Hortic. Res. 2019, 6, 68. [Google Scholar] [CrossRef] [Green Version]
- Mehta, A.; Brasileiro, A.C.M.; Souza, D.S.L.; Romano, E.; Campos, M.A.; Grossi-de-Sá, M.F.; Silva, M.S.; Franco, O.L.; Fragoso, R.R.; Bevitori, R.; et al. Plant–pathogen interactions: What is proteomics telling us? FEBS J. 2008, 275, 3731–3746. [Google Scholar] [CrossRef] [Green Version]
- Marra, R.; Li, H.; Barbetti, M.J.; Sivasithamparam, K.; Vinale, F.; Cavallo, P.; Lorito, M. Proteomic analysis of the interaction between Brassica napus cv. Surpass 400 and virulent or avirulent isolates of Leptosphaeria maculans. J. Plant Pathol. 2010, 92, 89–101. [Google Scholar]
- Sharma, N.; Hotte, N.; Rahman, M.H.; Mohammadi, M.; Deyholos, M.K.; Kav, N.N.V. Towards identifying Brassica proteins involved in mediating resistance to Leptosphaeria maculans: A proteomics-based approach. Proteomics 2008, 8, 3516–3535. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, B.; Bansal, V.K.; Kav, N.N. Proteome-level investigation of Brassica carinata-derived resistance to Leptosphaeria maculans. J. Agric. Food Chem. 2005, 53, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Chu, M.; Lahlali, R.; Yu, F.; Peng, G. Shotgun label-free proteomic analysis of Clubroot (Plasmodiophora brassicae) resistance conferred by the gene Rcr1 in Brassica rapa. Front. Plant Sci. 2016, 7, 1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, J.Y.; Kim, S.T.; Choi, G.J.; Kwon, S.-Y.; Cho, H.S.; Kim, H.-S.; Moon, J.S.; Park, J.M. Comparative proteomic analysis of host responses to Plasmodiophora brassicae infection in susceptible and resistant Brassica oleracea. Plant Biotech. Rep. 2020, 14, 263–274. [Google Scholar] [CrossRef]
- Lan, M.; Li, G.; Hu, J.; Yang, H.; Zhang, L.; Xu, X.; Liu, J.; He, J.; Sun, R. iTRAQ-based quantitative analysis reveals proteomic changes in Chinese cabbage (Brassica rapa L.) in response to Plasmodiophora brassicae infection. Sci. Rep. 2019, 9, 12058. [Google Scholar] [CrossRef]
- Sun, C.; Wang, L.; Hu, D.; Riquicho, A.R.M.; Liu, T.; Hou, X.; Li, Y. Proteomic analysis of non-heading Chinese cabbage infected with Hyaloperonospora parasitica. J. Proteom. 2014, 98, 15–30. [Google Scholar] [CrossRef]
- Rustagi, A.; Singh, G.; Agrawal, S.; Gupta, P.K. Proteomic studies revealing enigma of plant–pathogen interaction. In Molecular Aspects of Plant-Pathogen Interaction; Singh, A., Singh, I.K., Eds.; Springer: Singapore, 2018; pp. 239–264. [Google Scholar] [CrossRef]
- Kaur, P.; Jost, R.; Sivasithamparam, K.; Barbetti, M.J. Proteome analysis of the Albugo candida-Brassica juncea pathosystem reveals that the timing of the expression of defence-related genes is a crucial determinant of pathogenesis. J. Exp. Bot. 2011, 62, 1285–1298. [Google Scholar] [CrossRef]
- Shrivastava, N.; Jiang, L.; Li, P.; Sharma, A.K.; Luo, X.; Wu, S.; Pandey, R.; Gao, Q.; Lou, B. Proteomic approach to understand the molecular physiology of symbiotic interaction between Piriformospora indica and Brassica napus. Sci. Rep. 2018, 8, 5773. [Google Scholar] [CrossRef] [Green Version]
- Dutreux, F.; Da Silva, C.; d’Agata, L.; Couloux, A.; Gay, E.J.; Istace, B.; Lapalu, N.; Lemainque, A.; Linglin, J.; Noel, B.; et al. De novo assembly and annotation of three Leptosphaeria genomes using Oxford Nanopore MinION sequencing. Sci. Data 2018, 5, 180235. [Google Scholar] [CrossRef]
- Derbyshire, M.; Denton-Giles, M.; Hegedus, D.; Seifbarghy, S.; Rollins, J.; van Kan, J.; Seidl, M.F.; Faino, L.; Mbengue, M.; Navaud, O.; et al. The complete genome sequence of the phytopathogenic fungus Sclerotinia sclerotiorum reveals insights into the genome architecture of broad host range pathogens. Genome Biol. Evol. 2017, 9, 593–618. [Google Scholar] [CrossRef]
