Fluorescence-Activated Cell Sorting Using the D-Root Device and Optimization for Scarce and/or Non-Accessible Root Cell Populations

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
3. Protocol: Isolation of Fluorescently Marked Arabidopsis Root Protoplasts from Roots Grown under Darkness Conditions Using D-Root
- Weigh 50 mg of Arabidopsis seeds carrying the desired fluorescent cell type marker. In this case, GFP or mCherry markers were used. Please note that protoplasting and sorting procedure might have effects on gene expression. It is recommended to perform a comparison between whole roots and processed roots to determine these effects under your experimental settings.
- Add 2 mL of 70% bleach solution and mix continuously for 5 min in a laminar flow hood. Pipette off the bleach.
- Wash the seeds five times with sterile water until clean.
- Prepare squared plates (12 × 12 cm) filled with 60 mL of an agar-containing medium. This will render similar thickness to all plates and the aperture between the agar and the comb will be about 2 mm, which is enough to permit the growth of roots from the Arabidopsis seeds. You can find additional information of interest in the original publication [17] and in Video S1: Assembling the D-Root system.
- Place a sterile Nitex mesh (SEFAR Nitex 03-100/44) (10 × 5-mm) into a half Murashige and Skoog basal medium plate. The mesh must be autoclaved before use. We recommend autoclaving the mesh twice, especially for long treatments.
- Plate the seeds onto the mesh at a density of 3 seeds thickness (90–120 seeds per plate) with a Pasteur pipette.
- Introduce the petri dish into the D-Root device and draw a line at the top of the D-Root system. Insert a methacrylate comb into the agar in the line you have previously drawn.
- Close the lid of the plate with micropore tape and store the plates in dark conditions for 3 days at 4 °C to ensure a uniform germination.
- Put the plates inside the D-Root device at the germination chamber with a growth period of 16 h light (107.45 µmol/m²s)/8 h dark at 22 °C. Note the D-Root device and the methacrylate comb (8 mm thick) have to be sterilized. For this sterilization, we used ozone in an airtight container for 30 min.
- Grow the roots along the mesh for 6 days.
- 11.
- Using a surgical blade, make a first cut that covers all the material and harvest it. Subsequently, and depending on the tissue, make an additional 2 or 3 more cuts to increase the contact between the enzymes and tissues. This additional step increases the protoplast extraction yield and enzyme accessibility to inner tissues, such as pericycle or vascular tissues, that otherwise are hardly reached during the digestion process.
- 12.
- Transfer the collected tissue to a small Enlermeyer flask and submerge the pieces into 10 mL protoplasting solution for 1 h and 45 min in an orbital shaker at 100 rpm. Arabidopsis protoplasting solution (1): (10 mL): 400 mM mannitol (0.73 g), 20 mM MES hydrate (39 mg), 20 mM KCl (200 μL of 1 M KCl), 1.25% Cellulase R10 (125 mg), and 0.3% Macerozyme R10 (30 mg) in 10 mL water. Adjust pH to 5.7 with 1 M Tris pH 8. Heat 10 min in 55 °C water bath until solution is clear, cool down to room temperature while stirring. Add 10 mM CaCl2 (100 μL of 1 M CaCl2), 0.1% BSA (10 mg), and 1.79 μL of β-mercapoethanol. Arabidopsis protoplasting solution (2): This solution only differs from solution 1 in the addition of an extra enzyme, Pectolyase Y-23 0.35%. Both solutions should be made fresh, the same day that the protoplasts are prepared. If the material was previously treated with chemicals or hormones, such as auxins, cytokinins, estradiol, etc., the protoplasting solution is recommended to be supplemented with the same chemicals and hormones. This does not affect protoplast extraction and should ensure effectiveness of the treatment.
- 13.
- After 1 h and 45 min of incubation, pipette up and down vigorously using a P1000 micropipette set to 800 μL. This step is critical for extracting scarce and non-accessible root cell populations.
