Rooibos (Aspalathus linearis) Genome Size Estimation Using Flow Cytometry and K-Mer Analyses

 , ,
, ,
Abstract
:1. Introduction
2. Results
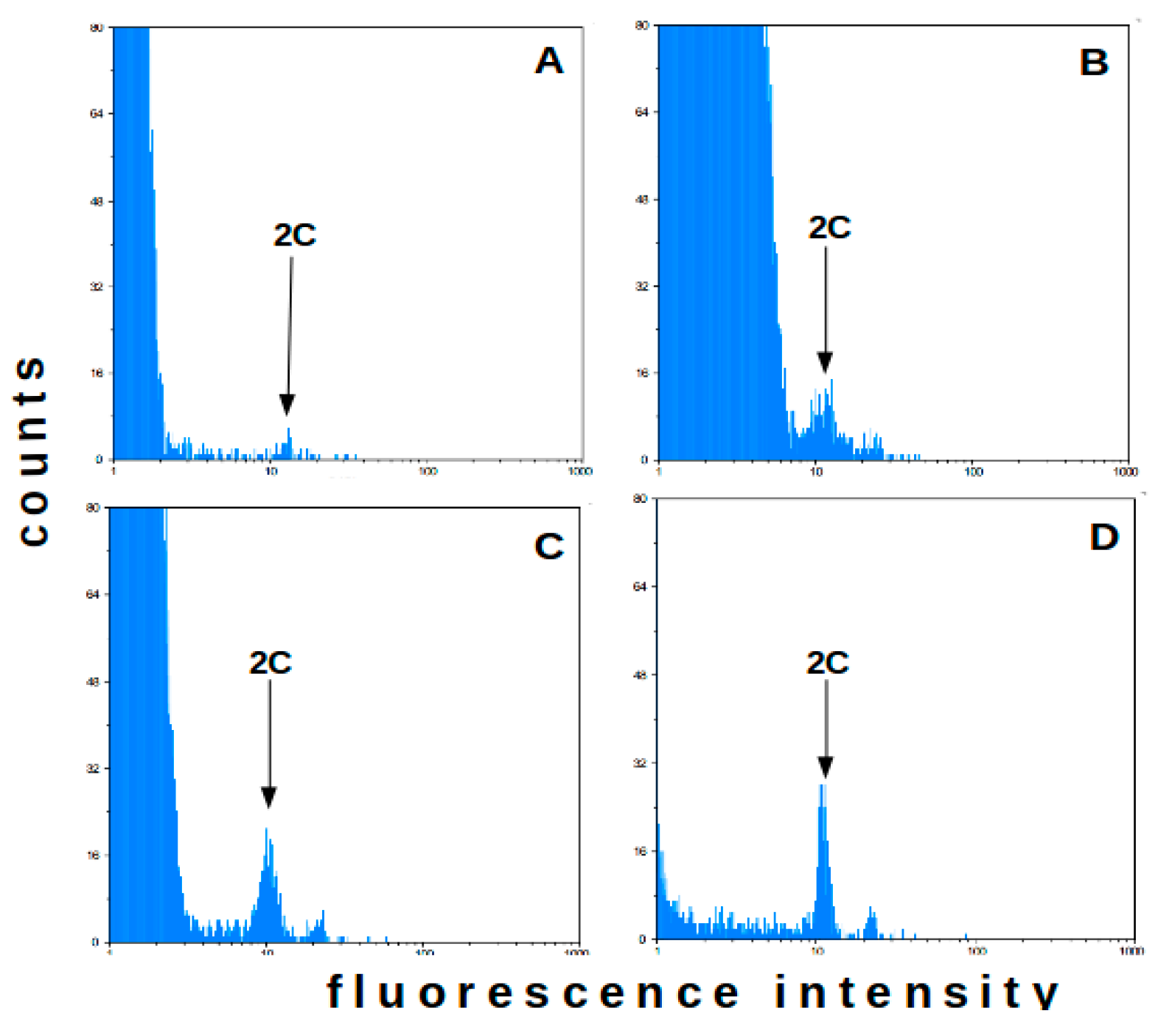
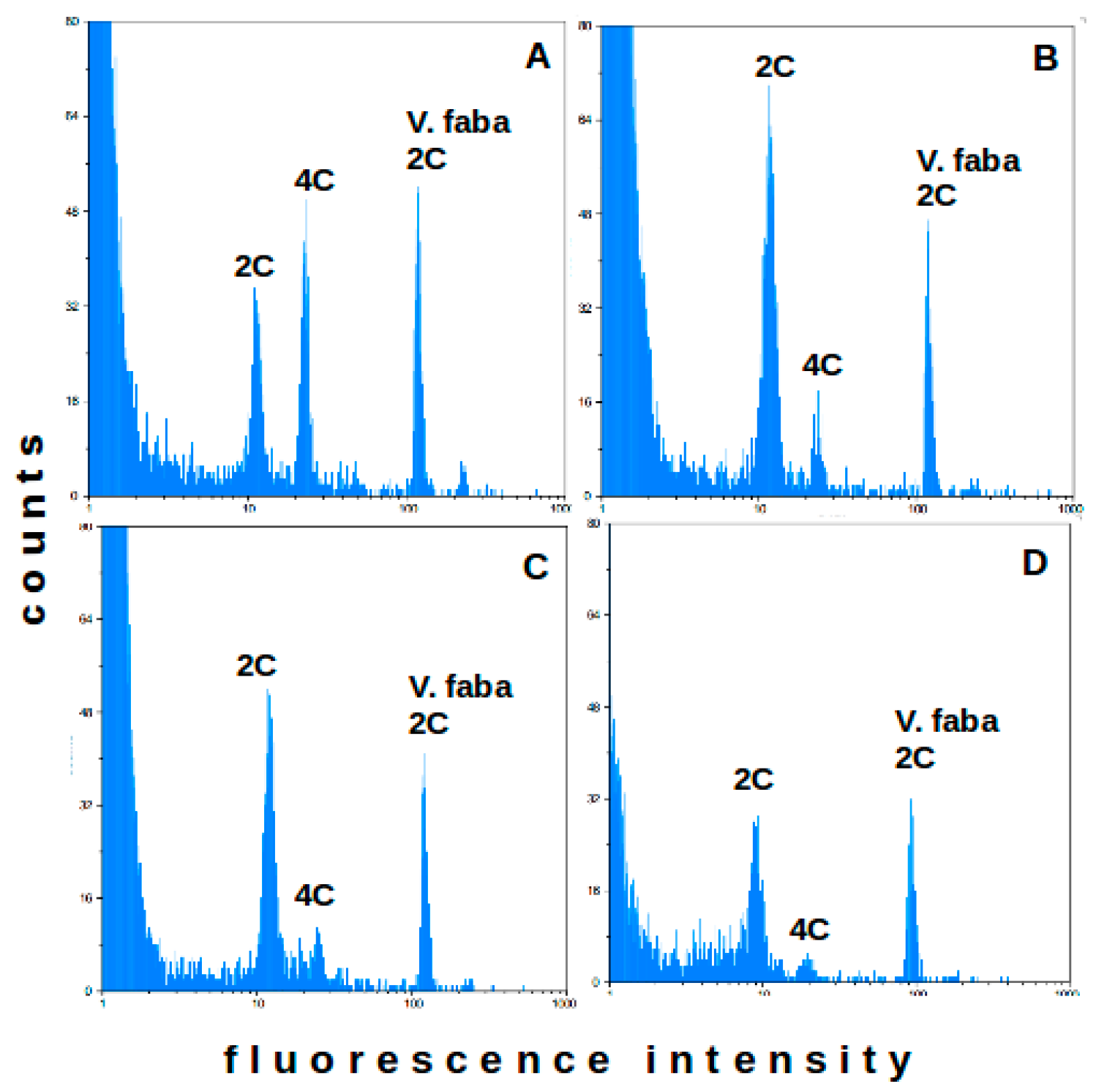
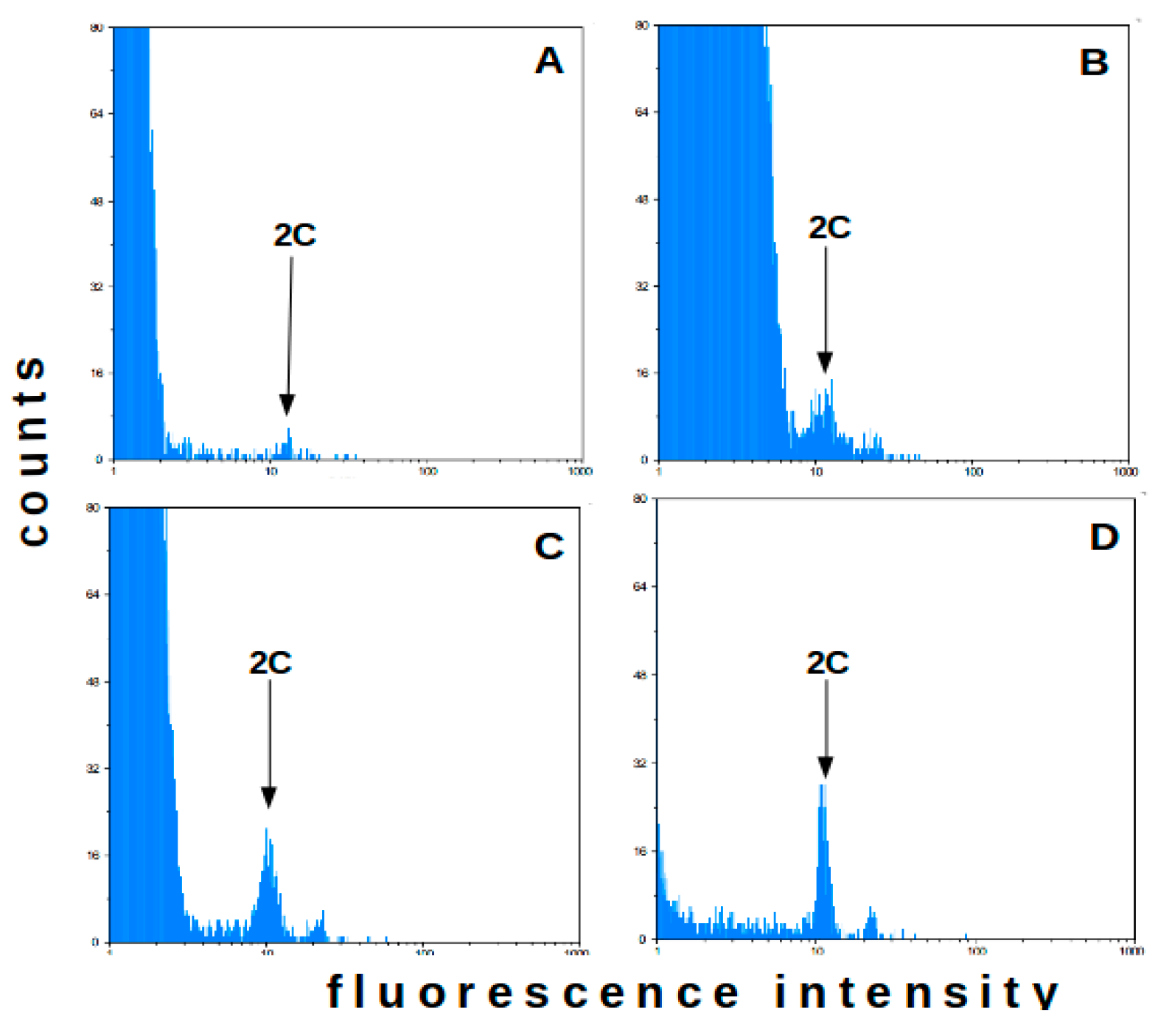
2.1. Flow Cytometry-Method Adaptation
2.2. Flow Cytometry—Analysis of Field Plants
2.3. Illumina Sequencing
2.4. K-Mer Analysis
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. Flow Cytometry Analyses
4.3. DNA Extraction, PCR Amplification and Sequencing
4.4. K-Mer Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
References
- Brink, C.; Postma, A.; Jacobs, K. Rhizobial diversity and function in rooibos (Aspalathus linearis) and honeybush (Cyclopia spp.) plants: A review. S. Afr. J. Bot. 2017, 110, 80–86. [Google Scholar] [CrossRef]
- Malgas, R.; Potts, A.J.; Oettlé, N.M.; Koelle, B.; Todd, S.W.; Verboom, G.A.; Hoffman, M.T. Distribution quantitative morphological variation and preliminary molecular analysis of different growth forms of wild rooibos (Aspalathus linearis) in the northern Cederberg and on the Bokkeveld Plateau. S. Afr. J. Bot. 2010, 76, 72–81. [Google Scholar] [CrossRef]
- Biénabe, E.; Marie-Vivien, D. Institutionalizing Geographical Indications in Southern Countries: Lessons Learned from Basmati and Rooibos. World Dev. 2017, 98, 58–67. [Google Scholar] [CrossRef]
- Street, R.A.; Prinsloo, G. Commercially Important Medicinal Plants of South Africa: A Review. J. Chem. 2013, 2013, 1–16. [Google Scholar] [CrossRef]
- Muller, C.; Malherbe, C.; Chellan, N.; Yagasaki, K.; Miura, Y.; Joubert, E. Potential of rooibos, its major C-glucosyl flavonoids, and Z-2-(β-D-glucopyranosyloxy)-3-phenylpropenoic acid in prevention of metabolic syndrome. Crit. Rev. Food Sci. Nutr. 2018, 58, 227–246. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, H.-J.; Malgas, R.; Biénabe, E. Ecotypes of wild rooibos (Aspalathus linearis (Burm. F) Dahlg., Fabaceae) are ecologically distinct. S. Afr. J. Bot. 2011, 77, 360–370. [Google Scholar] [CrossRef]