- Rajarammohan, S.; Pental, D.; Kaur, J. Near-complete genome assembly of Alternaria brassicae—A necrotrophic pathogen of Brassica crops. Mol. Plant Microbe Interact. 2019, 32, 928–930. [Google Scholar] [CrossRef] [Green Version]
- Rajarammohan, S.; Paritosh, K.; Pental, D.; Kaur, J. Comparative genomics of Alternaria species provides insights into the pathogenic lifestyle of Alternaria brassicae—A pathogen of the Brassicaceae family. BMC Genom. 2019, 20, 1036. [Google Scholar] [CrossRef] [Green Version]
- Daval, S.; Belcour, A.; Gazengel, K.; Legrand, L.; Gouzy, J.; Cottret, L.; Lebreton, L.; Aigu, Y.; Mougel, C.; Manzanares-Dauleux, M.J. Computational analysis of the Plasmodiophora brassicae genome: Mitochondrial sequence description and metabolic pathway database design. Genomics 2019, 111, 1629–1640. [Google Scholar] [CrossRef] [PubMed]
- Links, M.G.; Holub, E.; Jiang, R.H.Y.; Sharpe, A.G.; Hegedus, D.; Beynon, E.; Sillito, D.; Clarke, W.E.; Uzuhashi, S.; Borhan, M.H. De novo sequence assembly of Albugo candida reveals a small genome relative to other biotrophic oomycetes. BMC Genom. 2011, 12, 503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsushima, A.; Gan, P.; Kumakura, N.; Narusaka, M.; Takano, Y.; Narusaka, Y.; Shirasu, K. Genomic Plasticity Mediated by Transposable Elements in the Plant Pathogenic Fungus Colletotrichum higginsianum. Genome Biol. Evol. 2019, 11, 1487–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi-Kunne, X.; Faino, L.; van den Berg, G.C.M.; Thomma, B.P.H.J.; Seidl, M.F. Evolution within the fungal genus Verticillium is characterized by chromosomal rearrangement and gene loss. Environ. Microbiol. 2018, 20, 1362–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mousavi-Derazmahalleh, M.; Chang, S.; Thomas, G.; Derbyshire, M.; Bayer, P.E.; Edwards, D.; Nelson, M.N.; Erskine, W.; Lopez-Ruiz, F.J.; Clements, J.; et al. Prediction of pathogenicity genes involved in adaptation to a lupin host in the fungal pathogens Botrytis cinerea and Sclerotinia sclerotiorum via comparative genomics. BMC Genom. 2019, 20, 385. [Google Scholar] [CrossRef]
- Bertazzoni, S.; Williams, A.H.; Jones, D.A.; Syme, R.A.; Tan, K.-C.; Hane, J.K. Accessories Make the Outfit: Accessory Chromosomes and Other Dispensable DNA Regions in Plant-Pathogenic Fungi. Mol. Plant Microbe Interact. 2018, 31, 779–788. [Google Scholar] [CrossRef] [Green Version]
- Covo, S. Genomic Instability in Fungal Plant Pathogens. Genes 2020, 11, 421. [Google Scholar] [CrossRef] [Green Version]
- Peng, Z.; Oliveira-Garcia, E.; Lin, G.; Hu, Y.; Dalby, M.; Migeon, P.; Tang, H.; Farman, M.; Cook, D.; White, F.F.; et al. Effector gene reshuffling involves dispensable mini-chromosomes in the wheat blast fungus. PLoS Genet. 2019, 15, e1008272. [Google Scholar] [CrossRef] [Green Version]
- Möller, M.; Stukenbrock, E.H. Evolution and genome architecture in fungal plant pathogens. Nat. Rev. Microbiol. 2017, 15, 756. [Google Scholar] [CrossRef] [PubMed]
- Feurtey, A.; Stevens, D.M.; Stephan, W.; Stukenbrock, E.H. Interspecific Gene Exchange Introduces High Genetic Variability in Crop Pathogen. Genome Biol. Evol. 2019, 11, 3095–3105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stjelja, S.; Fogelqvist, J.; Tellgren-Roth, C.; Dixelius, C. The architecture of the Plasmodiophora brassicae nuclear and mitochondrial genomes. Sci. Rep. 2019, 9, 15753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chittem, K.; Yajima, W.R.; Goswami, R.S.; Del Río Mendoza, L.E. Transcriptome analysis of the plant pathogen Sclerotinia sclerotiorum interaction with resistant and susceptible canola (Brassica napus) lines. PLoS ONE 2020, 15, e0229844. [Google Scholar] [CrossRef] [Green Version]