- 14.
- Filter the contents of the Enlermeyer flasks through a 70 μm strainer (Fisher scientific # 352350, Whaltham, MA, USA) into a 50 mL Falcon. For meristematic cell types, it is also recommended to additionally filter the protoplast preparation through a 40 μm strainer (Fisher scientific # 352340, Whaltham, MA, USA) into a 50 mL Falcon tube.
- 15.
- Centrifuge the Falcon tubes at 500× g for 10 min at room temperature.
- 16.
- Aspirate and discard the supernatant except 500 μL. Carefully re-suspend protoplast pellet into the remaining supernatant using a P1000 micropipette.
- 17.
- Use 10 μL of protoplast preparation to check the protoplast integrity under the microscope and appropriate fluorescence settings (e.g., GFP or mCherry setting) before sorting. It is recommended to use a Neubauer counting chamber to avoid protoplast smashing. If the protocol has been performed successfully, individual isolated protoplasts should be observed.
- 18.
- Separate the GFP or Cherry protoplasts by passing them through a FACS (for instance FACS-Aria (Becton Dickinson) or other) fitted with a 100 μm nozzle and a 20 psi sheath pressure at a flow rate of 1000–2000 events per second.
- 19.
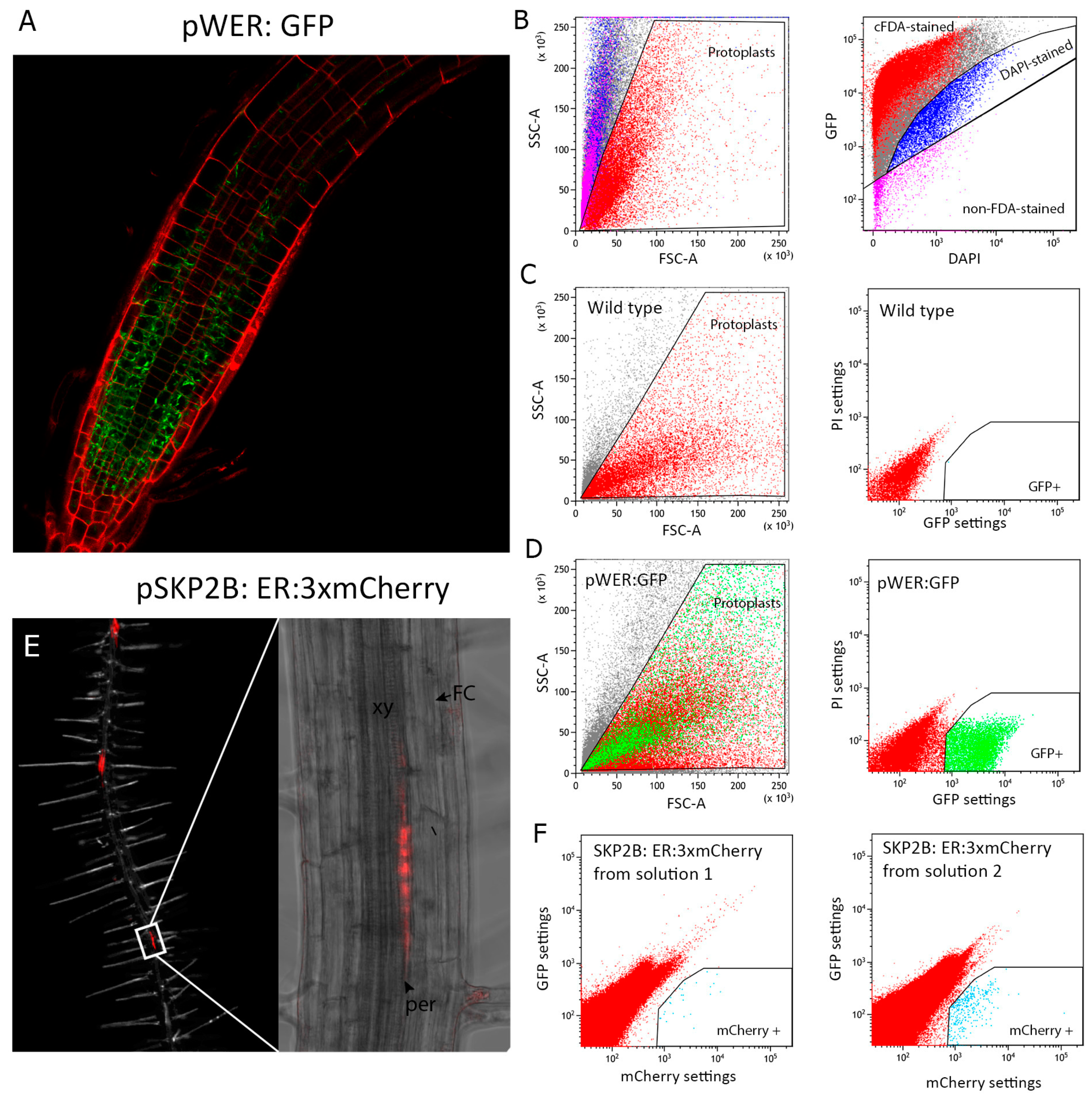
- Depending on the tissue, the protoplasts obtained may have different shape and complexity, which is important when you are defining your population gate for the first time. Create a graph of forward scatter area FSC-A (x-axis) versus side scatter area SSC-A (y-axis) and define your population gate taking into account the following: (1) the area where most the viable protoplasts localize based on the absence of DAPI staining or presence of cFDA, (2) the lowest possible complexity (SSC-A), and (3) where most of the fluorescently-marked protoplasts are located. The ideal gate should fulfill these three criteria while corresponding to a cluster of cells in the graph (Figure 2B–D).
- 20.