- Lötter, D.; Maitre, D. Modelling the distribution of Aspalathus linearis (Rooibos tea): Implications of climate change for livelihoods dependent on both cultivation and harvesting from the wild. Ecol. Evol. 2014, 4, 1209–1221. [Google Scholar] [CrossRef]
- Bester, C.; Joubert, M.E.; Joubert, E. A breeding strategy for South African indigenous herbal teas. Acta Hortic. 2016, 15–22. [Google Scholar] [CrossRef]
- van Heerden, F.R.; van Wyk, B.-E.; Viljoen, A.M.; Steenkamp, P.A. Phenolic variation in wild populations of Aspalathus linearis (rooibos tea). Biochem. Syst. Ecol. 2003, 31, 885–895. [Google Scholar] [CrossRef]
- van Wyk, B.-E.; Gorelik, B. The history and ethnobotany of Cape herbal teas. S. Afr. J. Bot. 2017, 110, 18–38. [Google Scholar] [CrossRef]
- van der Bank, M.; van der Bank, F.H.; van Wyk, B.-E. Evolution of sprouting versus seeding in Aspalathus linearis. Plant Syst. Evol. 1999, 219, 27–38. [Google Scholar] [CrossRef]
- Stander, M.A.; Van Wyk, B.-E.; Taylor, M.J.C.; Long, H.S. Analysis of Phenolic Compounds in Rooibos Tea (Aspalathus linearis) with a Comparison of Flavonoid-Based Compounds in Natural Populations of Plants from Different Regions. J. Agric. Food Chem. 2017, 65, 10270–10281. [Google Scholar] [CrossRef] [PubMed]
- Rolf, D.; Karlsson, T.; Lassen, P. Studies on the flora of the Balearic Islands. I. Chromosome numbers in Balearic angiosperms. Bot. Not. 1971, 6, 249–269. [Google Scholar]
- Dolezel, J.; Bartos, J. Plant DNA flow cytometry and estimation of nuclear genome size. Ann. Bot. 2005, 95, 99–110. [Google Scholar] [CrossRef] [Green Version]
- Mapleson, D.; Garcia Accinelli, G.; Kettleborough, G.; Wright, J.; Clavijo, B.J. KAT: A K-mer analysis toolkit to quality control NGS datasets and genome assemblies. Bioinformatics 2017, 33, 574–576. [Google Scholar] [CrossRef]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bushnell, B. BBTools: A Suite of Fast, Multithreaded Bioinformatics Tools Designed for Analysis of DNA and RNA Sequence Data; Joint Genome Institute: Walnut Creek, CA, USA, 2018. [Google Scholar]