- Sonah, H.; Deshmukh, R.K.; Bélanger, R.R. Computational prediction of effector proteins in fungi: Opportunities and challenges. Front. Plant Sci. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Pérez-López, E.; Waldner, M.; Hossain, M.; Kusalik, A.J.; Wei, Y.; Bonham-Smith, P.C.; Todd, C.D. Identification of Plasmodiophora brassicae effectors—A challenging goal. Virulence 2018, 9, 1344–1353. [Google Scholar] [CrossRef]
- Chen, W.; Li, Y.; Yan, R.; Xu, L.; Ren, L.; Liu, F.; Zeng, L.; Yang, H.; Chi, P.; Wang, X.; et al. Identification and characterization of Plasmodiophora brassicae primary infection effector candidates that suppress or induce cell death in host and nonhost plants. Phytopathology 2019, 109, 1689–1697. [Google Scholar] [CrossRef]
- Büttner, D.; Bonas, U. Regulation and secretion of Xanthomonas virulence factors. FEMS Microbiol. Rev. 2010, 34, 107–133. [Google Scholar] [CrossRef] [Green Version]
- Charkowski, A.; Blanco, C.; Condemine, G.; Expert, D.; Franza, T.; Hayes, C.; Hugouvieux-Cotte-Pattat, N.; Solanilla, E.L.; Low, D.; Moleleki, L.; et al. The role of secretion systems and small molecules in Soft-Rot Enterobacteriaceae pathogenicity. Ann. Rev. Phytopathol. 2011, 50, 425–449. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.H.; Lim, J.-A.; Lee, J.; Roh, E.; Jung, K.; Choi, M.; Oh, C.; Ryu, S.; Yun, J.; Heu, S. Characterization of genes required for the pathogenicity of Pectobacterium carotovorum subsp. carotovorum Pcc21 in Chinese cabbage. Microbiology 2013, 159, 1487–1496. [Google Scholar] [CrossRef]
- Seifbarghi, S.; Borhan, M.H.; Wei, Y.; Coutu, C.; Robinson, S.J.; Hegedus, D.D. Changes in the Sclerotinia sclerotiorum transcriptome during infection of Brassica napus. BMC Genom. 2017, 18, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, D.; Plummer, K.M.; Solomon, P.S.; Lebrun, M.-H.; Job, D.; Rafiqi, M. Editorial: How can secretomics help unravel the secrets of plant-microbe interactions? Front. Plant Sci. 2016, 7, 1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.-T.; Jeon, J.; Choi, J.; Cheong, K.; Song, H.; Choi, G.; Kang, S.; Lee, Y.-H. Kingdom-wide analysis of fungal small secreted proteins (SSPs) reveals their potential role in host association. Front. Plant Sci. 2016, 7, 186. [Google Scholar] [CrossRef] [Green Version]
- Van de Wouw, A.P.; Idnurm, A. Biotechnological potential of engineering pathogen effector proteins for use in plant disease management. Biotechnol. Adv. 2019, 37, 107387. [Google Scholar] [CrossRef]
- Depotter, J.R.L.; Doehlemann, G. Target the core: Durable plant resistance against filamentous plant pathogens through effector recognition. Pest Manag. Sci. 2020, 76, 426–431. [Google Scholar] [CrossRef] [Green Version]
- Sperschneider, J.; Dodds, P.N.; Gardiner, D.M.; Singh, K.B.; Taylor, J.M. Improved prediction of fungal effector proteins from secretomes with EffectorP 2.0. Mol. Plant Pathol. 2018, 19, 2094–2110. [Google Scholar] [CrossRef] [Green Version]
- Franceschetti, M.; Maqbool, A.; Jiménez-Dalmaroni, M.J.; Pennington, H.G.; Kamoun, S.; Banfield, M.J. Effectors of filamentous plant pathogens: Commonalities amid diversity. Microbiol. Mol. Biol. Rev. 2017, 81. [Google Scholar] [CrossRef] [Green Version]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef]