- Once your population gate is generated, create a new dot plot, linked to the population gate, of GFP settings (530 nm) versus PI settings (695 nm) of fluorescence for GFP-expressing lines. Set up a new gate (GFP+) using a negative control (protoplast population without markers) to establish the boundaries of an area without protoplasts. When the protoplasts from your GFP line are loaded into the FACS system, positive GFP protoplasts will fall into the gate you previously generated. Note that this plot enables you to differentiate false GFP-positive protoplasts from the positive ones, as autofluorescence will emit in both axis. For lines expressing red fluorescent proteins, plot mCherry settings (610 nm) versus GFP settings (530 nm). We decided to use red in the case of GFP-marker lines or GFP in the case of mCherry lines, but you can use other wave lengths such as blue or ultraviolet to check the autofluorescence.
- 21.
- Using low binding tubes, collect the sorted protoplasts directly into 300 μL of lysis buffer (Qiagen RLT buffer, Germantown, MD, USA) plus 3 μL of ß-mercaptoethanol. Mix well the tube before freezing on dry ice. Although sorted protoplasts may remain in the −80 °C freezer for months, we recommend extracting the RNA as soon as possible.
- 22.
- To isolate total RNA from sorted cells, gently flick the tubes to mix the cells. Note: We performed a successful RNA extraction using 100 positive protoplasts.
- 23.
- Add 300 μL of 70% cold ethanol to the homogenized lysate. Mix well by pipetting.
- 24.
- Apply 600 μL of the sample including any precipitate to a pink RNEasy column (Quiagen, Germantown, MD, USA) sitting in a 2 mL collection tube. Spin at 1000× g for 1 min, then 10,000× g for 30 s. Reapply flow-through and spin at 10,000× g for 30 s. Reuse collection tube in the next step. Note that other extraction methods such as miRVana (Ambion) or quick-RNA plant Kit (Zymo Research) can be used instead.
- 25.
- Pipette 700 μL of buffer RW1 (component of the Quiagen kit ID 74104, Germantown, MD, USA) onto the RNEasy column and centrifuge for 30 s at 10,000× g. Discard flow-through.