- Vurture, G.W.; Sedlazeck, F.J.; Nattestad, M.; Underwood, C.J.; Fang, H.; Gurtowski, J.; Schatz, M.C. GenomeScope: Fast reference-free genome profiling from short reads. Bioinformatics 2017, 33, 2202–2204. [Google Scholar] [CrossRef] [Green Version]
- Ranallo-Benavidez, T.R.; Jaron, K.S.; Schatz, M.C. GenomeScope 2.0 and Smudgeplots: Reference-free profiling of polyploid genomes. BioRxiv 2019, 1–16. [Google Scholar] [CrossRef]
- Sun, H.; Ding, J.; Piednoël, M.; Schneeberger, K. findGSE: Estimating genome size variation within human and Arabidopsis using k-mer frequencies. Bioinformatics 2018, 34, 550–557. [Google Scholar] [CrossRef] [Green Version]
- Boatwright, J.S.; leRoux, M.M.; Wink, M.; Morozova, T.; van Wyk, B.-E. Phylogenetic relationships of tribe Crotalarieae (Fabaceae) inferred from DNA sequences and morphology. Syst. Bot. 2008, 33, 752–761. [Google Scholar] [CrossRef]
- Lander, E.S.; Waterman, M.S. Genomic mapping by fingerprinting random clones: A mathematical analysis. Genomics 1988, 2, 231–239. [Google Scholar] [CrossRef]
- Li, X.; Waterman, M.S. Estimating the Repeat Structure and Length of DNA Sequences Using ℓ-Tuples. Genome Res. 2003, 13, 1916–1922. [Google Scholar] [CrossRef] [PubMed]
- Ajuwon, O.R.; Marnewick, J.L.; Davids, L.M. Rooibos (Aspalathus linearis) and its Major Flavonoids—Potential Against Oxidative Stress-Induced Conditions. In Basic Principles and Clinical Significance of Oxidative Stress; Intech: Rijeka, Croatia, 2015; pp. 171–217. [Google Scholar] [CrossRef] [Green Version]