- Sperschneider, J.; Dodds, P.N.; Singh, K.B.; Taylor, J.M. ApoplastP: Prediction of effectors and plant proteins in the apoplast using machine learning. New Phytol. 2017, 217, 1764–1778. [Google Scholar] [CrossRef] [Green Version]
- Carreón-Anguiano, K.G.; Islas-Flores, I.; Vega-Arreguín, J.; Sáenz-Carbonell, L.; Canto-Canché, B. EffHunter: A tool for prediction of effector protein candidates in fungal proteomic databases. Biomolecules 2020, 10, 712. [Google Scholar] [CrossRef]
- Collemare, J.; O’Connell, R.; Lebrun, M.-H. Nonproteinaceous effectors: The terra incognita of plant–fungal interactions. New Phytol. 2019, 223, 590–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruano, G.; Scheuring, D. Plant cells under attack: Unconventional endomembrane trafficking during plant defense. Plants 2020, 9, 389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanveer, T.; Shaheen, K.; Parveen, S.; Kazi, A.G.; Ahmad, P. Plant secretomics. Plant Signal. Behav. 2014, 9, e29426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, D.; Rafiqi, M.; Job, D. The multiple facets of plant-fungal interactions revealed through plant and fungal secretomics. Front. Plant Sci. 2020, 10, 1626. [Google Scholar] [CrossRef]
- Gupta, R.; Lee, S.E.; Agrawal, G.K.; Rakwal, R.; Park, S.; Wang, Y.; Kim, S.T. Understanding the plant-pathogen interactions in the context of proteomics-generated apoplastic proteins inventory. Front. Plant Sci. 2015, 6, 352. [Google Scholar] [CrossRef] [Green Version]
- Deboever, E.; Deleu, M.; Mongrand, S.; Lins, L.; Fauconnier, M.-L. Plant–Pathogen Interactions: Underestimated Roles of Phyto-oxylipins. Trends Plant Sci. 2020, 25, 22–34. [Google Scholar] [CrossRef] [Green Version]
- Genva, M.; Obounou Akong, F.; Andersson, M.X.; Deleu, M.; Lins, L.; Fauconnier, M.-L. New insights into the biosynthesis of esterified oxylipins and their involvement in plant defense and developmental mechanisms. Phytochem. Rev. 2019, 18, 343–358. [Google Scholar] [CrossRef] [Green Version]
- Granér, G.; Hamberg, M.; Meijer, J. Screening of oxylipins for control of oilseed rape (Brassica napus) fungal pathogens. Phytochemistry 2003, 63, 89–95. [Google Scholar] [CrossRef]
- Fischer, G.J.; Keller, N.P. Production of cross-kingdom oxylipins by pathogenic fungi: An update on their role in development and pathogenicity. J. Microbiology 2016, 54, 254–264. [Google Scholar] [CrossRef] [Green Version]
- Patkar, R.N.; Naqvi, N.I. Fungal manipulation of hormone-regulated plant defense. PLoS Pathog. 2017, 13, e1006334. [Google Scholar] [CrossRef] [Green Version]
- Thatcher, L.F.; Manners, J.M.; Kazan, K. Fusarium oxysporum hijacks COI1-mediated jasmonate signaling to promote disease development in Arabidopsis. Plant J. 2009, 58, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Spallek, T.; Gan, P.; Kadota, Y.; Shirasu, K. Same tune, different song—Cytokinins as virulence factors in plant–pathogen interactions? Curr. Opin. Plant Biol. 2018, 44, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Darma, R.; Lutz, A.; Elliott, C.E.; Idnurm, A. Identification of a gene cluster for the synthesis of the plant hormone abscisic acid in the plant pathogen Leptosphaeria maculans. Fungal Genet. Biol. 2019, 130, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Ludwig-Müller, J.; Jülke, S.; Geiß, K.; Richter, F.; Mithöfer, A.; Šola, I.; Rusak, G.; Keenan, S.; Bulman, S. A novel methyltransferase from the intracellular pathogen Plasmodiophora brassicae methylates salicylic acid. Mol. Plant Pathol. 2015, 16. [Google Scholar] [CrossRef] [PubMed]