- 26.
- Pipette 500 μL buffer RPE (component of the Quiagen kit ID 74104, Germantown, MD, USA) onto the RNEasy column, and centrifuge for 30 s at 10,000× g. Discard flow-through.
- 27.
- Pipette 500 μL buffer 80% ethanol. Spin at 10,000× g for 2 min to dry the RNEasy membrane. Place column in fresh tube.
- 28.
- Centrifuge for 5 min at maximum speed. Put the centrifuge at 18 °C.
- 29.
- Place the RNEasy spin column in a new 1.5 mL collection tube and discard the collection tube with the filtrate. Spin at 17,000× g for 1 min until you see a drop in the bottom of the tube.
- 30.
- Transfer the RNEasy column onto a label (on the top and lateral sides) in a 1.5 mL collection tube and pipette 20 μL of RNAse-free water directly on the RNEasy membrane. Centrifuge for 1 min at 1000× g plus, 1 min 17,000× g to elute. Repeat this step 1 min 17,000× g with the same eluted RNA to increase yield.
- 31.
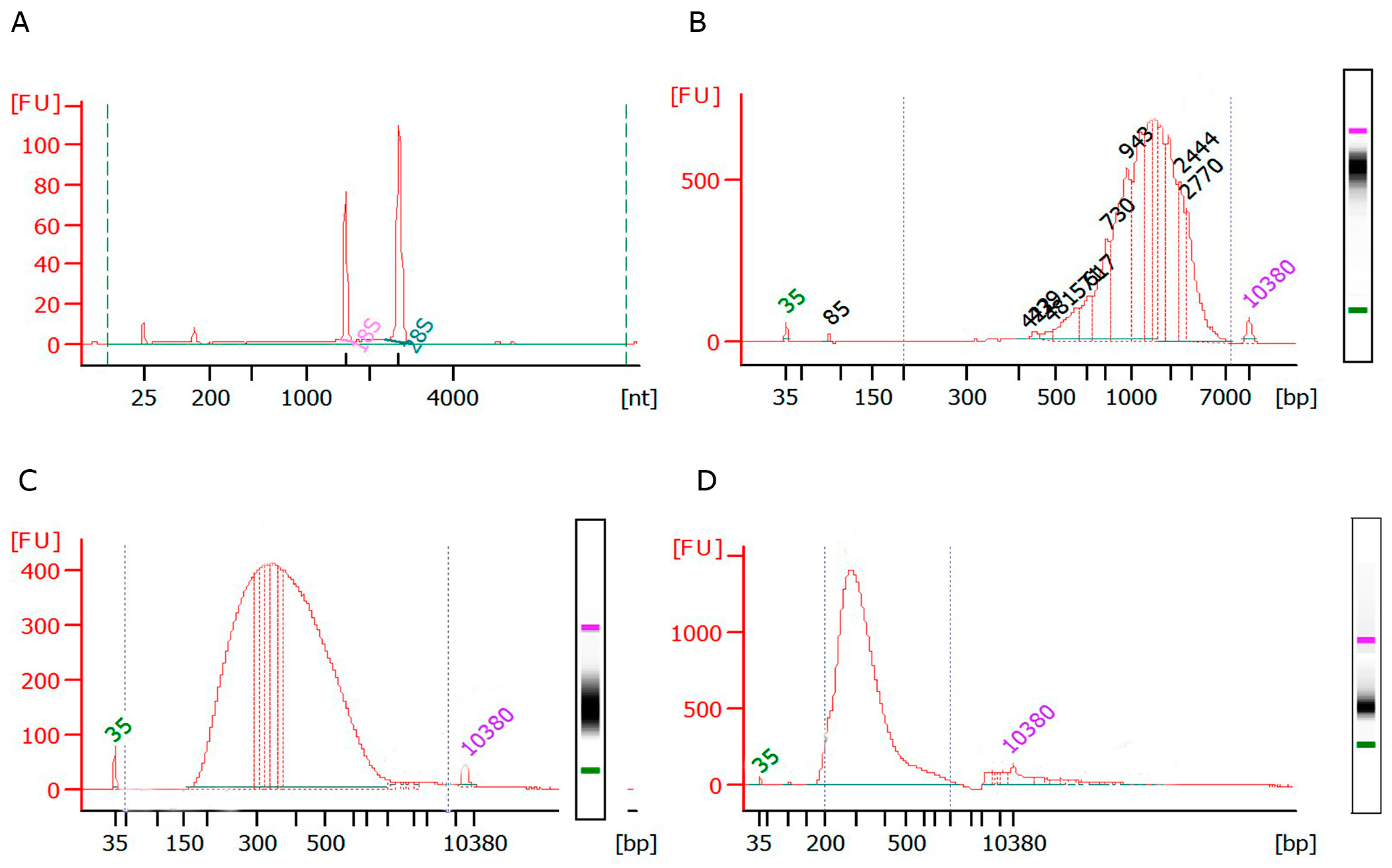
- Determine RNA integrity using the Agilent 6000 RNA Nanokit (Agilent, Santa Clara CA, USA) on an Agilent 2100 Bioanalyzer (Agilent, Santa Clara CA, USA) and Qubit™ RNA Assay Kits (Fisher scientific, Whaltham, MA, USA) on Qubit® 2.0 Fluorometer (Fisher scientific, Whaltham, MA, USA) according to the manufacturer’s protocol.
- 32.
- cDNA synthesis and library preparation can then be performed with available kits such as SMART-Seq v4 Ultra Low Input RNA kit (Takara, Mountain View, CA, USA) and The SMARTer® ThruPLEX® DNA-Seq Kit (Takara) or others.
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Castrillo, G.; Teixeira, P.J.; Paredes, S.H.; Law, T.F.; de Lorenzo, L.; Feltcher, M.E.; Finkel, O.M.; Breakfield, N.W.; Mieczkowski, P.; Jones, C.D.; et al. Root microbiota drive direct integration of phosphate stress and immunity. Nature 2017, 543, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Koevoets, I.T.; Venema, J.H.; Elzenga, J.T.; Testerink, C. Roots Withstanding their Environment: Exploiting Root System Architecture Responses to Abiotic Stress to Improve Crop Tolerance. Front. Plant Sci. 2016, 7, 1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aloni, R.; Aloni, E.; Langhans, M.; Ullrich, C.I. Role of cytokinin and auxin in shaping root architecture: Regulating vascular differentiation, lateral root initiation, root apical dominance and root gravitropism. Ann. Bot. 2006, 97, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Bhosale, R.; Giri, J.; Pandey, B.K.; Giehl, R.F.H.; Hartmann, A.; Traini, R.; Truskina, J.; Leftley, N.; Hanlon, M.; Swarup, K.; et al. A mechanistic framework for auxin dependent Arabidopsis root hair elongation to low external phosphate. Nat. Commun. 