- Jedrzejczyk, I.; Sliwinska, E. Leaves and Seeds as Materials for Flow Cytometric Estimation of the Genome Size of 11 Rosaceae Woody Species Containing DNA-Staining Inhibitors. J. Bot. 2010, 2010, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Tong, Y.; Zhang, Q.; Gao, L. Genome Size Variation among and within Camellia Species by Using Flow Cytometric Analysis. PLoS ONE 2013, 8, e64981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivakumaran, K.; Amarakoon, S. Bioactivity of fruit teas and tisanes—A review. J. Pharmacogn. Phytochem. 2018, 7, 323–327. [Google Scholar]
- Freitas Neto, M.; Pereira, T.N.S.; Geronimo, I.G.C.; Azevedo, A.O.N.; Ramos, S.R.R.; Pereira, M.G. Coconut genome size determined by flow cytometry: Tall versus Dwarf types. Genet. Mol. Res. 2016, 15, 1–9. [Google Scholar] [CrossRef]
- Motsa, M.M.; Bester, C.; Slabbert, M.M.; Hannweg, K.; Booyse, M. Flow cytometry: A quick method to determine ploidy levels in honeybush (Cyclopia spp.). Genet. Resour. Crop Evol. 2018, 65, 1711–1724. [Google Scholar] [CrossRef]
- Hajdera, I.; Siwinska, D.; Hasterok, R.; Maluszynska, J. Molecular cytogenetic analysis of genome structure in Lupinus angustifolius and Lupinus cosentinii. Theor. Appl. Genet. 2003, 107, 988–996. [Google Scholar] [CrossRef]
- Atri, C.; Banga, S. A protocol for flow cytometric determination of expected chromosome number of Brassica juncea L. introgression lines. J. Oilseed Brassica 2014, 5, 170–174. [Google Scholar]
- Sadhu, A.; Bhrada, S.; Bandyopadhyay, M. Novel nuclei isolation buffer for flow cytometric genome size estimation of Zingiberaceae: A comparison with common isolation buffers. Ann. Bot. 2016, 118, 1057–1170. [Google Scholar] [CrossRef] [Green Version]
- Inceer, H.; Garnatje, T.; Hayırlıoğlu-Ayaz, S.; Pascual-Díaz, J.P.; Vallès, J.; Garcia, S. A genome size and phylogenetic survey of Mediterranean Tripleurospermum and Matricaria (Anthemideae, Asteraceae). PLoS ONE 2018, 13, e0203762. [Google Scholar] [CrossRef] [PubMed]
- Loureiro, J.; Rodriguez, E.; Dolezel, J.; Santos, C. Two new nuclear isolation buffers for plant DNA flow cytometry: A test with 37 species. Ann. Bot. 2007, 100, 875–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bino, R.J.; Lanteri, S.; Verhoeven, H.A.; Kraak, H.L. Flow cytometry determination of nuclear replication stages in seed tissues. Ann. Bot. 1993, 72, 181–187. [Google Scholar] [CrossRef]