- Ciaghi, S.; Schwelm, A.; Neuhauser, S. Transcriptomic response in symptomless roots of clubroot infected kohlrabi (Brassica oleracea var. gongylodes) mirrors resistant plants. BMC Plant Biol. 2019, 19, 288. [Google Scholar] [CrossRef] [Green Version]
- Djavaheri, M.; Ma, L.; Klessig, D.F.; Mithöfer, A.; Gropp, G.; Borhan, H. Mimicking the host regulation of salicylic acid: A virulence strategy by the Clubroot pathogen Plasmodiophora brassicae. Mol. Plant Microbe Interact. 2018, 32, 296–305. [Google Scholar] [CrossRef] [Green Version]
- Pathak, R.K.; Baunthiyal, M.; Shukla, R.; Pandey, D.; Taj, G.; Kumar, A. In silico identification of mimicking molecules as defense inducers triggering jasmonic acid mediated immunity against Alternaria Blight disease in Brassica species. Front. Plant Sci. 2017, 8, 609. [Google Scholar] [CrossRef] [Green Version]
- Romero, F.M.; Rossi, F.R.; Gárriz, A.; Carrasco, P.; Ruíz, O.A. A bacterial endophyte from apoplast fluids protects canola plants from different phytopathogens via antibiosis and induction of host resistance. Phytopathology 2018, 109, 375–383. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.; Wang, D.; Mao, Z.; Pan, L.; Liao, J.; Cai, Z. Infection of Plasmodiophora brassicae changes the fungal endophyte community of tumourous stem mustard roots as revealed by high-throughput sequencing and culture-dependent methods. PLoS ONE 2019, 14, e0214975. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, H.; Howton, T.C.; Sun, Y.; Weinberger, N.; Belkhadir, Y.; Mukhtar, M.S. Network biology discovers pathogen contact points in host protein-protein interactomes. Nat. Commun. 2018, 9, 2312. [Google Scholar] [CrossRef]
- Urban, M.; Cuzick, A.; Rutherford, K.; Irvine, A.; Pedro, H.; Pant, R.; Sadanadan, V.; Khamari, L.; Billal, S.; Mohanty, S.; et al. PHI-base: A new interface and further additions for the multi-species pathogen–host interactions database. Nucleic Acids Res. 2016, 45, D604–D610. [Google Scholar] [CrossRef] [PubMed]
- Janowska-Sejda, E.I.; Lysenko, A.; Urban, M.; Rawlings, C.; Tsoka, S.; Hammond-Kosack, K.E. PHI-Nets: A network resource for Ascomycete fungal pathogens to annotate and identify putative virulence interacting proteins and siRNA targets. Front. Microbiol. 2019, 10, 2721. [Google Scholar] [CrossRef] [PubMed]
- Castro-Moretti, F.R.; Gentzel, I.N.; Mackey, D.; Alonso, A.P. Metabolomics as an emerging tool for the study of plant-pathogen interactions. Metabolites 2020, 10, 52. [Google Scholar] [CrossRef] [Green Version]
- Tortosa, M.; Cartea, M.E.; Rodríguez, V.M.; Velasco, P. Unraveling the metabolic response of Brassica oleracea exposed to Xanthomonas campestris pv. campestris. J. Sci. Food Agric. 2018, 98, 3675–3683. [Google Scholar] [CrossRef] [PubMed]
- Pathak, R.K.; Baunthiyal, M.; Pandey, N.; Pandey, D.; Kumar, A. Modeling of the jasmonate signaling pathway in Arabidopsis thaliana with respect to pathophysiology of Alternaria blight in Brassica. Sci. Rep. 2017, 7, 16790. [Google Scholar] [CrossRef] [Green Version]
- Botero, D.; Alvarado, C.; Bernal, A.; Danies, G.; Restrepo, S. Network analyses in plant pathogens. Front. Microbiol. 2018, 9, 35. [Google Scholar] [CrossRef] [PubMed]
- Peyraud, R.; Mbengue, M.; Barbacci, A.; Raffaele, S. Intercellular cooperation in a fungal plant pathogen facilitates host colonization. Proc. Natl. Acad. Sci. USA 2019, 116, 3193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, G.; Laperche, A.; Lariagon, C.; Marnet, N.; Renault, D.; Guitton, Y.; Bouchereau, A.; Delourme, R.; Manzanares-Dauleux, M.J.; Gravot, A. Resolution of quantitative resistance to clubroot into QTL-specific metabolic modules. J. Exp. Bot. 2019, 70, 5375–5390. [Google Scholar] [CrossRef]