2018, 9, 1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Moore, S.; Chen, C.; Lindsey, K. Crosstalk Complexities between Auxin, Cytokinin, and Ethylene in Arabidopsis Root Development: From Experiments to Systems Modeling, and Back Again. Mol. Plant 2017, 10, 1480–1496. [Google Scholar] [CrossRef] [Green Version]
- Himanen, K.; Vuylsteke, M.; Vanneste, S.; Vercruysse, S.; Boucheron, E.; Alard, P.; Chriqui, D.; Van Montagu, M.; Inze, D.; Beeckman, T. Transcript profiling of early lateral root initiation. Proc. Natl. Acad. Sci. USA 2004, 101, 5146–5151. [Google Scholar] [CrossRef] [Green Version]
- Brady, S.M.; Orlando, D.A.; Lee, J.Y.; Wang, J.Y.; Koch, J.; Dinneny, J.R.; Mace, D.; Ohler, U.; Benfey, P.N. A high-resolution root spatiotemporal map reveals dominant expression patterns. Science 2007, 318, 801–806. [Google Scholar] [CrossRef]
- Iyer-Pascuzzi, A.; Simpson, J.; Herrera-Estrella, L.; Benfey, P.N. Functional genomics of root growth and development in Arabidopsis. Curr. Opin. Plant. Biol. 2009, 12, 165–171. [Google Scholar] [CrossRef] [Green Version]
- Silva-Navas, J.; Moreno-Risueno, M.A.; Manzano, C.; Tellez-Robledo, B.; Navarro-Neila, S.; Carrasco, V.; Pollmann, S.; Gallego, F.J.; Del Pozo, J.C. Flavonols Mediate Root Phototropism and Growth through Regulation of Proliferation-to-Differentiation Transition. Plant Cell 2016, 28, 1372–1387. [Google Scholar] [CrossRef] [Green Version]
- Tellez-Robledo, B.; Manzano, C.; Saez, A.; Navarro-Neila, S.; Silva-Navas, J.; de Lorenzo, L.; Gonzalez-Garcia, M.P.; Toribio, R.; Hunt, A.G.; Baigorri, R.; et al. The polyadenylation factor FIP1 is important for plant development and root responses to abiotic stresses. Plant J. Cell Mol. Biol. 2019, 99, 1203–1219. [Google Scholar] [CrossRef]
- Swarup, R.; Parry, G.; Graham, N.; Allen, T.; Bennett, M. Auxin cross-talk: Integration of signalling pathways to control plant development. Plant Mol. Biol. 2002, 49, 411–426. [Google Scholar] [CrossRef] [PubMed]
- Halliday, K.J.; Martinez-Garcia, J.F.; Josse, E.M. Integration of light and auxin signaling. Cold Spring Harb. Perspect. Biol. 2009, 1, a001586. [Google Scholar] [CrossRef] [Green Version]