- Kolano, B.; Siwinska, D.; Maluzsynska, J. Endopolyploidy patterns during development of Chenopodium quiona. Acta Biol. Cravoc. Bot. 2008, 51, 85–92. [Google Scholar]
- Sliwinska, E.; Zielinska, E.; Jedrzejczyk, I. Are seeds suitable for flow cytometric estimation of plant genome size? Cytom. Part J. Int. Soc. Anal. Cytol. 2005, 64, 72–77. [Google Scholar] [CrossRef]
- de Beer, D.; Miller, N.; Joubert, E. Production of dihydrochalcone-rich green rooibos (Aspalathus linearis) extract taking into account seasonal and batch-to-batch variation in phenolic composition of plant material. S. Afr. J. Bot. 2017, 110, 138–143. [Google Scholar] [CrossRef]
- Stander, E.A.; Williams, W.; Rautenbach, F.; Le Roes-Hill, M.; Mgwatyu, Y.; Marnewick, J.; Hesse, U. Visualization of Aspalathin in Rooibos (Aspalathus linearis) Plant and Herbal Tea Extracts Using Thin-Layer Chromatography. Molecules 2019, 24, 938. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Resende, M.F.R.; Bombarely, A.; Brym, M.; Bassil, E.; Chambers, A.H. Genomics-based diversity analysis of Vanilla species using a Vanilla planifolia draft genome and Genotyping-By-Sequencing. Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef]
- Edwards, R.J.; Tuipulotu, D.E.; Amos, T.G.; O’Meally, D.; Richardson, M.F.; Russell, T.L.; Vallinoto, M.; Carneiro, M.; Ferrand, N.; Wilkins, M.R.; et al. Draft genome assembly of the invasive cane toad, Rhinella marina. GigaScience 2018, 7, 1–13. [Google Scholar] [CrossRef]
- Hedgecock, D.; Gaffney, P.M.; Goulletquer, P.; Guo, X.; Reece, K.; Warr, G.W. The case for sequencing the Pacific oyster genome. J. Shellfish Res. 2005, 24, 429–442. [Google Scholar] [CrossRef] [Green Version]