- Singh, A. Glucosinolates and Plant Defense. In Glucosinolates; Mérillon, J.-M., Ramawat, K.G., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 237–246. [Google Scholar] [CrossRef]
- Ranjan, A.; Westrick, N.M.; Jain, S.; Piotrowski, J.S.; Ranjan, M.; Kessens, R.; Stiegman, L.; Grau, C.R.; Smith, D.L.; Kabbage, M. Integrated soybean transcriptomics, metabolomics, and chemical genomics reveal the importance of the phenylpropanoid pathway and antifungal activity in resistance to the broad host range pathogen Sclerotinia sclerotiorum. bioRxiv 2018. [Google Scholar] [CrossRef]
- Fikere, M.; Barbulescu, D.M.; Malmberg, M.M.; Shi, F.; Koh, J.C.O.; Slater, A.T.; MacLeod, I.M.; Bowman, P.J.; Salisbury, P.A.; Spangenberg, G.C.; et al. Genomic prediction using prior Quantitative Trait Loci information reveals a large reservoir of underutilised Blackleg resistance in diverse canola (Brassica napus L.) lines. Plant Genome 2018, 11, 170100. [Google Scholar] [CrossRef] [Green Version]
- Poland, J.; Rutkoski, J. Advances and challenges in genomic selection for disease resistance. Ann. Rev. Phytopathol. 2016, 54, 79–98. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zheng, F.; Wei, S.; Zhang, S.; Li, G.; Cao, P.; Zhao, S. Evolution of disease defense genes and their regulators in plants. Int. J. Mol. Sci. 2019, 20, 335. [Google Scholar] [CrossRef] [Green Version]
- Tirnaz, S.; Batley, J. DNA Methylation: Toward crop disease resistance improvement. Trends Plant Sci. 2019, 24, 1137–1150. [Google Scholar] [CrossRef] [PubMed]
- Alonso, C.; Ramos-Cruz, D.; Becker, C. The role of plant epigenetics in biotic interactions. New Phytol. 2019, 221, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Liégard, B.; Baillet, V.; Etcheverry, M.; Joseph, E.; Lariagon, C.; Lemoine, J.; Evrard, A.; Colot, V.; Gravot, A.; Manzanares-Dauleux, M.J.; et al. Quantitative resistance to Clubroot infection mediated by transgenerational epigenetic variation in Arabidopsis. New Phytol. 2019, 222, 468–479. [Google Scholar] [CrossRef]
- Tirnaz, S.; Merce, C.; Bayer, P.E.; Severn-Ellis, A.A.; Edwards, D.; Batley, J. Effect of Leptosphaeria maculans infection on promoter DNA methylation of defence genes in Brassica napus. Agronomy 2020, 10, 1072. [Google Scholar] [CrossRef]
- Spring, O.; Gomez-Zeledon, J.; Hadziabdic, D.; Trigiano, R.N.; Thines, M.; Lebeda, A. Biological characteristics and assessment of virulence diversity in pathosystems of economically important biotrophic oomycetes. Crit. Rev. Plant Sci. 2018, 37, 439–495. [Google Scholar] [CrossRef]
- Sun, Q.; Lin, L.; Liu, D.; Wu, D.; Fang, Y.; Wu, J.; Wang, Y. CRISPR/Cas9-mediated multiplex genome editing of the BnWRKY11 and BnWRKY70 Genes in Brassica napus L. Int. J. Mol. Sci. 2018, 19, 2716. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S.Y.; Ahn, H.; Ryu, J.; Oh, Y.; Sivanandhan, G.; Won, K.-H.; Park, Y.D.; Kim, J.-S.; Kim, H.; Lim, Y.P.; et al. Generation of early-flowering Chinese cabbage (Brassica rapa spp. pekinensis) through CRISPR/Cas9-mediated genome editing. Plant Biotechnol. Rep. 2019, 13, 491–499. [Google Scholar] [CrossRef]
- Liu, F.; Selin, C.; Zou, Z.; Dilantha Fernando, W.G. LmCBP1, a secreted chitin-binding protein, is required for the pathogenicity of Leptosphaeria maculans on Brassica napus. Fungal Genet. Biol. 2020, 136, 103320. [Google Scholar] [CrossRef]
- Li, Q.; Yan, J. Sustainable agriculture in the era of omics: Knowledge-driven crop breeding. Genome Biol. 2020, 21, 154. [Google Scholar] [CrossRef] [PubMed]