- Dyachok, J.; Zhu, L.; Liao, F.; He, J.; Huq, E.; Blancaflor, E.B. SCAR mediates light-induced root elongation in Arabidopsis through photoreceptors and proteasomes. Plant Cell 2011, 23, 3610–3626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laxmi, A.; Pan, J.; Morsy, M.; Chen, R. Light plays an essential role in intracellular distribution of auxin efflux carrier PIN2 in Arabidopsis thaliana. PLoS ONE 2008, 3, e1510. [Google Scholar] [CrossRef] [PubMed]
- Sassi, M.; Lu, Y.; Zhang, Y.; Wang, J.; Dhonukshe, P.; Blilou, I.; Dai, M.; Li, J.; Gong, X.; Jaillais, Y.; et al. COP1 mediates the coordination of root and shoot growth by light through modulation of PIN1- and PIN2-dependent auxin transport in Arabidopsis. Development 2012, 139, 3402–3412. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Ding, G.; Yokawa, K.; Baluska, F.; Li, Q.F.; Liu, Y.; Shi, W.; Liang, J.; Zhang, J. An improved agar-plate method for studying root growth and response of Arabidopsis thaliana. Sci. Rep. 2013, 3, 1273. [Google Scholar] [CrossRef] [Green Version]
- Silva-Navas, J.; Moreno-Risueno, M.A.; Manzano, C.; Pallero-Baena, M.; Navarro-Neila, S.; Téllez-Robledo, B.; Garcia-Mina, J.M.; Baigorri, R.; Javier Gallego, F.; del Pozo, J.C. D-Root: A system to cultivate plants with the root in darkness or under different light conditions. Plant J. 2015, 84, 244–255. [Google Scholar] [CrossRef] [Green Version]
- Birnbaum, K.; Shasha, D.E.; Wang, J.Y.; Jung, J.W.; Lambert, G.M.; Galbraith, D.W.; Benfey, P.N. A gene expression map of the Arabidopsis root. Science 2003, 302, 1956–1960. [Google Scholar] [CrossRef] [Green Version]
- Nawy, T.; Lee, J.Y.; Colinas, J.; Wang, J.Y.; Thongrod, S.C.; Malamy, J.E.; Birnbaum, K.; Benfey, P.N. Transcriptional profile of the Arabidopsis root quiescent center. Plant Cell 2005, 17, 1908–1925. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Colinas, J.; Wang, J.Y.; Mace, D.; Ohler, U.; Benfey, P.N. Transcriptional and posttranscriptional regulation of transcription factor expression in Arabidopsis roots. Proc. Natl. Acad. Sci. USA 2006, 103, 6055–6060. [Google Scholar] [CrossRef] [Green Version]
- Levesque, M.P.; Vernoux, T.; Busch, W.; Cui, H.; Wang, J.Y.; Blilou, I.; Hassan, H.; Nakajima, K.; Matsumoto, N.; Lohmann, J.U.; et al. Whole-genome analysis of the SHORT-ROOT developmental pathway in Arabidopsis. PLoS Biol. 2006, 4, e143. [Google Scholar] [CrossRef]
- Clark, N.M.; Fisher, A.P.; Sozzani, R. Identifying Differentially Expressed Genes Using Fluorescence-Activated Cell Sorting (FACS) and RNA Sequencing from Low Input Samples. Methods Mol. Biol. (Clifton N.J.) 2018, 1819, 139–151. [Google Scholar] [CrossRef]