- Jansen, H.J.; Liem, M.; Jong-Raadsen, S.A.; Dufour, S.; Weltzien, F.-A.; Swinkels, W.; Koelewijn, A.; Palstra, A.P.; Pelster, B.; Spaink, H.P.; et al. Rapid de novo assembly of the European eel genome from nanopore sequencing reads. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doležel, J.; Binarová, P.; Lucretti, S. Analysis of Nuclear DNA content in plant cells by Flow cytometry. Biol. Plant. 1989, 31, 113–120. [Google Scholar] [CrossRef]
- Richards, E.; Riechardt, M.; Rogers, S. Preparation of genomic DNA from plant tissue. In Current Protocols in Molecular Biology; Ausubel, F.M., Brent, R., Kingston, R.E., Moore, D.D., Seidman, J.G., Smith, J.A., Struhl, K., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2003; pp. 203–207. ISBN 047150338X. [Google Scholar]
- Moyo, M.; Amoo, S.O.; Bairu, M.W.; Finnie, J.F.; Van Staden, J. Optimising DNA isolation for medicinal plants. S. Afr. J. Bot. 2008, 74, 771–775. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Skinner, D.Z.; Liang, G.H.; Hulbert, S.H. Phylogenetic analysis of Sorghum and related taxa using internal transcribed spacers of nuclear ribosomal DNA. Theor. Appl. Genet. 1994, 89, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment SoftwareVersion 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGAX: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Florea, L.; Langmead, B. Lighter: Fast and memory-efficient sequencing error correction without counting. Genome Biol. 2014, 15, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Bioinformatics, Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Build Software Better, Together. Available online: https://github.com (accessed on 18 October 2019).
- Hane, J.K.; Ming, Y.; Kamphuis, L.G.; Nelson, M.N.; Garg, G.; Atkins, C.A.; Bayer, P.E.; Bravo, A.; Bringans, S.; Cannon, S.; et al. A comprehensive draft genome sequence for lupin (Lupinus angustifolius), an emerging health food: Insights into plant–microbe interactions and legume evolution. Plant Biotechnol. J. 2017, 15, 318–330. [Google Scholar] [CrossRef]