| Approach | Disease Type | Brassica Sample | Main Findings | Reference |
|---|---|---|---|---|
| WGRS, SNP genotyping | - | 991 B. napus worldwide accessions | Selective-sweep regions enriched with genes related to stress response | Wu, et al. [82] |
| WGRS, SNP genotyping | - | 588 B. napus worldwide accessions | A sub-genomic-specific selection contributes towards biotic stress response with several candidate genes identified | Lu, et al. [83] |
| WGRS, QTL mapping | Black rot | Mapping population of cabbage B. oleracea var. capitata inbred lines “C1234” (resistant) and “C1184” (susceptible) | 21 candidate NBS-LRR genes associated with black rot resistance in B. oleracea | Lee, et al. [84] |
| GBS, GWAS | Blackleg | 243 B. napus accessions from Canada and China | Significant SNPs were found on chromosome A08 with 25 RGAs identified consisting of NBS, RLK, RLP and TM-CC type R genes | Fu, et al. [73] |
| GBS, GWAS | Sclerotinia | B. juncea–B. fruticulosa introgression lines | 20 candidate genes mostly located on the A sub-genome of B. juncea | Atri, et al. [75] |
| GBS, GWAS | Sclerotinia | B. juncea–Erucastrum cardaminoides introgression lines | QTL region on chromosomes A03 and B03 and candidate genes being LRR-RLK, LRR-PK and TIR-NBR-LRR | Rana, et al. [76] |
| tGBS®, GWAS | - | 135 B. oleracea accessions including var. broccoli, Brussels sprout, cabbage, cauliflower, Chinese kala, kale, kohlrabi and savoy cabbage | Resistant phenotype mostly found in kale. Candidate genes encoding pathogenesis-related proteins were mainly found on chromosome C07 | Farid, et al. [85] |
| Brassica 60K SNP array, GWAS | Clubroot | Mapping population of cabbage B. oleracea inbred lines “263” and “GZ87” | Significant QTL and novel loci found on C sub-genome | Peng, et al. [78] |
| Brassica 60K SNP array, linkage disequilibrium (LD) analysis | - | 327 B. napus worldwide accessions comprising three ecotypes | Selective-sweep regions enriched with Blackleg and Sclerotinia resistance QTLs | Wei, et al. [86] |
| Brassica 60K SNP array, GWAS | Clubroot | 472 B. napus worldwide accessions | Most candidate genes were found on C sub-genome with novel QTLs and TIR-NBS gene clusters | Li, et al. [77] |
| Brassica 60K SNP array, GWAS | Sclerotinia | 448 worldwide B. napus accessions | Two novel loci with 39 candidate genes on C sub-genome | Wu, et al. [74] |
| Brassica 60K SNP array | Blackleg | Seven B. napus seven donor parents for introgression lines | Genomic background of individual varieties and multiple defence-related gene interactions influence the resistance levels | Larkan, et al. [87] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neik, T.X.; Amas, J.; Barbetti, M.; Edwards, D.; Batley, J. Understanding Host–Pathogen Interactions in Brassica napus in the Omics Era. Plants 2020, 9, 1336. https://doi.org/10.3390/plants9101336
Neik TX, Amas J, Barbetti M, Edwards D, Batley J. Understanding Host–Pathogen Interactions in Brassica napus in the Omics Era. Plants. 2020; 9(10):1336. https://doi.org/10.3390/plants9101336
Chicago/Turabian StyleNeik, Ting Xiang, Junrey Amas, Martin Barbetti, David Edwards, and Jacqueline Batley. 2020. "Understanding Host–Pathogen Interactions in Brassica napus in the Omics Era" Plants 9, no. 10: 1336. https://doi.org/10.3390/plants9101336
APA StyleNeik, T. X., Amas, J., Barbetti, M., Edwards, D., & Batley, J. (2020). Understanding Host–Pathogen Interactions in Brassica napus in the Omics Era. Plants, 9(10), 1336. https://doi.org/10.3390/plants9101336