- Redenbaugh, K.; Ruzin, S.; Bartholomew, J.; Bassham, J.A. Characterization and separation of plant protoplasts via flow cytometry and cell sorting. Z. Für Pflanzenphysiol. 1982, 107, 65–80. [Google Scholar] [CrossRef] [Green Version]
- Hammatt, N.; Lister, A.; Blackhall, N.; Gartland, J.; Ghose, T.; Gilmour, D.; Power, J.; Davey, M.; Cocking, E. Selection of plant heterokaryons from diverse origins by flow cytometry. Protoplasma 1990, 154, 34–44. [Google Scholar] [CrossRef]
- Galbraith, D.W.; Lambert, G.M.; Grebenok, R.J.; Sheen, J. Flow cytometric analysis of transgene expression in higher plants: Green-fluorescent protein. Methods Cell Biol. 1995, 50, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Galbraith, D.W. Flow cytometry and sorting in Arabidopsis. Methods Mol. Biol. (Clifton N.J.) 2014, 1062, 509–537. [Google Scholar] [CrossRef]
- Amor, K.B.; Breeuwer, P.; Verbaarschot, P.; Rombouts, F.M.; Akkermans, A.D.; De Vos, W.M.; Abee, T. Multiparametric flow cytometry and cell sorting for the assessment of viable, injured, and dead Bifidobacterium cells during bile salt stress. Appl. Environ. Microbiol. 2002, 68, 5209–5216. [Google Scholar] [CrossRef] [Green Version]
- Guzzo, F.; Cantamessa, K.; Portaluppi, P.; Levi, M. Flow cytometry and sorting of protoplasts from carrot cell cultures reveal two cell subpopulations with different morphogenetic potential. Plant Cell Rep. 2002, 21, 214–219. [Google Scholar]
- Vilarrasa-Blasi, J.; Gonzalez-Garcia, M.P.; Frigola, D.; Fabregas, N.; Alexiou, K.G.; Lopez-Bigas, N.; Rivas, S.; Jauneau, A.; Lohmann, J.U.; Benfey, P.N.; et al. Regulation of plant stem cell quiescence by a brassinosteroid signaling module. Dev. Cell 2014, 30, 36–47. [Google Scholar] [CrossRef] [Green Version]
- Dinneny, J.R.; Long, T.A.; Wang, J.Y.; Jung, J.W.; Mace, D.; Pointer, S.; Barron, C.; Brady, S.M.; Schiefelbein, J.; Benfey, P.N. Cell identity mediates the response of Arabidopsis roots to abiotic stress. Science 2008, 320, 942–945. [Google Scholar] [CrossRef] [Green Version]
- Bargmann, B.O.; Birnbaum, K.D. Fluorescence activated cell sorting of plant protoplasts. J. Vis. Exp. Jove 2010. [Google Scholar] [CrossRef] [PubMed]