- Gali, K.K.; Tar’an, B.; Madoui, M.-A.; van der Vossen, E.; van Oeveren, J.; Labadie, K.; Berges, H.; Bendahmane, A.; Lachagari, R.V.B.; Burstin, J.; et al. Development of a Sequence-Based Reference Physical Map of Pea (Pisum sativum L.). Front. Plant Sci. 2019, 10, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Raw Sequencing Data | Quality Processed Sequencing Data | |||
|---|---|---|---|---|
| MiSeq | # read pairs | read length (bp) | # read pairs | read length (bp) |
| Flow cell 1 | 455045310 | 119 | 446988504 | 120 |
| Flow cell 2 | 153531660 | 119 | 150862318 | 120 |
| HiSeq | # read pairs | read length (bp) | # read pairs | read length (bp) |
| Lane 1 | 284728634 | 120 | 278778534 | 120 |
| Lane 2 | 299563658 | 119 | 292564436 | 120 |
| Lane 3 | 311852326 | 119 | 227020170 | 118 |
| Lane 4 | 355362948 | 119 | 347367568 | 120 |
| Lane 5 | 374476482 | 119 | 366378088 | 120 |
| Lane 6 | 321173550 | 119 | 314166814 | 120 |
| K19 | K23 | K27 | K47 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MiSEQ (0.6 Billion read pairs) | Raw | QP | Raw | QP | Raw | QP | Raw | QP | Average | SD |
| GenomeScope v1 (CovMax 1k) | 0.60 | 0.60 | 0.64 | 0.64 | 0.67 | 0.67 | 0.74 | 0.75 | 0.66 | 0.06 |
| GenomeScope v2 (CovMax 1k) | 0.59 | 0.59 | 0.63 | 0.63 | 0.66 | 0.66 | 0.74 | 0.74 | 0.66 | 0.06 |
| GenomeScope v1 (CovMax 10k) | 0.76 | 0.76 | 0.79 | 0.79 | 0.81 | 0.81 | 0.84 | 0.85 | 0.80 | 0.03 |
| GenomeScope v2 (CovMax 10k) | 0.76 | 0.76 | 0.79 | 0.79 | 0.80 | 0.80 | 0.83 | 0.84 | 0.80 | 0.03 |
| GenomeScope v1 (CovMax 900k) | 0.97 | 0.97 | 0.97 | 0.97 | 0.97 | 0.97 | 0.95 | 0.95 | 0.97 | 0.01 |
| GenomeScope v2 (CovMax 900k) | 0.97 | 0.96 | 0.97 | 0.96 | 0.96 | 0.96 | 0.94 | 0.94 | 0.96 | 0.01 |
| FindGSE | 1.05 | 1.06 | 1.08 | 1.06 | 1.08 | 1.09 | 1.13 | 1.11 | 1.08 | 0.03 |
| BBNorm | 1.05 | 1.06 | 1.08 | 1.06 | 1.08 | 1.09 | 1.14 | 1.13 | 1.09 | 0.03 |
| Formula | 1.07 | 1.03 | 0.98 | 0.97 | 1.08 | 1.06 | 1.01 | 1.02 | 1.03 | 0.04 |