- Laplaze, L.; Parizot, B.; Baker, A.; Ricaud, L.; Martiniere, A.; Auguy, F.; Franche, C.; Nussaume, L.; Bogusz, D.; Haseloff, J. GAL4-GFP enhancer trap lines for genetic manipulation of lateral root development in Arabidopsis thaliana. J. Exp. Bot. 2005, 56, 2433–2442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramakrishna, P.; Ruiz Duarte, P.; Rance, G.A.; Schubert, M.; Vordermaier, V.; Vu, L.D.; Murphy, E.; Vilches Barro, A.; Swarup, K.; Moirangthem, K.; et al. EXPANSIN A1-mediated radial swelling of pericycle cells positions anticlinal cell divisions during lateral root initiation. Proc. Natl. Acad. Sci. USA 2019, 116, 8597–8602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naseer, S.; Lee, Y.; Lapierre, C.; Franke, R.; Nawrath, C.; Geldner, N. Casparian strip diffusion barrier in Arabidopsis is made of a lignin polymer without suberin. Proc. Natl. Acad. Sci. USA 2012, 109, 10101–10106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzano, C.; Ramirez-Parra, E.; Casimiro, I.; Otero, S.; Desvoyes, B.; De Rybel, B.; Beeckman, T.; Casero, P.; Gutierrez, C.; Del Pozo, J.C. Auxin and epigenetic regulation of SKP2B, an F-box that represses lateral root formation. Plant Physiol. 2012, 160, 749–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva-Navas, J.; Conesa, C.M.; Saez, A.; Navarro-Neila, S.; Garcia-Mina, J.M.; Zamarreno, A.M.; Baigorri, R.; Swarup, R.; Del Pozo, J.C. Role of cis-zeatin in root responses to phosphate starvation. New Phytol 2019, 224, 242–257. [Google Scholar] [CrossRef] [PubMed]
- Perez-Garcia, P.; Moreno-Risueno, M.A. Stem cells and plant regeneration. Dev. Biol. 2018, 442, 3–12. [Google Scholar] [CrossRef]
- De Smet, I.; Vanneste, S.; Inze, D.; Beeckman, T. Lateral root initiation or the birth of a new meristem. Plant Mol. Biol. 2006, 60, 871–887. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-García, M.-P.; Bustillo-Avendaño, E.; Sanchez-Corrionero, A.; del Pozo, J.C.; Moreno-Risueno, M.A. Fluorescence-Activated Cell Sorting Using the D-Root Device and Optimization for Scarce and/or Non-Accessible Root Cell Populations. Plants 2020, 9, 499. https://doi.org/10.3390/plants9040499
González-García M-P, Bustillo-Avendaño E, Sanchez-Corrionero A, del Pozo JC, Moreno-Risueno MA. Fluorescence-Activated Cell Sorting Using the D-Root Device and Optimization for Scarce and/or Non-Accessible Root Cell Populations. Plants. 2020; 9(4):499. https://doi.org/10.3390/plants9040499
Chicago/Turabian StyleGonzález-García, Mary-Paz, Estéfano Bustillo-Avendaño, Alvaro Sanchez-Corrionero, Juan C. del Pozo, and Miguel A. Moreno-Risueno. 2020. "Fluorescence-Activated Cell Sorting Using the D-Root Device and Optimization for Scarce and/or Non-Accessible Root Cell Populations" Plants 9, no. 4: 499. https://doi.org/10.3390/plants9040499