| MiSEQ + HiSEQ (1.9 Billion read pairs) | Raw | QP | Raw | QP | Raw | QP | Raw | QP | Average | SD |
| GenomeScope v1 (CovMax 1k) | 0.59 | 0.52 | 0.59 | 0.58 | 0.63 | 0.62 | 0.74 | 0.74 | 0.62 | 0.08 |
| GenomeScope v2 (CovMax 1k) | 0.58 | 0.51 | 0.58 | 0.56 | 0.61 | 0.60 | 0.73 | 0.73 | 0.61 | 0.08 |
| GenomeScope v1 (CovMax 10k) | 0.71 | 0.69 | 0.75 | 0.74 | 0.78 | 0.77 | 0.85 | 0.85 | 0.77 | 0.06 |
| GenomeScope v2 (CovMax 10k) | 0.70 | 0.69 | 0.74 | 0.72 | 0.76 | 0.75 | 0.84 | 0.83 | 0.76 | 0.06 |
| GenomeScope v1 (CovMax 900k) | 1.00 | 0.97 | 1.01 | 0.97 | 1.01 | 0.98 | 1.00 | 0.97 | 0.99 | 0.02 |
| GenomeScope v2 (CovMax 900k) | 1.00 | 0.96 | 0.99 | 0.96 | 0.99 | 0.96 | 0.99 | 0.96 | 0.98 | 0.02 |
| FindGSE | 1.01 | 1.04 | 1.01 | 1.04 | 1.01 | 1.05 | 1.04 | 1.06 | 1.03 | 0.02 |
| BBNorm | 1.07 | 1.04 | 1.08 | 1.04 | 1.08 | 1.05 | 1.11 | 1.07 | 1.07 | 0.02 |
| Formula | 1.07 | 1.02 | 1.06 | 1.01 | 1.06 | 1.02 | 1.00 | 0.97 | 1.03 | 0.03 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mgwatyu, Y.; Stander, A.A.; Ferreira, S.; Williams, W.; Hesse, U. Rooibos (Aspalathus linearis) Genome Size Estimation Using Flow Cytometry and K-Mer Analyses. Plants 2020, 9, 270. https://doi.org/10.3390/plants9020270
Mgwatyu Y, Stander AA, Ferreira S, Williams W, Hesse U. Rooibos (Aspalathus linearis) Genome Size Estimation Using Flow Cytometry and K-Mer Analyses. Plants. 2020; 9(2):270. https://doi.org/10.3390/plants9020270
Chicago/Turabian StyleMgwatyu, Yamkela, Allison Anne Stander, Stephan Ferreira, Wesley Williams, and Uljana Hesse. 2020. "Rooibos (Aspalathus linearis) Genome Size Estimation Using Flow Cytometry and K-Mer Analyses" Plants 9, no. 2: 270. https://doi.org/10.3390/plants9020270
APA StyleMgwatyu, Y., Stander, A. A., Ferreira, S., Williams, W., & Hesse, U. (2020). Rooibos (Aspalathus linearis) Genome Size Estimation Using Flow Cytometry and K-Mer Analyses. Plants, 9(2), 270. https://doi.org/10.3390/plants9020270